

Creutzfeldt-Jakob disease (CJD) is a rare and invariably fatal neurologic disorder caused by an abnormal prion protein. Perceptual disturbances, including visual and auditory agnosias, have been reported in the course of CJD but are not typically the initial complaint. We report a case of hyperacusis as the initial presentation of sporadic CJD.
Case.
A 52-year-old man with diabetes presented with 1 month of hyperacusis and trouble discerning noises, including speech and music. He reported that all sounds, both complex and simple, seemed “jarring” and unfamiliar. He denied tinnitus, falls, or imbalance. Over the ensuing 3 months he developed diffuse myalgias and neck pain.
Medical and neurologic examination was normal as was audiometry. Laboratory investigations including erythrocyte sedimentation rate, C-reactive protein, antinuclear antibody, anti-Ro, anti-La, anti-Jo, creatine kinase, aldolase, and thyroid function testing were normal. An HIV ELISA test was negative. Brain MRI demonstrated abnormal T2 hyperintensity and gyriform restricted diffusion with corresponding signal on apparent diffusion coefficient images in the bilateral parieto-occipital regions (figure, A). CSF analysis demonstrated white blood cell count 0, protein 60 mg/dL, and no oligoclonal bands. CSF was negative for Lyme antibodies and herpes simplex virus PCR. CSF 14-3-3 protein was negative and the total tau level was normal at 631 pg/mL. Ten months after the onset of hyperacusis, he developed hypophonia, dysarthria, and ataxia. Two months later, examination revealed psychomotor slowing and delayed recall, alexia, agraphia, and difficulty with walking. He retained the ability to discuss current events but could no longer work.
Figure. Brain MRI and brain pathology in Creutzfeldt-Jakob disease.
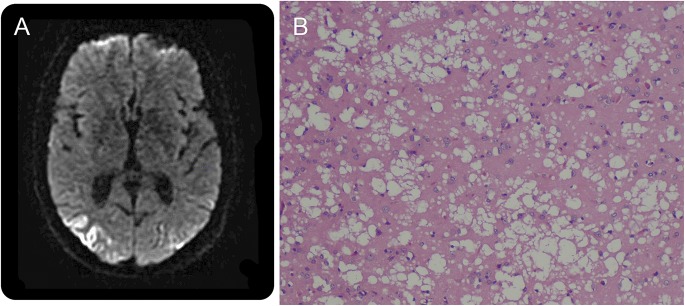
Axial diffusion-weighted image sequence (A) shows abnormal gyriform restricted diffusion in the bilateral parieto-occipital lobes. Cortical brain biopsy (B) shows gray matter with diffuse spongiform changes, neuronal dropout, and gliosis.
A brain biopsy 13 months into his course revealed spongiform changes predominantly affecting the gray matter with marked neuronal dropout and associated gliosis (figure, B). There was no evidence of inflammation, demyelination, vasculitis, or amyloid deposition. The patient died 21 months after initial symptom onset. Immunochemistry performed at the National Prion Surveillance Center at Case Western Reserve University confirmed the presence of the abnormal prion protein and the diagnosis of CJD with the MM2 molecular subtype.
Discussion.
CJD is the most prevalent human prion disease and the sporadic form accounts for about 85% of all cases.1 According to the prion hypothesis, progressively spreading misfolding of this protein from the normal alpha helix to the pathogenic beta pleated sheet causes neuronal damage and spongiform changes within the brain.
Sporadic CJD occurs worldwide with an estimated annual incidence of 1 case per million people.1 There is considerable variability in the presenting symptoms of CJD, but typical complaints include behavioral changes, memory impairment, visual loss, aphasia, motor deficits, ataxia, or depression.1 The disease progresses rapidly with prominent cognitive decline, the development of myoclonus, and gait impairment. The median time to death from symptom onset is 5 months and 90% of patients with sporadic CJD die within 1 year.1
Although auditory symptoms are unusual in patients with CJD, there are a few case reports of hearing loss as the initial complaint in CJD.2,3 In most cases, the hearing loss was thought to be due to cortical deafness from damage to the bilateral temporal cortices including the primary auditory cortex on bilateral transverse gyri of Heschl, auditory radiation, and insulae. Hyperacusis is typically defined as discomfort to noises below 90 dB on audiometry, which our patient did not exhibit. Instead, he complained that all sounds were jarring and unfamiliar to him, thus suggesting that his sense of hyperacusis may also have been a manifestation of a central auditory processing disorder.
Diagnostic tests, including CSF analysis, EEG, and MRI, are useful in supporting the diagnosis of CJD, but none are 100% sensitive or specific. MRI characteristically shows abnormal increased signal on T2-weighted images in the caudate, putamen, and involved cortex with accompanying restricted diffusion on diffusion-weighted imaging.4 EEG may demonstrate periodic sharp wave complexes.5 Routine CSF studies are typically normal, but CSF 14-3-3 protein and total tau are often elevated.6
The definitive diagnosis of CJD requires histologic examination of the brain and immunostaining for the prion protein. The crucial features are spongiform change accompanied by neuronal loss and gliosis. On autopsy, this patient had the MM2 molecular subtype, which accounts for less than 10% of all cases of sporadic CJD and is often associated with longer disease duration (average 16 months).7 The clinical presentation of the MM2 molecular subtype is dominated by cognitive impairment, aphasia, and pyramidal signs.7 The 14-3-3 protein is often positive, but, as seen in our patient, no single test is absolutely conclusive.7
In summary, CJD is a rare and fatal disorder caused by an abnormal prion protein. This case highlights the wide spectrum of initial symptoms in patients with spongiform encephalopathy, including auditory perceptual disturbances such as hyperacusis.
Footnotes
Author contributions: Alexander E. Merkler conceived and designed the study and drafted and revised the manuscript for intellectual content. Mukesh Prasad designed the study and revised the article critically for important intellectual content. Ehud Lavi revised the article critically for important intellectual content. Jospeh Safdieh designed the study and revised the article critically for important intellectual content.
Study funding: No targeted funding reported.
Disclosure: A. Merkler and M. Prasad report no disclosures. E. Levi is on the editorial board for Journal of Neurovirology, has consulted as a medico-legal expert, and has been an expert witness for defense and plaintiff. J. Safdieh has received publishing royalties from Elsevier and has done consulting work for legal proceedings. Go to Neurology.org/nn for full disclosures. The Article Processing Charge was paid by the authors.
References
- 1.Johnson RT. Prion diseases. Lancet Neurol 2005;4:635–642 [DOI] [PubMed] [Google Scholar]
- 2.Orimo S, Ozawa E, Uematsu M, et al. A case of Creutzfeldt-Jakob disease presenting with auditory agnosia as an initial manifestation. Eur Neurol 2000;44:256–258 [DOI] [PubMed] [Google Scholar]
- 3.Krishna P, Bauer C. Hearing loss as the initial presentation of Creutzfeldt-Jakob disease. Ear Nose Throat J 2004;83:535, 538, 540 passim [PubMed] [Google Scholar]
- 4.Tschampa HJ, Zerr I, Urbach H. Radiological assessment of Creutzfeldt-Jakob disease. Eur Radiol 2007;17:1200–1211 [DOI] [PubMed] [Google Scholar]
- 5.Steinhoff BJ, Räcker S, Herrendorf G, et al. Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch Neurol 1996;53:162–166 [DOI] [PubMed] [Google Scholar]
- 6.Hsich G, Kenney K, Gibbs CJ, Lee KH, Harrington MG. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med 1996;335:924–930 [DOI] [PubMed] [Google Scholar]
- 7.Krasnianski A, Meissner B, Schulz-Schaeffer W, et al. Clinical features and diagnosis of the MM2 cortical subtype of sporadic Creutzfeldt-Jakob disease. Arch Neurol 2006;63:876–880 [DOI] [PubMed] [Google Scholar]