

Abstract
The pathogenesis of non-alcoholic steatohepatitis (NASH) is not fully understood. In the present study, both in vitro and in vivo vimentin expression and secretion in NASH were investigated. The exposure of palmitate and lipopolysaccharide (LPS) to HepG2 cells enhanced caspase-3 activity and vimentin expression, respectively. The combined effects of both treatments on vimentin expression and caspase-3 activation appeared to be synergic. In contrast, blockade of caspase-3 activity by zVADfmk resulted in a significant reduction of cleaved vimentin and secreted vimentin into the culture supernatant. Similarly, lipid accumulation and inflammation occurred in mice fed a methionine-choline-deficient diet; thus, vimentin expression and serum cleaved vimentin levels were increased. However, vimentin was not significantly upregulated, and no cleavage occurred in mice fed a high-fat diet. It was conclusively determined that lipid accumulation in hepatocytes induces apoptosis through a caspase-3 dependent pathway; whereas, LPS stimulates vimentin expression, leading to its cleavage and secretion. Increased vimentin fragment levels indicated the existence of substantial hepatocellular death via an apoptotic mechanism. [BMB Reports 2014; 47(8): 457-462]
Keywords: Vimentin, Caspase-3, Secretion, NASH, MCD diet
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) often refers to liver damage, ranging from simple hepatic steatosis, to non-alcoholic steatohepatitis (NASH). Simple hepatic steatosis usually presents a benign and non-progressive condition. Conversely, NASH is characterized by inflammation, and may progress to cirrhosis (1). The mechanisms of progression of a simple fatty liver to NASH are not yet fully understood; however, a two-hit progression has been proposed. Specifically, the first hit is considered to be accumulation of fat in hepatocytes; while the second hit consists of a potent stimulus, such as lipid toxicity, mitochondrial dysfunction, adipocytokines, pro-inflammatory cytokines (e.g. TNF α), or oxidative stress and subsequent lipid peroxidation (2, 3). The second hit may lead to hepatocyte injury, inflammation, fibrosis, and cellular damage, characteristic of NASH (4). It should be noted that exposure to lipopolysaccharides (LPS) can also act as a second hit, resulting in progressive liver injury.
Emerging data has suggested that apoptosis of liver cells plays an important role in liver injury and disease progression in NAFLD (5). Recent evidence suggests that intracellular accumulation of saturated free fatty acids (FFA) is associated with cellular dysfunction, in a condition known as lipotoxicity. Diverse mechanisms have been proposed to explain lipotoxicity, including induction of a proinflammatory state, and apoptotic cell death. An essential consequence of the apoptotic process is activation of the effector caspase. Caspases are activated during apoptosis, after which the proteases cleave certain critical cellular proteins, leading to functional modifications, or dramatic morphological changes in cell structure.
Several intermediate filament proteins (IF) are known to be cleaved by specific proteases in the liver; however, few studies have investigated the role of IF in apoptosis (6, 7). IF proteins are classified into several groups, according to their molecular structure. In epithelial cells, the most common type I and type II cytokeratins (CK) are CK-18 and 19, and CK-8, respectively. Vimentin is one of the most widely expressed type III IF proteins, and is the only cytoplasmic IF found in lymphocytes, neutrophils, endothelial cells, and fibroblasts (8). In recent years, vimentin has been considered a predominant IF protein in the cytoplasm of mesenchymal cells; thus, this protein has been identified as a mesenchymal marker of epithelial-mesenchymal transition (9). Moreover, vimentin has been found in various cancers, including human breast cancer, and hepatocellular carcinoma. Caspase-mediated degradation of keratins and vimentin takes place during apoptosis. The apoptotic cleavage of CK-18 and 19 occurs through caspases, at conserved aspartate residues. Similarly, vimentin is cleaved in vitro and during apoptosis by multiple caspases at distinct sites, including Asp85 by caspase-3 and caspase-7, and Asp259 by caspase-6 (10). The proteolysis of vimentin promotes apoptosis, by dismantling IF, and amplifying the cell death signal.
Here, the involvement of vimentin in the progression of NASH was explored in human hepatocarcinoma cells (HepG2) and mice that were fed a high-fat diet (HFD) or methionine-choline-deficient (MCD) diet. The results demonstrated that apoptosis by lipid accumulation and the LPS pathway stimulates vimentin expression in NASH, suggesting that vimentin can be used as a marker for NASH.
RESULTS
Lipid accumulation was associated with caspase-3 activity in HepG2 cells
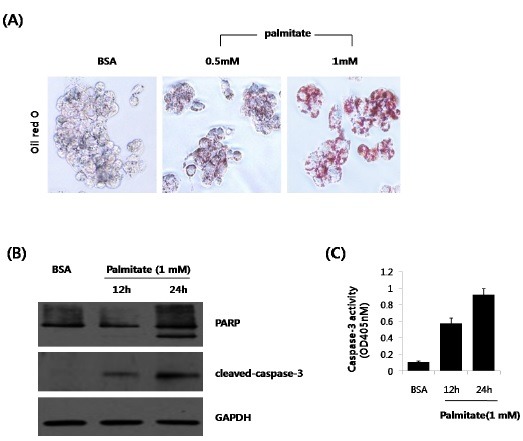
Intracellular accumulation of lipids is a hallmark of hepatosteatosis, which may lead to lipotoxicity. Thus, we explored whether lipid accumulation participates in the apoptosis process, using HepG2 cells. Briefly, HepG2 cells were treated with palmitate, and cellular steatosis was confirmed by oil red-O staining (Fig. 1A). The results showed a dose-dependent increase in intracellular lipid vacuoles, in response to palmitate. Procaspase-3 is commonly cleaved to active caspase-3, during apoptosis. The activation of caspases, which are key mediators of apoptosis, was analyzed upon exposure of the HepG2 cells to palmitate. Caspase-3 activity was measured in cells exposed to 1mM palmitate for 12 or 24 h. The levels of caspase activation in HepG2 cells exposed to palmitate were then compared to those of 1% BSA treated cells. Western blotting showed that palmitate treatment markedly increased caspase-3 activity in a time-dependent manner (Fig. 1B). Moreover, palmitate induced PARP cleavage, as indicated by the diminution of its parent band (116 kDa), and the concomitant accumulation of its cleavage fragment (89 kDa). Treatment with fatty acids gradually increased the amount of cleaved fragments in a time-dependent manner. These findings were confirmed using a spectrophotometer (Fig. 1C). Based on these results, we specified the contribution of lipid accumulation to caspase-3 activity.
Fig. 1. Effect of palmitate treatment on intracellular lipid accumulation and apoptosis. (A) Human hepatocarcinoma HepG2 cells were incubated with increasing palmitate concentrations (0.5 mM and 1 mM), for 24 h. Cells treated with 1% (w/v) FFA-free BSA served as controls. Lipid droplet accumulation was visualized by Oil-Red-O staining (Original magnification ×200). (B) Time course of PARP and cleaved caspase-3 levels, after treatment with 1 mM of palmitate, as assessed by Western blot analysis. (C) Levels of cleaved caspase-3 activity, as assessed by ELISA assay. The densitometric analyses from n = 3/group are presented; *P < 0.05.
LPS stimulated vimentin expression and its cleavage in HepG2 cells
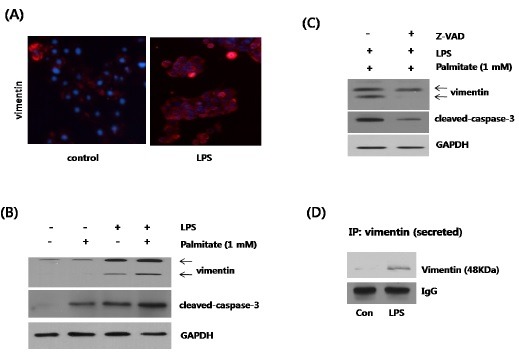
Apoptosis is characterized by caspase activation, and subsequent cleavage of cellular proteins essential for cell viability. Immunofluorescence analysis of LPS-treated hepG2 cells showed an increase in vimentin expression, when compared to the control cells (Fig. 2A). We then used Western blot analysis to evaluate the effects of LPS and palmitate on vimentin expression and caspase-3 activity (Fig. 2B). Treatment with palmitate induced caspase activation, but had no effect on vimentin expression. However, LPS significantly increased the cleaved caspase-3 levels, as well as the vimentin expression. Vimentin cleavage commonly occurs through activation of the caspase pathway. Thus, a significant increase in vimentin fragments was also detected in the LPS treatment lane. The intact vimentin band appeared at a molecular weight of about 57 kDa; whereas, the fragment band was detected at 48 kDa. It should be noted that the caspase activity was positively correlated with the levels of vimentin fragments. In the presence of both LPS and palmitate, the levels of the cleaved caspase-3 and vimentin were dramatically increased. To evaluate whether LPS-induced vimentin fragmentation was the result of caspase cleavage, we pretreated the samples with zVADfmk, a broad-spectrum caspase-inhibitor., In conjunction with the levels of cleaved caspase-3, zVADfmk pretreatment resulted in a dramatic reduction of vimentin cleavage (Fig. 2C). Moreover, a vimentin specific antibody was used to immunoprecipitate the vimentin secreted from the culture supernatant of HepG2 cells (Fig. 2D). Treatment with LPS resulted in a significant increase in secreted fragments (about 48 kDa) in the cell culture media.
Fig. 2. Up-regulation of vimentin expression by LPS treatment in HepG2 cells. (A) Cells were treated either with or without 1 ng LPS, for 24 h. Immunofluorescence staining shows the intensity of vimentin in red; whereas, blue is nuclear counterstaining, using DAPI. (B) To assess the role of LPS and palmitate in vimentin regulation, HepG2 cells were treated with either 1 ng LPS, 1 mM palmitate, or both. The levels of vimentin and cleaved caspase-3 were then measured by Western blot. Cells were treated with either 1% (w/v) FFA-free BSA or 1 mM palmitate for 24 h, either with or without 1 ng LPS for 24 h. GAPDH blot served as a loading control. (C) Cells were treated with 1 mM palmitate and 1 ng LPS for 24 h, either with or without pretreatment with the caspase-3 inhibitor, zVADfmk (10 μM). Vimentin and cleaved caspase-3 were assessed by Western blot. (D) HepG2 cells were treated with 1 mM palmitate for 24 h, either with or without 1 ng LPS. The culture media were then collected, and the levels of secreted vimentin were measured by immunoprecipitation.
The increase of vimentin expression during hepatic steatosis progression to NASH in MCD mice
Next, we evaluated the effects of lipid accumulation on hepatic vimentin expression and caspase-dependent apoptosis, in HFD fed mice and LPS-injected MCD mice. To induce hepatic steatosis, C57BL/6 mice were fed HFD from 4 to 16 weeks. At 4 weeks after HFD treatment, some hepatic parenchymal cells were filled with vesicular fat (data not shown); but at 16 weeks, most hepatic cells had multivesicular fat. In addition, we initially fed C57BL/6 mice a MCD diet for 3 weeks, to investigate the role of caspase-3 activation in the pathogenesis of NASH. The mice were then injected intraperitoneally with a single dose of 2.5 mg/kg LPS. LPS further amplified the pathological changes observed in mice fed the MCD diet, which may be associated with progressive steatohepatitis, and be pathologically similar to severe human steatohepatitis.
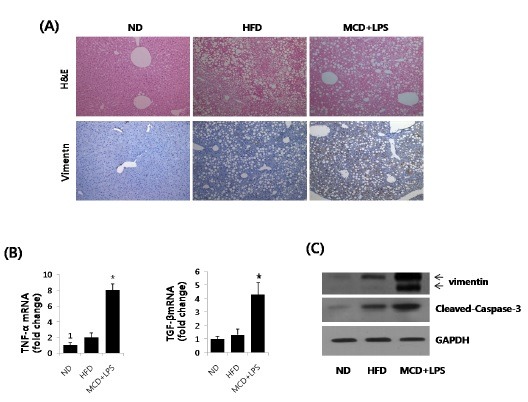
For closer observation of fatty liver development, histological sections were stained with H&E (Fig. 3A). We observed significant hepatic fat accumulation in HFD and MCD mice. Immunohistochemistry analyses showed that the number of vimentin positive cells significantly increased in LPS-injected MCD mice, when compared to ND mice and HFD mice. To define the effects of HFD and LPS-stimulated MCD on the progression of hepatitis, hepatic messenger RNA (mRNA) expression levels of inflammatory genes TNF-α and TGF β were analyzed by qRT-PCR (Fig. 3B). The levels of TNFα and TGFβ1 significantly increased in the LPS-injected MCD mice, when compared to the ND or HFD mice. Recent studies showed that stimulation of the TGFβ1 dependent signaling pathway in hepatocytes upon injury was closely associated with the progression of steatohepatitis (12). Hepatic tissue extracts were analyzed by Western blot, to define the effects of lipid or LPS treatment on hepatic vimentin expression and apoptosis (Fig. 3C). HFD resulted in an increase in the circulating levels of hepatic FFA and triglycerides, as well as the levels of active caspase-3. In contrast, vimentin expression was only slightly increased, even at 16 weeks, and the vimentin cleavage was hardly detectable. However, in LPS injected mice that received the MCD diet, the amount of vimentin cleaved by caspase-3 dramatically increased. These findings suggest that the level of caspase-generated vimentin fragments was positively correlated with the progression of inflammation.
Fig. 3. Hepatic cellular accumulation of vimentin and active caspase-3 in hepatosteatosis progression to steatohepatitis. (A) Mice were provided a normal diet (ND) or a high fat diet (HFD) for 16 weeks, and MCD for 3 weeks. MCD mice were subsequently injected with 2.5 mg/kg LPS. The liver sections were then analyzed with H&E staining (upper panel), and immunohistochemistry staining for vimentin (lower panel). (B) Hepatic levels of TNF-α and TGF-β mRNA were measured by qRT-PCR in ND, HFD, and LPS-injected MCD mice. Genes were normalized to18S rRNA as an internal standard, and data shown as the fold increase. Data plots represent the mean ± S.D. of three independent experiments. Asterisks indicate statistical significance (*P < 0.05). (C) Western blots of vimentin and cleaved caspase-3 in ND, HFD, or LPS-injected MCD diet fed mice. Liver vimentin and cleaved caspase-3 levels were quantified by Western blot, using an equal amount of total liver proteins. Expression levels were normalized, relative to GAPDH. Liver extracts from a pool of three mice in each group were analyzed by Western blot, using specific antibodies.
Caspase-cleaved vimentin fragment was secreted in NASH
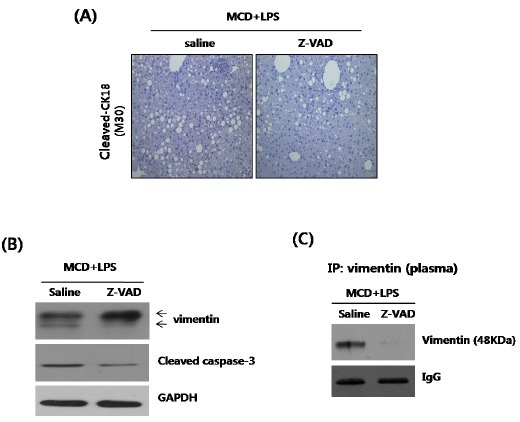
The levels of the cytokeratin-18 (CK-18) fragments are closely related to the extent of caspase activity. Indeed, the levels of caspase-cleaved CK-18 fragments have been found to increase in patients with NASH, when compared to those with simple steatosis (13). zVADfmk was injected into LPS-treated MCD mice, to test whether or not caspase-3 activation is implicated in hepatic apoptosis (Fig. 4A). Biotinylated mAb M30 staining showed that apoptosis was induced in the LPS-injected MCD diet mice; whereas, the zVADfmk preinjected mice showed a significant reduction in hepatic apoptosis. To confirm these findings, Western blot analyses were performed, to compare the expression of vimentin and cleaved caspase-3 in LPS-injected MCD diet mice (Fig. 4B). The results showed that LPS-treated MCD diet mice injected with saline had markedly increased caspase-3 levels. In contrast, pretreatment with zVADfmk led to a significant decrease in the levels of cleaved caspase-3 and vimentin fragment. The correlation between the levels of caspase and vimentin fragments was similar to that observed in HepG2 cells. In addition, an immunoprecipitation assay was performed, using the plasma from LPS-treated MCD diet mice injected with either saline or zVADfmk (Fig. 4C), and secreted vimentin fragments were detected in the LPS-treated MCD diet mice; whereas, no obvious band was observed in the mice injected with zVADfmk.
Fig. 4. Effects of zVADfmk in LPS-injected MCD mice. (A) Representative liver tissue sections from LPS-MCD mice injected with either saline or zVADfmk (10 mg/kg) were analyzed for cleaved CK18 (M30), by immunohistochemistry staining (original magnification 200×). (B) Liver tissue extracts were prepared from LPS-MCD mice injected with either saline or zVADfmk. The vimentin and cleaved-caspase-3 were quantified by Western blot, using equal amounts of total liver proteins. Expression levels were normalized, relative to GAPDH. (C) The levels of plasma vimentin were determined by immunoprecipitation. Expression levels were normalized, relative to immunoglobulin G.
DISCUSSION
A causative link between lipid accumulation and the pathology of NASH has yet to be elucidated. Products of oxidative stress, including lipid peroxides and cytokines, have been found in patients with NASH, suggesting their involvement in the transition from steatosis to steatohepatitis (14). Therefore, production of cytokines, alternative lipid metabolic pathways, and activation of cellular proapoptotic machinery are putative mechanisms, through which lipoapoptosis is thought to occur. In the current study, we showed that palmitate directly induced apoptosis in HepG2 hepatocytes. Moreover, accumulation of lipids in HFD mice and MCD mice also resulted in increased caspase 3 activity. We recently reported that during the development of hepatic steatosis, the level of M30 was significantly altered, in mice fed a HFD for 24 weeks (11). The M30 antibody generated from the hepatic caspase-cleaved CK-18 fragment has commonly been used as a hepatic apoptosis marker in steatohepatitis. Those results are in agreement with the finding in the present study, that progression of steatosis is positively correlated with apoptosis.
MCD diet caused steatohepatitis, and the pathological changes could be further amplified by the LPS signaling pathway. This regulation might explain the enhancement of hepatic TNF-α and TGF-β mRNA expression, in LPS injected MCD fed mice. Vimentin expression also increased, in response to these proinflammatory cytokines in LPS injected MCD fed mice, but not in HFD mice. Recent studies have shown that LPS induces inflammation in vitro and in vivo, where it may lead to many diseases (15-17). Here, we demonstrated that LPS stimulated caspase-dependent apoptosis and vimentin up-regulation. Pretreatment with the non-specific caspase inhibitor zVADfmk protects endothelium against LPS-induced apoptosis. Therefore, this finding suggests that FFA lipoapoptosis can be attenuated by caspase inhibition.
It is interesting to investigate whether the increase in expression and cleavage of vemintin was associated with hepatic steatosis leading to NASH. Similar to other intermediate filament proteins, such as nuclear lamins and CK-18 and CK-19, vimentin is cleaved by caspases, during apoptotic cell death (18, 19). It has been suggested that caspase proteolysis of vimentin plays an active role in the execution of apoptosis. Cleavage of vimentin at Asp85 by caspase-3 generates a pro-apoptotic amino-terminal product that is sufficient to induce the cell death signal (10). Our results demonstrated that FFA cytotoxicity resulted in lipoapoptosis, via a caspase-dependent mechanism. Thus, it is likely that both vimentin expression and truncation are upregulated by LPS signaling and caspase-3 activity. zVADfmk inhibited the formation of cleaved caspase-3, which resulted in blockage of the formation of vimentin fragments, indicating that caspases are involved in vimentin proteolysis during lipoapoptosis. A previous study has demonstrated that caspase cleavage of vimentin promotes apoptosis by generating the truncated product, which subsequently amplifies caspase activity, thereby creating a positive feedback loop (10).
The truncated vimentin (48 kDa) is likely to be secreted in HepG2 cells and a NASH model. Neither the specific function of secreted vimentin, nor the mechanism of secretion, has been defined. We believe that vimentin is secreted into the extracellular space by activated macrophages during inflammation, and that this is related to the promotion of innate immunity. Several studies recently suggested that extracellular vimentin might be involved in the inflammation signaling pathway (20, 21). Accordingly, the secreted vimentin levels may be correlated with the extent of inflammation. Interestingly, most epithelial cancers express vimentin during EMT; therefore, extracellular vimentin may serve as a possible target for diagnosis.
In summary, we investigated the expression and secretion of vimentin in NASH, in both in vitro and in vivo models. Levels of CK18 fragments as biomarkers of apoptosis in NASH were found to increase proportionally with lipid accumulation in serum. However, the mechanisms by which vimentin expression is upregulated and the truncated vimentin is secreted, have yet to be elucidated. Here, we demonstrated that vimentin expression was mediated by the LPS pathway. Significant vimentin expression with the truncated product was observed in LPS injected MCD mice, but not HFD mice. In contrast, activation of caspase-3 was stimulated by both LPS treatment and HFD. These results suggested that the up-regulation of vimentin was associated with inflammation induced by LPS, but not with lipid accumulation. Moreover, the truncated vimentin by caspase-3 activation was secreted into blood. Therefore, the vimentin fragments may serve as a promising marker to differentiate NASH from simple steatosis, as well as a potential indicator of the severity of liver disease.
MATERIALS AND METHODS
Animals
We obtained male C57BL/6J mice from Orient Bio, Inc. (Seoul, Korea). The mice were then divided into three groups: (1) ND (n = 10), in which animals were treated with a standard diet for 16 weeks, (2) HFD (n = 12), in which animals were provided a HFD for 16 weeks, as previously described [11], and (3) MCD, in which animals were treated with the MCD diet for 3 weeks (n = 8). All studies were conducted in accordance with guidelines provided by the Animal Care and Use Committee of Kyungpook National University (approval no. KNU 2011-1-15). The mice were then intraperitoneally (i.p.) injected with either a single dose of 2.5 mg/kg LPS (Sigma Chemical, St. Louis, MO), or normal saline. zVADfmk was injected intraperitoneally, 1 h prior to LPS treatment.
HepG2 cell culture and treatment
HepG2 from ATCC were used in these experiments from passages three to eight. HepG2 cells were grown under a humidified 5% CO2 atmosphere at 37℃, in DMEM culture medium amended with 10% FBS, 2 mM L-glutamine, penicillin/streptomycin (100 U/ml), and antibiotic-antimycotic solution (500 U/ml). Cells were then treated for 24 h with 0.5-1 mM palmitate, or 10 ng LPS, or LPS plus palmitate.
Western blot analysis
For Western blot analysis, HepG2 cell lysates (50 μg of protein) were separated by 12% SDS-PAGE, after which the transfer membranes were exposed to specific antibodies against vimentin (1:1,000, Abcam (Cambridge, UK)), cleaved caspase-3 (1:1,000, Cell Signaling Technology (Danver MA, USA)), and poly (ADP-ribose) polymerase (PARP) (1:1,000, Santa Cruz Biotechnology (Dallas, TX, USA)). Anti-GAPDH antibody (1 : 1,000, Santa Cruz Biotechnology (Dallas, TX, USA)) was used as a loading control.
Quantitative PCR analysis
Total RNA was isolated from mouse livers using TRIzol (Invitrogen), according to the manufacturer’s directions. Quantitative RT-PCR was performed using SuperScript III RNase H- (Invitrogen). The PCR primer sequences used were as follows: TNF-α forward 5’-CAC CAC CAT CAA GGA CTC AA-3’, reverse 5’-AGG CAA CCT GAC CAC TCT CC-3’; TGF-β forward 5’-ATT CCT GGC GTT ACC TTG-3’, reverse 5’-CTG TAT TCC GTC TCC TTG GTT-3’.
Immunohistochemistry
4 μm paraffin sections were deparaffinized in Histo-clear (National Diagnostics, Merck Eurolab S.A.), and rehydrated in a graded alcohol series. The primary antibodies applied to the sections were a mouse antibody against vimentin (dilution 1 : 200, Abcam, Cambridge, MA), and anti-caspase-cleaved CK- 18 (M30, 1 : 100, Roche). Peroxidase activity was revealed using DAB substrate, after which slides were counterstained with hematoxylin (Sigma), then dehydrated, and mounted in Safemount embedding medium (Labonord, France).
Oil Red O staining
After incubation in the presence or absence of palmitate for 24 h, the confluent cell monolayer was fixed in phosphate-buffered formalin (10%) for 10 min, rinsed with water followed by 70% ethanol, and stained with Oil Red O solution for 15 min. The cell monolayer was then incubated for 5 min with 1.5 ml of 4% Nonidet P-40 in isopropyl alcohol, which dissolved the stained oil droplets.
Immunofluorescence staining
Vimentin antibody (dilution 1 : 200, Abcam, Cambridge, MA) was used for immunofluorescence staining. Cells were further incubated with a 5% goat serum/1xPBS solution, containing a 1 : 500 dilution of a species-specific Alexa-488 labeled secondary antibody. VectashieldⓇ with DAPI (Vector Laboratories, Burlingame, CA, USA) was used as the mounting medium.
Analysis of caspase-3 activity
The nitrogen-frozen hepatic or hepG2 cell lysates were incubated with the caspase-3 substrate, acetyl-DEVD-p-nitroanilide (Ac-DEVDpNA; Sigma) at 37℃ for 1 h, in buffer containing 30mM HEPES, 0.3 mM EDTA, 100 mM NaCl, 0.15% Triton X-100, and 10 mM DTT. The changes in optical density at 405 nm were then measured, using a spectrophotometer.
Statistical analysis
All values reported are the means ± S.D. of the indicated number of measurements. A one-way analysis of variance test was used to determine significance, with P < 0.05 indicating statistical significance.
Acknowledgments
This research was supported by the Basic Science Research Program, through the National Research Foundation of Korea (NRF), funded by the Korean Ministry of Education, Science and Technology (2011-0014420).
References
- 1.Schreuder T. C., Verwer B. J., van Nieuwkerk C. M., Mulder C. J. Nonalcoholic fatty liver disease: an overview of current insights in pathogenesis, diagnosis and treatment. World J. Gastroenterol. (2007);14:2474–2486. doi: 10.3748/wjg.14.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Day C. P., James O. F. Steatohepatitis: a tale of two "hits"? Gastroenterology. (1998);114:842–845. doi: 10.1016/S0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 3.Marra F., Gastaldelli A., Svegliati Baroni G., Tell G., Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends. Mol. Med. (2008);14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Carter-Kent C., Zein N. N., Feldstein A. E. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: implications for treatment. Am. J. Gastroenterol. (2008);103:1036–1042. doi: 10.1111/j.1572-0241.2007.01709.x. [DOI] [PubMed] [Google Scholar]
- 5.Alkhouri N., Carter-Kent C., Feldstein A. E. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert. Rev. Gastroenterol. Hepatol. (2011);5:201–212. doi: 10.1586/egh.11.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wieckowska A., Zein N. N., Yerian L. M., Lopez A. R., McCullough A. J., Feldstein A. E. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. (2006);44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 7.Bantel H., Ruck P., Gregor M., Schulze-Osthoff K. Detection of elevated caspase activation and early apoptosis in liver diseases. Eur. J. Cell Biol. (2001);80:230–239. doi: 10.1078/0171-9335-00154. [DOI] [PubMed] [Google Scholar]
- 8.Evans R. M. Vimentin: the conundrum of the intermediate filament gene family. Bioessays. (1998);20:79–86. doi: 10.1002/(SICI)1521-1878(199801)20:1<79::AID-BIES11>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Thiery J. P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. (2002);2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 10.Byun Y., Chen F., Chang R., Trivedi M., Green K. J., Cryns V. L. Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death. Differ. (2001);8:443–450. doi: 10.1038/sj.cdd.4400840. [DOI] [PubMed] [Google Scholar]
- 11.Park J. E., Kim H. T., Lee S., Lee Y. S., Choi U. K., Kang J. H., Choi S. Y., Kang T. C., Choi M. S., Kwon O. S. Differential expression of intermediate filaments in the process of developing hepatic steatosis. Proteomics. (2011);11:2777–2789. doi: 10.1002/pmic.201000544. [DOI] [PubMed] [Google Scholar]
- 12.Cong M., Iwaisako K., Jiang C., Kisseleva T. Cell signals influencing hepatic fibrosis. Int. J. Hepatol. (2012);2012:158547. doi: 10.1155/2012/158547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldstein A. E., Wieckowska A., Lopez A. R., Liu Y. C., Zein N. N., McCullough A. J. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology. (2009);50:1072–1078. doi: 10.1002/hep.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Day C. P., James O. F. Hepatic steatosis: innocent bystander or guilty party? Hepatology. (1998);27:1463–1466. doi: 10.1002/hep.510270601. [DOI] [PubMed] [Google Scholar]
- 15.Seehase S., Lauenstein H. D., Schlumbohm C., Switalla S., Neuhaus V., Forster C., Fieguth H. G., Pfennig O., Fuchs E., Kaup F. J., Bleyer M., Hohlfeld J. M., Braun A., Sewald K., Knauf S. LPS-induced lung inflammation in marmoset monkeys - an acute model for anti-inflammatory drug testing. PLoS One. (2012);7:e43709. doi: 10.1371/journal.pone.0043709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatia M., Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. (2004);202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 17.Park J. H., Kim K. H., Lee W. R., Han S. M., Park K. K. Protective effect of melittin on inflammation and apoptosis in acute liver failure. Apoptosis. (2012);17:61–69. doi: 10.1007/s10495-011-0659-0. [DOI] [PubMed] [Google Scholar]
- 18.Caulin C., Salvesen G. S., Oshima R. G. Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J. Cell Biol. (1997);138:1379–1394. doi: 10.1083/jcb.138.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao L., Perez D., White E. Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. (1996);135:1441–1455. doi: 10.1083/jcb.135.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mor-Vaknin N, Punturieri A., Sitwala K., Markovitz D. M. Vimentin is secreted by activated macrophages. Nat. Cell Biol. (2003);5:59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 21.Xu B., deWaal R. M., Mor-Vaknin N., Hibbard C., Markovitz D. M., Kahn M. L. The endothelial cell-specific antibody PAL-E identifies a secreted form of vimentin in the blood vasculature. Mol. Cell Biol. (2004);24:9198–9206. doi: 10.1128/MCB.24.20.9198-9206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]