

Abstract
Hevea brasiliensis hydroxynitrile lyase (HbHNL) and salicylic acid binding protein 2 (SABP2, an esterase) share 45% amino acid sequence identity, the same protein fold, and even the same catalytic triad of Ser-His-Asp. However, they catalyze different reactions: cleavage of hydroxynitriles and hydrolysis of esters, respectively. To understand how other active site differences in the two enzymes enable the same catalytic triad to catalyze different reactions, we substituted amino acid residues in HbHNL with the corresponding residues from SABP2, expecting hydroxynitrile lyase activity to decrease and esterase activity to increase. Previous mechanistic studies and x-ray crystallography suggested that esterase activity requires removal of an active site lysine and threonine from the hydroxynitrile lyase. The Thr11Gly Lys236Gly substitutions in HbHNL reduced hydroxynitrile lyase activity for cleavage of mandelonitrile 100-fold, but increased esterase activity only threefold to kcat ~ 0.1 min−1 for hydrolysis of p-nitrophenyl acetate. Adding a third substitution – Glu79His – increased esterase activity more than tenfold to kcat ~ 1.6 min−1. The specificity constant (kcat/KM) for this triple substitution variant versus wild type HbHNL shifted more than one million-fold from hydroxynitrile lyase activity (acetone cyanohydrin substrate) to esterase activity (p-nitrophenyl acetate substrate). The contribution of Glu79His to esterase activity was surprising since esterases and lipases contain many different amino acids at this position, including glutamate. Saturation mutagenesis at position 79 showed that 13 of 19 possible amino acid substitutions increased esterase activity, suggesting that removal of glutamate, not addition of histidine, increased esterase activity. Molecular modeling indicates that Glu79 disrupts esterase activity in HbHNL when its negatively charged side chain distorts the orientation of the catalytic histidine. Naturally occurring glutamate at the corresponding location of Candida lipases is uncharged due to other active site differences and does not cause the same distortion. This example of the fine tuning of the same catalytic triad for different types of catalysis by subtle interactions with other active site residues shows how difficult it is to design new catalytic reactions of enzymes.
Keywords: hydroxynitrile lyase, esterase, α/β-hydrolase, catalytic promiscuity, protein engineering, divergent evolution
Introduction
Although protein engineering to improve enzyme properties like stability and selectivity is increasingly common, protein engineering to change catalytic mechanisms remains difficult. The transition states for different reactions are understood, but assembling an active site to stabilize these transition states has rarely succeeded. Protein engineering to change the catalytic mechanism may use rational design, copying key residues from homologous enzymes, metal substitutions, advanced computational design as well as directed evolution. A survey in 2010 (Table S2 in supporting information of reference 1) shows that values of kcat for the eighteen examples were typically 1−10 min−1, but ranged from 0.01 min−1 to 50,000 min−1. Natural enzymes typically show kcat values of > 107 min−1. A success reported after this survey is the directed evolution of an enzyme that catalyzes the Kemp elimination with a kcat of 42,000 min−1.2
One way to identify how enzymes catalyze different reactions is to compare homologous enzymes that catalyze different reactions. These enzymes differ both within the active site and outside the active site. Some of these differences may be to accommodate different substrates, some to stabilize the protein fold, but some must cause the switch in catalytic activities. Our goal is to identify these differences to better understand how similar enzymes catalyze different reactions and shed light onto the possible paths natural evolution follows to arrive at new modes of catalysis. This understanding of natural evolution may help future design or engineering of new catalytic activities.
One of the best understood class of enzymes is the serine hydrolases. These hydrolases use a catalytic triad of Ser-His-Asp and an oxyanion hole to catalyze hydrolyses of various substrates as well as related reactions. Researchers have uncovered some of the fine tuning that determines which reaction is favored. For example, although lipases and esterases have the same catalytic triad and oxyanion hole as serine proteases, they are poor catalysts for hydrolysis of amides. One hypothesis is that a hydrogen bond from the amide disrupts the catalytic histidine orientation in esterases, but not in proteases.3, 4 Other subtleties in the mechanism of serine hydrolases include questions of whether there is a tunnel to allow water access to the active site,5, 6 how acyl transferases avoid hydrolysis to favor acyl transfer7 and how perhydrolases favor hydrogen peroxide over water.8 Nevertheless, all these reactions are similar and involve similar transitions states.
An example of enzymes with a serine esterase catalytic triad, but a very different mechanism and transition state is the hydroxynitrile lyases (HNL’s) in the α/β-hydrolase family. Hydroxynitrile lyases evolved in plants to generate cyanide as a defense against insects. HNL’s occur in five different protein families, one of which is the α/β-hydrolase family. HNL’s in the α/β-hydrolase family contain a serine-histidine-aspartate catalytic triad which superimposes closely with the catalytic triad of esterases. However, HNL’s catalyze the cleavage of hydroxynitriles to generate cyanide and do not catalyze ester hydrolysis. For example, Hevea brasiliensis hydroxynitrile lyase (HbHNL) catalyzes the cleavage of acetone cyanohydrin to hydrogen cyanide and acetone in nature.9
X-ray crystal structures and detailed mechanism and kinetic studies showed that the catalytic mechanisms of hydroxynitrile lyases and esterases differ in at least four ways,10 Figure 1. First, the reaction intermediate and transition state differs. Ester hydrolysis involves formation and hydrolysis of an acyl enzyme intermediate, but hydroxynitrile cleavage by HNL’s has a single transition state without an acyl enzyme intermediate. Second, esterases use an oxyanion hole to stabilize tetrahedral intermediates, but HNL’s oxyanion hole is blocked by a threonine or asparagine residue. Esterases have a glycine at this location. This blocking of the oxyanion hole causes the substrate to bind differently. Third, because of this different substrate binding, the active site serine interacts with different parts of the substrate: carbonyl carbon in esterases versus carbonyl oxygen in HNL’s. In esterases, the catalytic serine Oγ–H is the nucleophile that attacks the carbonyl carbon, but in hydroxynitrile lyases it interacts with the hydroxyl group of the cyanohydrin. This hydroxyl group becomes the carbonyl oxygen in the product aldehyde. Fourth, a positively charged lysine residue in the hydroxynitrile lyase, not present in esterases, stabilizes the formation of the negative charge on the cyano group in the transition state.
Figure 1.

Reactions, arrow-pushing mechanisms and an overlay of active sites for SABP2 and HbHNL. A) In the first step of methyl salicylate hydrolysis catalyzed by SABP2, the oxygen of Ser80 acts as a nucleophile to attack the carbonyl carbon of the ester. Hydrogen bonds from the oxyanion hole (shown as two amide N–H) stabilize the negative charge that will form on the carbonyl oxygen. After formation of the tetrahedral intermediate, it loses the methanol leaving group to form a salicyl serine intermediate (not shown). B) Cleavage of (S)-mandelonitrile catalyzed by HbHNL occurs in one step. The side chain of Thr11 blocks the oxyanion hole, so the cyanohydrin orients in the active site differently. The oxygen of Ser80 removes a proton from the cyanohydrin hydroxyl group. The positively charged lysine stabilizes loss of the negatively charged cyanide. C) Overlay of the active site residues (sticks representation, Asp residues not shown) and product salicylate and substrate mandelonitrile (line representation) from x-ray crystal structures of SABP2 (1Y7I, green carbon atoms)10 and HbHNL (1YB6, gold carbon atoms)9 shows a similar orientation of the active site residues and substrate or product. Dashed lines indicate hydrogen bonds, but hydrogen atoms are hidden for clarity.
In this paper, we describe protein engineering to convert HbHNL into an esterase, which is a major change in the catalytic mechanism. No esterase has been designed from first principles. We hypothesize that starting from a closely related enzyme with correctly oriented active site residues will enable a switch in activity since natural evolution is believed to have made similar switches. The esterase with the highest amino acid sequence identity to HbHNL is esterase Nicotiana tabacum salicylic acid binding protein 2 (SABP2; see Figure S1). This natural function of this esterase is hydrolysis of methyl salicylate to salicylic acid, 11 which is a signal for plant defense. Forty-five percent of the amino acids in this esterase are identical to those in HbHNL and the two enzyme’s x-ray structures superimpose with a root-mean-square deviation between 256 equivalent α-carbon atoms of only 0.4 Å. This paper shows that substituting only three of the active site residues in HbHNL switch its catalytic activity to an esterase.
Results
Rational for HbHNL mutagenesis
The active sites of HbHNL and SABP2 contain 18 amino acid residues within 4 Å of the substrates. Five of these residues are identical: Ser80, His235 of the catalytic triad and His14, Trp128, Leu157 (HNL numbering). Asp207 of the catalytic triad is also identical, but lies more than 4 Å from the substrate. This leaves thirteen potential substitutions to interconvert the two active sites. Previous research identified a mechanistic role for two of these potential substitutions – Thr11Gly and Lys236Met. Replacing Thr11 with glycine will allow the ester substrate to bind to the oxyanion hole. Lys236 likely stabilizes the negatively charge buildup on the cyano group in HNL and should be removed because esterases bind an uncharged alcohol in this region. Indeed, either of these substitutions reduced HNL activity to < 1% of the wild type activity and increased esterase activity, see below.
None of the other eleven single substitutions eliminated HNL activity or increased esterase activity,12 so were not further investigated in this paper. Two substitutions yielded variants with up to 4.2-fold higher HNL activity than wild type HNL (Leu121Tyr, Phe125Thr), five showed similar HNL activity (within a factor of two: Cys81Leu, Tyr133Phe, Leu146Met, Leu148Phe, Phe210Ile), three showed a four to eight-fold decrease in HNL activity (Val106Phe, Ile109Gly, Leu152Phe). One substitution yielded only insoluble protein. Ile12Ala. None of these residues had previously been identified as important for catalysis, so we did not expect large effects from these substitutions.
Since SABP2 is a relatively slow esterase, we also used Pseudomonas fluorescence esterase (PFE) as a reference esterase.13 This esterase has more than 10-fold higher esterase activity than SABP2, but is not a first choice because it is less similar to HbHNL; only 26% of its amino acids are identical. Like SABP2, PFE also has a glycine at the position corresponding to Thr11 in HbHNL. Thus, we tested only the Thr11Gly substitution at this location. But at position Lys236 in HbHNL, PFE has a glycine, while SABP2 has a methionine. We tested both Lys236Gly and Lys236Met variants.
Another amino acid position, Glu79 in HbHNL, was the most important for high esterase activity, see below. This location is more than 4 Å from the substrate. In HbHNL, Glu79 forms a salt bridge with Lys236 to position it for catalysis. Esterases PFE and SABP2 have a Phe and His, respectively, at this location, but some esterases and lipases have a Glu. We replaced Glu79 with His initially and later used saturation mutagenesis to test all possible amino acids.
Site-directed mutagenesis of HbHNL created all eleven possible combinations of these four substitutions: Thr11Gly, Lys236Gly or Lys236Met, and Glu79His. The combinations yield four single amino acid substitutions, five double substitutions and two triple substitutions, Table 1. The variant proteins were expressed in E. coli BL21 yielding soluble protein for ten of the eleven variants. Variant HbHNL Lys236Gly did not express as soluble protein so it was not characterized. All soluble variants were purified by anion exchange chromatography followed by size exclusion chromatography. See supplemental information Figures S3-S5. Crude extracts of E. coli BL21 without the plasmid encoding HbHNL variants also contained a protein (mol. wt. ~17,000 kDa) with low esterase activity. The purification procedure removed this protein. See supplemental information Figures S5 and S6.
Table 1.
Catalytic activity of HbHNL variants, SABP2, and PFE toward cleavage of hydroxynitrile and hydrolysis of p-nitrophenyl acetate
| Variant | Hydroxyni trile lyasea |
Esterase (p-nitrophenyl acetate)b | ||||
|---|---|---|---|---|---|---|
| Specific Activity (mU/mg) |
Specific Ac- tivity (mU/mg) |
kcat (min−1) |
Km (mM) |
kcat/Km( M−1 min−1) |
Fold Esterase Increasec |
|
| wild type | 30000±100 | 0.88±0.2 | 0.11±0.02 | 1.0±0.07 | 110 | 1.0 |
| K236G | npd | npd | npd | npd | npd | npd |
| T11G | 240±30 | 1.3±0.05 | 0.09±0.005 | 0.20±0.04 | 450 | 4.1 |
| E79H | 1300±39 | 1.4±0.01 | 0.08±0.004 | 0.19±0.04 | 410 | 3.7 |
| K236M | 36±13 | 8.4±0.03 | 0.74±0.02 | 0.58±0.05 | 1,270 | 11.5 |
| T11G K236G | 300±14 | 2.7±0.2 | 0.13±0.004 | 0.15±0.02 | 860 | 7.8 |
| T11G K236M | ≤7 | 7.3±0.12 | 0.60±0.06 | 0.55±0.14 | 1,080 | 9.8 |
| E79H K236M | 46±10 | 5.8±0.07 | 0.32±0.02 | 0.21±0.04 | 1,540 | 14 |
| K236G E79H | ≤7 | 8.0±1.0 | 0.37±0.01 | 0.16±0.02 | 2,260 | 20.5 |
| T11G E79H | 540±40 | 14.±0.2 | 0.58±0.02 | 0.14±0.02 | 4,160 | 38 |
| T11G K236M E79H | 65±1 | 14.±0.6 | 0.59±0.02 | 0.14±0.02 | 4,200 | 38 |
| T11G K236G E79H (TM) | 145±30 | 35.±1.4 | 1.62±0.05 | 0.16±0.02 | 10,100 | 92 |
| SABP2 | ≤7 | 780.±30 | 120.±6 | 1.4±0.1 | 86,000 | nae |
| PFE | nae | 19500 | 1460 | 0.45 | 32,400,000 | nae |
Release of benzaldehyde (ε280 nm = 1380 M−1 cm−1) from mandelonitrile (5 mM) at pH 5.0 measured at 280 nm. Error limits are the standard deviation of three measurements.
Release of p-nitrophenoxide (ε404 nm = 11,580 M−1 cm−1 at pH 7.2) from p-nitrophenyl acetate (0.3 mM) at pH 7.2 measured at 404 nm. Steady state kinetics were measured by varying the p-nitrophenyl acetate concentration from 0.025 mM to 2 mM and fitting the data (5 to 10 data points) to the Michaelis-Menten equation. Errors limits are standard deviations.
Specificity constant (kcat/Km) relative to wt.
np = no protein; protein was expressed, but only as insoluble aggregates.
na = not applicable.
Converting a hydroxynitrile lyase into an esterase
The remaining hydroxynitrile lyase activity was measured spectrophotometrically at 280 nm, which monitored the release of benzaldehyde from mandelonitrile (5 mM, pH 5, solution contained 5 vol% acetonitrile). The natural substrate is acetone cyanohydrin, but cleavage of mandelonitrile is easier to measure and is the standard assay for this enzyme. The new esterase activity was measured spectrophotometrically at 404 nm, which monitors the release of p-nitrophenoxide from p-nitrophenyl acetate (0.3 mM, pH 7.2, solutions contained 7 vol% acetonitrile). The natural substrate of SABP2 is methyl salicylate and the natural substrate of PFE is unknown. Cleavage of p-nitrophenyl acetate is easier to measure and is a standard assay for esterases. Wild-type HNL showed a low esterase activity of 0.88 mU/mg, while SABP2 showed 780 mU/mg and PFE showed 19,500 mU/mg. The activity of the best variants with the natural substrates will also be measured below.
The three single amino acid substitution variants that expressed as soluble proteins showed at least a 10-fold drop in hydroxynitrile lyase activity, but only one showed a significant increase in esterase activity, Table 1. The largest drop in HNL activity was 840-fold for Lys236Met. This decrease is consistent with a previous report,14 that the Lys236Leu substitution eliminated the hydroxynitrile lyase activity. The esterase activity of variant HbHNL Lys236Met increased 9.5-fold to 8.4 mU/mg, but the esterase activity of variants HbHNL Thr11Gly and Glu79His increased only ~1.5-fold as compared to wild type HbHNL.
The double substitution variants showed additional large drops in hydroxynitrile lyase activity and only small increases in esterase activity. One double substitution that includes the Lys236Met substitution (Thr11Gly-Lys236Met) showed no detectable HNL activity. This is a > 4,000-fold decrease as compared to wild type HbHNL and at least a 20-fold decrease from the Lys236Met substitution alone. Another double substitution that includes the Lys236Met substitution, Glu79His-Lys236Met (46 mU/mg), shows ~1.3-fold higher HNL activity than the single HbHNL Lys236Met variant (36 mU/mg) indicating a non-additive negative effect of the Glu79His substitution. The Glu79His substitution alone decreased HNL activity 23-fold; but here, combined with the Lys236Met substitution, it caused a ~1.3-fold increase. Another double substitution (Glu79His-Lys236Gly) also shows no detectable HNL activity. The effect of the Lys236Gly substitution alone is unknown because that variant did not yield soluble protein. The remaining two double substitution variants (Thr11Gly-Lys236Gly and Thr11Gly-Glu79His) show the highest remaining HNL activities: 300 mU/mg (100-fold drop relative to wild type) and 540 mU/mg (~56-fold drop relative to wild type). The esterase activity of the double substitutions ranges from 2.7 to 14 mU/mg, which is a 3–16-fold increase over the wild type. The best variant is Thr11Gly-Glu79His with an esterase activity of 14 mU/mg.
The two triple substitution variants both show low HNL activity. Thr11Gly-Glu79His-Lys236Met shows 65 mU/mg (a 460-fold decrease relative to wild type) and Thr11Gly-Glu79His-Lys236Gly (called HbHNL-TM below) shows 150 mU/mg (a 200-fold decrease). These activities are lower than two of the double substitution variants, but, surprisingly, higher than three other double variants. The observation that HNL activity increases upon adding a substitution that by itself decreases HNL activity is another example of the non additive effect of the substitutions.
The esterase activity of triple substitution variant Thr11Gly-Glu79His-Lys236Met is 14 mU/mg, which is the same as that for the double variant Thr11Gly-Glu79His, indicating that adding the Lys236Met substitution did not increase esterase activity. In contrast, Lys236Met increased esterase activity 9.5-fold as a single substitution in HbHNL. The esterase specific activity of triple substitution variant HbHNL-TM is 35 mU/mg, which is the highest of all variants in this paper and 40-fold higher than wild type HbHNL.
Characterization of HbHNL-TM
The increase in esterase activity is higher – 92-fold – if one compares the specificity constants, kcat/KM, from steady state kinetics. The kcat value for HbHNL-TM-catalyzed hydrolysis of p-nitrophenyl acetate is 15-fold higher than the wild type HNL and the KM value is 6-fold lower, which corresponds to a specificity constant 92-fold higher than the wild type HNL. This value of kcat/KM is only 8.5-fold lower than the value for SABP2, which is the esterase closest in amino acid sequence to HbHNL. This esterase activity is > 3000-fold lower than PFE, a highly active esterase, but PFE is distantly related to HbHNL, so other amino acid differences contribute to higher esterase activity.
The specificity constant for cleavage of mandelonitrile decreased 176-fold from 440,000 M−1 min−1 for HbHNL-wt to 2,500 M−1 min−1 for HbHNL-TM, mainly due to changes in kcat, Table 2. The kcat value decreased 180 fold from 1440 to 8.1 min−1, but the KM values were similar – 3.3 and 3.2 mM. The decrease was even larger – 10,900-fold, from 9,700,000 to 890 M−1 min−1 – for acetone cyanohydrin, the natural substrate of HbHNL. The kcat value decreased 670 fold from 3900 to 5.8 min−1, and the KM value increased 13-fold from 0.4 to 6.5 mM.
Table 2.
Steady state kinetic constants for hydroxynitrile cleavage (entries 1–2) and ester hydrolysis (entries 3–7) catalyzed by HbHNL-wt and HbHNL-TM.a
| HbHNL wt | HbHNL-TM | ||||||
|---|---|---|---|---|---|---|---|
| entry | substrate | kcat(min−1) | KM (mM) |
kcat/KM(M−1 min−1) |
kcat (min−1) |
KM (mM) | kcat/KM (M−1 min−1) |
| 1 | Mandelonitrile | 1, 440.±80 | 3.3±0.5 | 440,000 | 8.1±1.0 | 3.2±0.9 | 2,500 |
| 2 | Acetone cyanohydrin | 3, 900.±20 | 0.4±0.07 | 9,700,000 | 5.8±0.5 | 6.5±1 | 890 |
| 3 | Phenyl acetate | <0.01 | ndb | <1 | 1.2±0.1 | 2.8±0,4 | 410 |
| 4 | Methyl benzoate | <0.01 | nd | <1 | 0.15 | 9.2±1.4c | 16.±1.7d |
| 5 | Methyl salicylate | <0.01 | nd | <1 | nde | >10e | 7.3±2d |
| 6 | Ethyl acetate | <0.01 | nd | <1 | <0.01 | nd | <1 |
| 7 | Phenylalanine methyl ester | <0.01 | nd | <1 | <0.01 | nd | <1 |
Steady state kinetics measured as described in the Experimental Section. Error limits are standard deviations of 5–10 measurements the fit to the Michaelis-Menten equation. Detection limits are for esterase activity using pH indicator assay.
nd = no data
Ki of methyl benzoate inhibition of pNPAc hydrolysis; this value is an estimate of KM.
The slope of a rate vs. ester substrate concentration plot gives kcat/KM for cases where substrate solubility is too low to reach enzyme saturation.
The minimum for the KM corresponds to the highest concentration tested due to limited substrate solubility.
This decrease in specificity constants for hydroxynitrile cleavage and corresponding increase specificity constants for ester hydrolysis show that the catalytic activity switched, Table 3, but the number depends on the substrates used in the comparison. The ratio of specificity constants comparing esterase activity with p-nitrophenyl acetate to hydroxynitrile lyase activity with mandelonitrile was 0.00025 for HbHNL-wt indicating a strong preference for hydroxynitrile activity, but this ratio increased to 4 for variant HbHNL-TM. That this value is more than one shows that this variant is now primarily an esterase - 10,100 M−1 min−1 for p-nitrophenyl acetate hydrolysis versus 2,500 M−1 min−1 for mandelonitrile cleavage. For p-nitrophenyl acetate compared to acetone cyanohydrin, the differences were even larger: from 10−5 for HbHNL-wt to 11 for HbHNL-TM, which is more than a million-fold change.
Table 3.
Ratio of esterase to hydroxynitrile lyase specificity constants for HbHNL-wt and HbHNL-TM.
| Esterase/HNL kcat/KMa | HbHNL-wt | HbHNL-TM | Fold Change |
|---|---|---|---|
| pNPAc/mandelonitrile | 0.00025 | 4 | 16,000 |
| pNPAc/acetone cyanohydrin | 10−5 | 11 | 1,100,000 |
| methyl salicylate/acetone cyanohydrin | <10−7 | 0.00820 | >82,000 |
Ratio of specificity constants for hydrolysis of the ester (pNPAc or methyl salicylate) and cleavage of the hydroxynitrile (mandelonitrile or acetone cyanohydrin).
The last substrate pair compares the natural substrates– methyl salicylate and acetone cyanohydrin. The ratio of specificity constants is less than one for both HbHNL-wt and HbHNL-TM, indicating that acetone cyanohydrin cleavage remains the favored reaction. The favored catalytic activity does not switch, but the change in activity ratio is > 82,000. No hydrolysis of methyl salicylate could be detected with HbHNL-wt, but is easily measured with HbHNL-TM.
The p-nitrophenol leaving group of p-nitrophenyl acetate makes it an unusually reactive ester. To confirm that HbHNL-TM can also catalyze hydrolysis of esters with poorer leaving groups, we also tested phenyl acetate and methyl benzoate, Table 2 above. Mandelonitrile is a good substrate for HbHNL, so we chose ester substrates with aromatic rings. An overlay of x-ray crystal structures shows that the aromatic ring of mandelonitrile bound to HbHNL in a catalytically competent orientation orients similarly as the aromatic ring of salicylic acid bound to SABP2, Figure 1C. The kcat value for hydrolysis of phenyl acetate, 1.2 min−1 is similar to that for pNPAc, 1.6 min−1 (Table 1), but phenyl acetate binds less tightly (KM = 2.8 mM) than pNPAc (KM = 0.16 mM), yielding a 17-fold lower specificity constant for phenyl acetate – 410 M−1 min−1 for phenyl acetate as compared to 10,100 M−1 min−1 for pNPAc. Methyl benzoate is a poor substrate. The kcat value, 0.15 min−1 is tenfold lower than the value for pNPAc and the KM value, 9.2 mM, indicates 57-fold poorer binding. The specificity constant for hydrolysis of methyl benzoate, 16 M−1 min−1 is 630-fold lower than the value for pNPAc. Methyl salicylate, whose structure is similar to methyl benzoate was also a poor substrate with a specificity constant of 7.3 M−1min−1. This value for specificity constant comes from the increase in rate with increasing methyl salicylate concentration. The solubility limit of methyl salicylate was 10 mM, which prevented measurement of kcat and KM individually.
Neither HbHNL-wt nor HbHNL-TM catalyzed the hydrolysis of ethyl acetate or phenylalanine methyl ester. Hickel reported ‘low activity‘ of HbHNL-wt catalyzed hydrolysis of phenylalanine methyl ester,15 but we did not detect any hydrolysis. A possible, but untested, explanation is that a contaminating esterase from E. coli was present in Hickel’s experiments.
Role of the Glu79His substitution
The Glu79His substitution contributed at least a factor of ten to the esterase activity. The addition of Glu79His to Thr11Gly increased esterase specificity constant 9.2 fold, and its addition to Thr11Gly-Lys236Gly increased it 11.6 fold.
Esterases in the α/β-hydrolase family are serine hydrolases where the active site serine occurs within a GXSXG motif; the X indicates any amino acid. The Glu79His substitution corresponds to the first X in this motif. The 3DM database of 11,901 α/β-hydrolase family enzymes16 shows that all 20 amino acids occur at this positions. The most common amino acid is histidine, followed by glutamate, Table 4. However, subgroups of α/β-hydrolase family enzymes may favor a particular amino acid. For example, the Pseudomonas fluorescens esterase group favors phenylalanine (27/28 sequences), the Candida antarctica lipase B group favors tryptophan (17/17 sequences) and the Candida rugosa lipase group favors glutamate.
Table 4.
Frequency of different amino acids at the residue before the active site serine and the effect of different amino acids at this position on esterase specific activity in HbHNL T11G K236G and SABP2.
| X= | % in α/β hydrolasesa |
HbHNL T11G K236G E79Xb (mU/mg) |
SABP2 H80Xb (mU/mg) |
|
|---|---|---|---|---|
| Negatively charged side chain | Glu | 15.5 | 2.7±0.2 | 3±0.2 |
| Asp | 12 | 11.8±0.6 | 123±6.4 | |
| avec = 7.2 | avec = 63 | |||
| Positively charged side chain | Lys | 0.6 | 11.7±0.5 | 6±0.1 |
| Arg | 0.9 | 6.2±0.4 | npd | |
| His | 25 | 35±1.4 (TM) | 780±1.3 (wt) | |
| avec = 18 | avec = 390 | |||
| Polar side chain | Ser | 1.9 | np | 390±20 |
| Thr | 0.9 | 14.5±1.5 | 544±37 | |
| Tyr | 2.8 | 19.4±1.2 | 14±0.12 | |
| Asn | 4.8 | 13.9±0.4 | 500±1.2 | |
| Gln | 5.1 | 9.7±0.8 | 520±1.2 | |
| Cys | 0.4 | 8.0±0.9 | 125±6.4 | |
| avec=13 | avec = 350 | |||
| Non-polar side chain | Pro | 0.4 | npd | 120±3.4 |
| Met | 0.9 | 4.9±0.2 | 19±3.4 | |
| Gly | 8.3 | <1.0 | 25±2.6 | |
| Ala | 2.3 | 9.7±0.7 | 580±8.6 | |
| Val | 1.2 | 5.6±0.8 | 20±2.5 | |
| Leu | 4.1 | npd | npd | |
| Ile | 1 | 3.2±0.1 | npd | |
| Trp | 5.6 | npd | npd | |
| Phee | 6.4 | npd | 69±1.7 | |
| avec = 4.9 | avec = 140 |
Occurrence of each amino acid at this position within the α/β hydrolase database of 11,901 sequences.
Specific activity for hydrolysis of pNPAc (0.3 mM) at pH 7.2; 1 U = 1 μmol of product formed per minute. Error limits are standard deviations for three measurements.
ave = average for each group.
np = no protein. Expression yielded no soluble protein, only aggregated insoluble protein, indicating that the protein could not fold or could not remain folded.
Pseudomonas fluorescens esterase contains Phe at this position.
To identify the molecular reasons for increase in esterase activity due to the Glu79His substitution, we measured the esterase activity for the eighteen other amino acid substitutions at this location, Table 4. The substitutions in HbHNL T11G K236G and in SABP2 gave similar results. Histidine at this position yielded the highest esterase activity in HbHNL T11G K236G, (35 mU/mg; this variant corresponds to HbHNL-TM) and in SABP2 (780 mU/mg; this corresponds to wild type SABP2). Many substitutions yielded higher activity than glutamate at this position. Positively charged side chains and polar side chain gave the highest average activities. The result that many amino acids at this position yielded good esterase activity suggests that removing glutamate is the reason for higher esterase activity.
One hypothesis is that Glu79 may affect the pKa of the catalytic histidine, but measurement of the pH dependence of esterase activity ruled out this possibility, Figure S11. As is typical for esterases, the esterase activity increased with increasing pH and reached a plateau. The esterase mechanism requires a deprotonated histidine and the kinetic pKa corresponds to the midpoint of this increase. The kinetic pKa’s of HbHNL-Thr11Gly-Lys236Gly, 6.0±0.2, and HbHNL-TM 5.8±0.2, are within the experimental errors and similar to that previously reported for the esterase PFE, kinetic pKa = 5.6.17 Thus, the lower activity of the variant with glutamate at position 79 is not due to an altered kinetic pKa of the catalytic histidine. However, Glu79 likely affects the thermodynamic pKa of the catalytic histidine, see below.
We used molecular dynamics to model how well a tetrahedral intermediate for ester hydrolysis fits in the active site. Quantum mechanics calculations show that this tetrahedral intermediate is a good mimic for the transition state for ester hydrolysis.18 In particular, we looked for five essential hydrogen bonds needed for catalysis. This approach is similar to the near attack complex approach developed by Bruice,19 but differs in using not a substrate for modeling, but the covalently linked tetrahedral intermediate. Using PFE and a tetrahedral intermediate for hydrolysis of ethyl acetate as a positive control, we looked for three hydrogen bonds involving the catalytic histidine 235 (His Nε-H to both Oγ of Ser80 and the alcohol oxygen from the tetrahedral intermediate and His Nδ-H to Oδ of Asp207) and two hydrogen bonds in the oxyanion hole – from the tetrahedral intermediate’s oxyanion to the amide backbone N–H of Cys81 and Ile12. This simulation yielded all five hydrogen bonds in 86% of the frames, consistent with a good esterase. As a negative control, we modeled the tetrahedral intermediate for hydrolysis of phenyl acetate in the active site of HbHNL-wt. None of the frames in the molecular dynamic simulation showed all five catalytically essential hydrogen bonds, consistent with the very poor esterase activity of this hydroxynitrile lyase. Thr11 blocked the oxyanion hole region causing the tetrahedral intermediate to shift its position so that its oxyanion accepted a hydrogen bond from the γ hydroxyl of Thr11. Besides preventing formation of hydrogen bonds in the oxyanion hole, this shift also moved the alcohol oxygen of the tetrahedral intermediate too far from His235 Nε-H to form a hydrogen bond.
Molecular dynamics simulation of phenyl acetate hydrolysis within HbHNL-Thr11Gly-Lys236Gly shows that Glu79 disrupts the orientation of the catalytic histidine. A simulation of the tetrahedral intermediate started with all five hydrogen bonds, but during the simulation, the two hydrogen bonds from Nε-H of His235 to oxygens of the tetrahedral intermediate broke. His235 moved away from the tetrahedral intermediate to form a hydrogen bond with Glu79, Figure 2. In contrast, a simulation of HbHNL-TM with the phenyl acetate tetrahedral intermediate showed all five catalytically essential hydrogen bonds in 56% of the frames over a 1.2 ns simulation (42% had only 4 essential bonds and 2% had only 3 essential bonds: the two in the oxyanion hole and one from His235 Nδ-H to Oδ of Asp207). There was no special interaction of the His79 in HbHNL-TM with the substrate or catalytic triad; the essential feature was the absence of Glu79. Among the many amino acids that increased activity over glutamate, histidine yielded the highest activity, but only slightly higher. Modeling did not reveal why histidine was best; it is likely that the reasons may be subtle.20
Figure 2.

Molecular dynamics simulation of the tetrahedral intermediate for hydrolysis of phenyl acetate in the active site of HbHNL T11G K236G. The initial model contains all five hydrogen bonds (red dotted lines) required for catalysis. After 24 ps of simulation the catalytic histidine moved, breaking two key hydrogen bonds to the tetrahedral intermediate and forming a new, nonproductive hydrogen bond with the carboxylate oxygen of Glu79.
The importance of removing the glutamate at position 79 to increase esterase activity in the HbHNL variants raises the question why glutamate is nevertheless the second most common residue after histidine at this position among lipases and esterases. For example, Candida rugosa lipase isoenzymes 1–5 all contain a glutamate at position 208, which corresponds to position 79 in HbHNL. The predicted pKa for this glutamate in HbHNL wt is 1.3 and in HbHNL Thr11Gly Lys236Gly is 3.9 (estimated using PROPKA 3.121) indicating that it is negatively charged near neutral pH. In contrast, the predicted pKa for the corresponding glutamate in the Candida rugosa lipases is 10.4 indicating that it remains uncharged at neutral pH. Molecular dynamics simulation of ester hydrolysis catalyzed by Candida rugosa lipase isozyme 1 confirmed that an uncharged glutamate does not interfere with the catalytic mechanism, presumably due to much lower electrostatic interactions with the catalytic histidine. Isoenzyme 1 exhibits all hydrogen bonds essential for ester hydrolysis in 62% of molecular dynamics frames, consistent with good catalytic activity. Thus, other amino acids within the Candida rugosa lipase favor the uncharged glutamate, thereby preventing the glutamate from interfering with catalysis. Presumably the many esterases and lipases that contain glutamate at this location similarly keep it uncharged to prevent the glutamate from interfering with catalysis.
Discussion
To compare the effect of different substitutions on catalysis, we compared the average esterase activity and average HNL activity for all HbHNL variants that contained a particular substitution. This approach minimizes the influence of any particular positive or negative cooperative effect because it averages the effect of the substitution in four to six combinations of other substitutions.
The most important substitution to increase esterase activity was the unexpected one, Glu79His (average esterase specificity constant = 3800 M−1 min−1), followed closely by the expected Thr11Gly (3500 M−1 min−1) and Lys236Gly (3300 M−1 min−1), Figure 3A. Consistent with the notion that the Glu79His substitution is most important for increasing esterase activity, but not decreasing hydroxynitrile lyase activity, the Glu79His variants had the highest average HNL activity, Figure 3A.
Figure 3.

Effect of substitutions in HbHNL on esterase and hydroxynitrile lyase activity. A) The average activity of variants containing the substitutions shown. The Glu79His substitution yields the largest increase in esterase activity (circles). The Lys236Met substitution caused the largest decrease in HNL activity (squares). B) Some multiple substitutions act cooperatively. The single substitutions (squares) increased esterase activity only slightly, except for Lys236Met. If these single substitutions acted additively when combined, then the esterase activity of the double and triple substitutions would be as indicated by the open diamonds. In contrast, the measured esterase activities (circles) are sometimes dramatically higher indicating positive cooperativity among the substitutions. Esterase activity refers to hydrolysis of p-nitrophenyl acetate (0.3 mM, pH 7.2, 25 °C); HNL activity refers to the cleavage of mandelonitrile (5 mM, pH 5.0, 25 °C). The single substitution Lys236Gly (open square) did not yield soluble protein and was assigned a low esterase activity of < 0.5 mU/mg.
The most important substitutions to decrease hydroxynitrile lyase activity were Lys236Met and Lys236Gly. The HbHNL Lys236Met and Lys236Gly variants showed the lowest average remaining HNL activity, 38 and 115 mU/mg, respectively, Figure 3A. This result is consistent with the hypothesis that Lys236 holds the cyanide substrate for reaction. Removing Lys236 caused the smallest average increase in esterase activity. Variants with the Lys236Met and Lys236Gly substitutions had the lowest average esterase activity, 2000 and 3300 M−1 min−1.
Substitutions to create a new catalytic mechanism are expected to show positive cooperativity since efficient catalysis requires all the components to function. Adding parts of the mechanism should not create a good catalyst, but adding the last piece will allow the entire mechanism to work. Indeed, some of the double and triple substitutions in HbHNL show this expected positive cooperativity, Figure 3B. The four single substitutions have little effect on esterase activity; only one – Lys236Met – increased it significantly. Three of the double substitutions – Thr11Gly-Lys236Gly, Thr11Gly-Lys236Met, and Glu79His-Lys236Met — showed esterase activity similar to the sum of the individual single substitutions; that is, almost no cooperativity. The remaining two double substitution variants – Glu79His-Lys236Gly and Thr11Gly-Glu79His – showed 5.8 to 7.4 fold higher esterase activity than expected from adding the effects of the individual single substitutions; that is, significant positive cooperativity. Both of the triple substitution variants showed positive cooperativity. The esterase activity of Thr11Gly-Glu79His-Lys236Met was 1.4-fold higher than expected from the sum of the effects of the single substitutions, while the esterase activity of HbHNL-TM (Thr11Gly-Glu79His-Lys236Gly) was 19-fold higher. This positive cooperativity in HbHNL-TM was essential to achieve high esterase activity.
The positive effect of the Glu79His substitution is due to the removal of the disruptive effect of the glutamate, not a positive effect of the adding the histidine. Saturation mutagenesis shows that many substitutions (13 of the 19 possible) at position 79 yield variants with higher esterase activity than the Glu79 variant. Only a glycine substitution at 79 yielded lower esterase activity. The five other unsuccessful substitutions caused protein misfolding, so some of these may have been suitable at position 79 if the protein had folded properly. Among the substitutions, the Glu79His substitution is the most effective one, but since so many variants yielded higher activity than glutamate, it is the removal of glutamate, not addition of histidine, that is the key change. Molecular dynamics simulation suggests that the glutamate can disrupt ester hydrolysis by accepting a hydrogen bond from the catalytic histidine, thereby preventing it from hydrogen bonding to the tetrahedral intermediate. This disruption does not occur in the hydroxynitrile lyase reaction of HbHNL presumably because the glutamate accepts hydrogen bonds from Lys236.
As a single substitution, the Lys236Met substitution gave the highest esterase specific activity – 8.4 mU/mg, a 9.5-fold increase over wild type, Table 1, Figure 3B. The Lys236Gly single substitution did not yield soluble protein so was assigned a low specific activity of < 0.5 mU/mg. However, when combined with the Thr11Gly and Glu79His substitutions, the triple substitution variant containing Lys236Met had lower esterase activity (14 mU/mg) than the triple substitution variant containing Lys236Gly (35 mU/mg). Adding the Lys236Met substitution to HbHNL-Thr11Gly-E79H had no effect on esterase activity. A possible rationalization is that the large size of the methionine partly blocks the esterase-hindering interaction between Glu79 and the catalytic histidine, but the large size also hinders the esterase reaction. Addition of Glu79His to HbHNL-Thr11Gly-Lys236Met only increased the specificity constant fourfold, but addition of Glu79His to HbHNL-Thr11Gly-Lys236Gly increased the specificity constant tenfold. This difference suggests that the unfavorable interaction between Glu79 and the catalytic histidine is less important when Lys236Met is present than when Lys236Gly is present. Similarly, adding the Thr11Gly substitution to HbHNL-Lys236Gly increased the esterase activity specificity constant at most 3 fold, presumably because the unfavorable interaction between Glu79 and the catalytic histidine prevented reaction. In contrast, adding Thr11Gly to HbHNL-Lys236Met increased esterase activity 9.8 fold. When the unfavorable interaction between Glu79 and the catalytic histidine is removed by the Glu79His substitution, adding the Lys236Met substitution has no greater esterase activity than Lys236Met alone.
Since the removal of glutamate at position 79 was important to create esterase activity, it is initially surprising that many esterases and lipases, for example, the Candida rugosa family of lipases, contain a glutamate at the corresponding position. However, the predicted pKa of this glutamate is high, indicating that it remains un-ionized at neutral pH. Molecular dynamics simulations also showed that the un-ionized glutamate does not interfere with the catalytic histidine in CRL. An electrostatic charge map of the active site of CRL shows a strong negative charge.22 The CRL family of lipases contain a fatty acid binding tunnel that holds the product fatty acid through many hydrophobic contacts. This negative charge may promote release of the product fatty acid. This negative charge also prevent ionization and potential disruption of the catalytic histidine by the glutamate.
While this paper considers the conversion of an HNL to an esterase, amino acid sequence analysis and phylogenetic trees indicate that HNL’s in plants diverged from esterases, not vice versa. HNL activity arose ~100 million years ago from esterases and several other enzyme classes when flowering plants and insects species proliferated. Below we consider the hypothetical reverse evolution of an esterase from HNL.
Divergent evolution of an enzyme can follow one of three paths as mutations accumulate: 1) a simultaneous monotonic decrease in the original activity and concomitant increase in new activity, 2) a specialists path where the original activity decreases followed by an increase in the new activity or 3) a generalist path where the new activity increases followed by a decrease in the second activity, Figure 4A.23
Figure 4.

Switching an enzyme from one activity to another. A) The hypothetical paths to replace the old catalytic activity with the new one. The triangles show the direction of evolution. Adapted from reference 18. B) The best path from a hydroxynitrile lyase (HbHNL) to an esterase (SABP2) lies below the dotted line connecting the activities of the two natural enzymes indicating that esterase activity drops more quickly than hydroxynitrile lyase activity increases. The axes mark the detection limit for each enzymatic activity. The black dots indicate all the variants and natural enzymes tested in this paper, but for clarity only some of them are labelled. Activities are log10 of specific activity in units of mU/mg; 5 mM mandelonitrile and 0.3 mM pNPAc. Adding an additional 142 amino acid substitutions and 3 insertions to HbHNL-TM would create the SABP2 sequence.
In some cases promiscuous activities can coexist with the native activities (the generalist path in Figure 4A), but these examples involve similar catalytic mechanisms for both the native and promiscuous activities.23 For example, during the initial stages of directed evolution of a phosphtriesterase into a carboxylic acid esterase, the variants maintained the original phosphotriesterase activity as the new activity increased.24 In the first six rounds, the phosphotriesterase activity dropped only 28-fold, while the carboxylic acid esterase activity increased 1,600-fold. These changes created a generalist enzyme that catalyzed both reactions. Although the transition states differ for the two reactions (pentavalent transition state for the phophotriesterases as compared to tetrahedral for carboxylic acid esterase), both are hydrolyses where the active site zinc ions stabilize negative charges in the transition state, so the same catalytic machinery can catalyze both reactions.
In another example, an x-ray crystal structure of a generalist enzyme showed how it accommodates different substrates.25 Isopropylmalate isomerase catalyzes the isomerization of 2-isopropyl and 3-isopropyl-L-malate. Catalysis involves a dehydration/hydration and requires recognition of the hydrophobic isopropyl group. The isopropylmalate isomerase from hyperthermophilic archaea Pyrococcus horikoshii catalyzes an additional reaction – a similar isomerization of homocitrate to homoisocitrate (homoaconitase activity) in the lysine biosynthesis pathway. Homocitrate contains a polar substituent (–CH2CH2COO−) at the spot where 2-isopropylmalate contains a nonpolar isopropyl group. The x-ray structure of this enzyme revealed a flexible loop in the active site that lacks an arginine present in many homoaconitases. The lack of an arginine likely reduces the affinity for homocitrate, but allows it to accept isopropylmalate. In addition, the flexible loop may adopt different conformations when acting on the two different substrates. In this second example, the catalytic mechanism is similar both reactions so that a generalist is possible. Our case differs from these two because the catalytic mechanisms differ and are incompatible.
The catalytic mechanisms of hydroxynitrile lyase and esterase contain incompatible elements. Esterases use an oxyanion hole, while HNL’s block the oxyanion hole. Esterases require space for hydrophobic alcohol moiety of the ester, while HNL‘s require a small charged cyanide in that region. It seems unlikely that one enzyme could efficiently catalyze both reactions, so the conversion of an HNL to an esterase is most likely to follow paths 1 or 2. The hypothetical mutation path of highest enzymatic activities, Figure 4B, shows a loss of HNL activity first, followed by a gain of esterase activity. Thus, the path is closest to path 2 for the first mutation, but the remaining two mutations lie closest to path 1. The final variant had 200 fold lower HNL specific activity and 36 fold higher in esterase specific activity, which is a 7,200-fold change. The same comparison using specificity constants (kcat/KM) shows 16,000-fold change.
Although we identified the Glu79His substitution as a major contributor to esterase activity, there are additional substitutions needed to make an efficient esterase. SABP2 differs from HbHNL-TM by 142 substitutions and 3 insertions and is an 8.6-fold better esterase. Some of these changes must be responsible for the higher esterase activity of SABP2 as compared to HbHNL-TM, but we do not know which ones. Adding to the uncertainty of the features needed to be a good esterase is the fact that SABP2 is a relatively poor esterase. The specificity constant of Pseudomonas fluorescens esterase is 380-fold higher than that for SABP2, so additional substitutions in HbHNL-TM could increase esterase activity ~3,300-fold. These differences in catalytic activity may be due to additional disruption of the catalytic machinery similar to the one discussed in this paper or may be due to poor substrate fit or undefined dynamic properties of the protein.
Experimental Section
General
Chemicals were bought from Sigma Aldrich and used as received unless otherwise noted. All Escherichia coli strains were grown in lysogeny broth (LB, 10 g tryptone, 5 g yeast extract, 10 g NaCl per liter of water) autoclaved for 20 min at 121 °C before use. If the bacterial strain had a selectable marker for ampicillin, ampicillin (100 µg/mL) was added once the media cooled to 50 °C (LB Amp). For agar plates 7.5 g/L of agar was added before autoclaving. Chemically competent cells26 were prepared from a single colony from a streak plate of commercial stock of E. coli DH5α (Life Technologies) for cloning plasmids and E. coli BL21 (Life Technologies) for protein expression. Protein concentrations were measured using the Bradford dye-binding assay (Bio-Rad; Coomassie Brilliant Blue G-250) and bovine serum albumin as the protein standard. Protein purity was established using SDS-PAGE using 4–12% polyacrylamide gradient gel (NuPage, Life Technologies) running in 50 mM MOPS, 50 mM Tris Base, 0.1% SDS, 1 mM EDTA, pH 7.7 and stained with Coomassie Blue (SimplyBlue SafeStain, Life Technologies).
Plasmids
The DNA encoding Hevea brasiliensis hydroxynitrile lyase (HbHNL, 774 base pairs)27 received from Prof. Dr. Helmut Schwab (Technische Universität Graz, Austria), was cloned into the vector pSE42028 (Life Technologies) at the Nco I-Hind III restriction sites. This vector contains an amp+ selectable marker and a lacO operator to regulate transcription. The salicylic acid binding protein 2 (SABP2) gene containing a C-terminal 6xHis tag was inserted in a pET21a plasmid (EM Biosciences), which contains an ampicillin selectable marker and a lacO operator to regulate transcription. Plasmid pJOE2792 contained the gene encoding esterase from Pseudomonas fluorescens (PFE) with an C-terminal 6xHis tag, an ampicillin selectable marker and a rhamnose inducible promoter.17 Plasmids were transformed into E. coli using heat shock. Plasmid (75 ng) was added to competent cells of E. coli DH5α (50 µL; 2.5×108 cells) cooled in an ice bath and incubated for 20 min. The mixture was warmed to 42 °C for 1 min, then returned to the ice bath for 2 min. LB media (1 mL) was added and the mixture was incubated for 1 h at 37 °C with shaking at 225 rpm. An aliquot (150 µL) was plated on an LB Amp plate and incubated overnight at 37 °C. The resulting bacterial colonies were picked and grown overnight in 5 mL of LB Amp media. Plasmids were isolated using the Qiaprep Spin Miniprep Kit (Qiagen Valencia, CA) following the procedure for 3 mL of culture. Plasmids were sequenced by ACGT Incorporated (Wheeling, IL) using a single pass with the sequencing primer.
Site Directed Mutagenesis
The QuickChange method (Stratagene) introduced mutations in pSE420 HbHNL by incorporating the mutagenesis primer via PCR. The polymerase chain reaction (total volume 50 µL) used ~60 ng of template, 1.25 units of Pfu DNA polymerase, 5 µL of Reaction Mix, and 50 nmoles of both forward and reverse mutagenesis primers, Table 4. The temperature program was: initial 94 °C for 2 min, then 25 cycles of denaturation at 94 °C for 15 s, annealing at 5 °C below the calculated Tm for the primer29 for 30 s, and extension at 68 °C for 5.5 min. The final step concluded with a 2 min extension time 68 °C followed by cooling to 10 °C for storage. The PCR product was digested with Dpn I (0.5 µL, 2.5 U, 1.5 h, 37 °C) to remove template DNA and the digest was transformed by heat shock into E. coli DH5α and plated on LB Amp plate. A colony was picked, grown overnight in 5 mL of LB Amp media, and the plasmid was isolated with QIAquick spin kit (Qiagen). The mutation was confirmed by DNA sequencing and the plasmid was transformed into E. coli BL21 for protein expression.
Table 4.
Polymerase chain reaction primers for mutagenesis and sequencing.a
| Primer Name | Sequence |
|---|---|
| HbHNL -E79H-F | GGT GAT TCT GGT TGG CCA CAG CTG TGG AGG ACT CAA TAT AGC |
| HbHNL -E79H-R | GCT ATA TTG AGT CCT CCA CAG CTG TGG CCA ACC AGA ATC ACC |
| HbHNL -T11G-F | GTT CTT ATT CAT GGC ATA TGC CAC GG |
| HbHNL -T11G-R | CCG TGG CAT ATG CCA TGA ATA AGA AC |
| HbHNL -K236G-F | GGT GGA GAT CAT GGC TTG CAG CTT AC |
| HbHNL-K236G-R | GTA AGC TGC AAG CCA TGA TCT CCA CC |
| HbHNL-K236M-F | GTG GAG ATC ATA TGT TGC AGC TTA C |
| HbHNL-K236M-R | GT AAG CTG CAA CAT ATG ATC TCC AC |
| HbHNL-E79Xb | GGT GAT TCT GGT TGG XXX CAG CTG TGG AGG ACT CAA TAT AGC |
| SABP2-H80Xb | G AAG GTT ATA TTA GTG GGG XXX AGT CTT GGT GGT ATG AAT TTG GG |
| T7 Plasmid Sequencing | TAA TAC GAC TCA CTA TAG GG |
| pSE420 Sequencing | CGA CTC ACT ATA GGG GAA TTG TGA GC |
All oligonucleotides are for mutagenesis except for the last two, which are for the single pass sequencing. F indicates forward primer; R indicates reverse primer. Bold indicates nucleotides that have been changed and the underline indicates codon changes.
Represents 20 different, separate primers each encoding one of the 20 amino acids. XXX represents the three nucleotides to encode each amino acid.
Expression and Purification
A 2-mL preculture of BL21 E. coli transformed with desired plasmid was added to 200 mL LB Amp media and incubated at 37 °C with shaking at 225 rpm until the OD600 reached 0.6 to 0.8. The culture was cooled to 18 °C, isopropyl β-D–1-thiogalactopyranoside was added to a final concentration of 0.75 mM, and the mixture was incubated for 22 h at 18 °C with shaking at 200 rpm. The culture was centrifuged at 9,000 rpm for 10 min at 4 °C and the supernatant discarded. The cells were resuspended in Tris-HCl (50 mM tris(hydroxymethyl)aminomethane, pH 7.5, 15 mL) and lysed by sonication (Branson 250) at 40% amplitude, 3 s ON, 6 s OFF, for a total of 5 min. The mixture was centrifuged at 15,000 rpm for 30 min and the supernatant was purified by one of the procedures below. A 200 mL culture of HbHNL typically yielded 1 mg of purified protein.
For purification of supernatant HbHNL from lysate, 15 mL from a 200 mL culture, was added to Sepharose-Q resin (Biorad, Hercules, CA, 5 mL) in a 1.5×12 cm column and washed with six resin volumes the running buffer (Tris HCl, 50 mM, pH 7.5). Contaminating proteins were eluted with six column volumes of running buffer containing 60 mM NaCl. The target protein was eluted with six column volumes of 110 mM NaCl while collecting 10-mL fractions. The fractions were analyzed by SDS-PAGE and the fractions containing the 30 kDa protein were combined and concentrated to ~5 mg protein/mL. A 500-µL sample of this solution was added to a gel filtration column (Sephadex G-75 in two XK–16 GE columns in series, 150 mL total) equilibrated with 50 mM Tris-HCl and 300 mM NaCl. The column was eluted at 0.5 mL/min. The first 40 mL were discarded and thereafter 1-mL fractions were collected. The fractions containing the purest HNL protein were combined.
For purification of SABP2 and PFE, 10 mL of lysate from a 100 mL culture was added to 1 mL of Ni-NTA resin (Qiagen) in a 30 ml column. The column was washed with 9 column volumes of 50 mM Na2HPO4, 300 mM NaCl, and 50 mM imidazole, pH 7. Protein was then eluted with 50 mM Na2HPO4, 300 mM NaCl, and 220 mM imidazole, pH 7. To decrease concentration of salts and imidazole, the protein was added to a Millipore spin column (10 kDa cutoff) and washed three times with 10 ml of 50 mM Na2HPO4, pH 7.2. SDS-PAGE gels analyzed purity of fractions and target protein was collected from corresponding fraction. A 100 mL culture of SABP2 and PFE typically yielded 5 mg of protein.
All proteins were prepared for assays by buffer exchange by concentrating with Millipore spin columns (10 kDa cutoff) and washing three times with BES buffer (5 mM N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid, pH 7.2, 10 mL) with a final concentration of 1–2 mg protein/mL. Typical purity analyzed by SDS-PAGE gels is shown in supporting information. Crude extracts of E. coli BL21 contained a small amount of esterase activity, but the combined ion exchange and gel filtration removed this impurity; see supporting information.
Enzyme Assays
Experimental data was fit to the Michaelis Menten equation by non-linear fitting using a spreadsheet (Solver function of Microsoft Excel).30 One unit of enzyme activity corresponds to one micromole of product formed per minute. The constant kcat (min−1) was determined by Vmax (in U/mg)·(29,460 g/mol HbHNL)/1000.
Hydroxynitrile lyase activity (mandelonitrile).31 HNL activity was measured by monitoring the release of benzaldehyde from racemic mandelonitrile at 280 nm at 25 °C, pH 5.0. The reaction mixture (200 µL) in a 96-well microtiter plate contained 10 µL of enzyme (10–20 µg protein), 10 µL 0.5–20 mM mandelonitrile in 4.9 mM citric acid buffer pH 2.3, and 180 µL 50 mM citrate buffer pH 5. The activity was calculated using the equation below from the linear increase in absorbance (R2 typically > 0.97) over the first 10 min. The activities are corrected for the spontaneous cleavage of mandelonitrile, typically 4 mAbs/min at 5 mM.
Hydrolysis of p-nitrophenyl acetate.32 Esterase activity was measured by monitoring the release of p-nitrophenoxide upon hydrolysis of p-nitrophenyl acetate. The assay solution (100 µL volume) contained buffer (4.2 mM BES pH 7.2) with p-nitrophenyl acetate (0.05–1.2 mM; 0.3 mM for specific activity measurements), 6.7 vol% acetonitrile and 10 µL of protein solution. The reaction was monitored at 404 nm for 10 min and slope with R2 above 0.99 was used in the calculation of U/mg from mAbs/min with the equation below. The activities are corrected for spontaneous hydrolysis of p-nitrophenyl acetate, typically 0.27 mAbs/min at 0.3 mM.
Hydrolysis of simple esters. The hydrolysis of ethyl acetate, methyl benzoate, ethyl acetoacetate, phenyl acetate, and methyl salicylate were measured using p-nitrophenol (pNP) as the pH indicator at pH 7.2. Release of carboxylic acid slightly decreases the pH of the solution leading to a decrease in the yellow p-nitrophenoxide (ε404 nm 6220 M−1 cm−1). Extinction coefficient was calculated with reaction conditions using a gradient of the respective released acid and measuring the change in abs at 404 nm for greater than 10 min. The reaction of 100 µL contained 0.4 mM pNP, 1.5 mM BES pH 7.2, 5% MeCN, varying concentrations of substrate, and 10 µL of enzyme.
Hydroxynitrile lyase activity (acetone cyanohydrin).33, 34 Cleavage of acetone cyanohydrin was measured by incubating a reaction mixture for 2 mins, and detecting cyanide released by forming a blue dye via the König reaction. The reaction mixture (160 µL) in a 96-well microtiter plate contained enzyme (10 µL, 10–20 µg protein), acetone cyanohydrin (10 µL, 0.5–20 mM in 4.9 mM citric acid buffer pH 2.3), and citrate buffer (140 µL 50 mM, pH 5). Cyanide anion is oxidized by addition of a solution (10 µL) of N-chlorosuccinimide (60 mM) andsuccinimide (800 mM). After 2 mins a solution (30 µL) of isonicotinic acid (6.5 mM), barbituric acid (125 mM), and NaOH (200 mM) was added and the increase in absorbance monitored at 600 nm for 15 min. The standard curve for sodium cyanide had a slope of 0.352 mAbs/min/M.
pH dependence of the rate of hydrolysis of p-nitrophenyl acetate. p-Nitrophenyl acetate hydrolysis was measured at pH 5.5 to 9.0 using the following buffers (20 mM): citrate (pH 3.5 to 6.0), BES (pH 6.5 to 7.5), and Tris HCl (pH 8.0 to 9.0). The p-nitrophenyl acetate concentration (1 mM) was 5-fold higher than its KM measured at pH 7.2 (0.2 mM), so the measured activities correspond to Vmax. The extinction coefficients for p-nitrophenoxide vary with pH, Table 5.
Table 5.
Extinction coefficient for p-nitrophenol at in 6.7 vol% acetonitrile at differing pH.a
| pH | ε404 nm (M−1cm−1) |
|---|---|
| 3.5 | 51.7 |
| 4 | 73.8 |
| 4.5 | 138 |
| 5 | 290 |
| 5.5 | 759 |
| 6 | 2410 |
| 6.5 | 5380 |
| 7 | 9410 |
| 7.5 | 13300 |
| 8 | 16200 |
| 8.5 | 17600 |
| 9 | 18100 |
The absorbance at 404 nm was measured for solutions of p-nitrophenol (0.1 −1 mM). The slope of a plot of absorbance versus concentration yielded the extinction coefficient.
The kinetic pKa values were determined by fitting the measured values of Vmax to the equations below to find Kes, the ionization constant of the catalytic species; –log Kes gave the kinetic pKa.35 For the enzymes that showed a decrease in activity a high pH, an equation including a second kinetic pKa was used to fit the data, Figure S9.
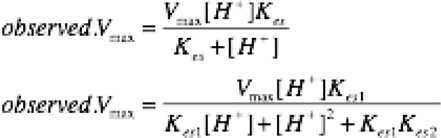 |
Molecular Modeling
Starting from the x-ray crystal structure of HbHNL (1YB6)10 and Candida rugosa lipase isoenzyme 1 (1CRL36) the Schrodinger Maestro software (Schrödinger Software Suite, Vol. 9.3; Maestro-Desmond Interoperability Tools, version 3.1) was used to add hydrogens, delete water molecules more than 5 Å from the protein, adjust the protonation state to match pH 7.0 with PROPKA 3.1, and optimize the side chain orientation by restrained minimization of heavy atoms to RMSD to 0.3 Å using OPLS (Optimized Potential for Liquid Simulations)-2005 force field.37 Esterase activity was modeled by molecular dynamics simulation of the first tetrahedral intermediate for phenyl acetate hydrolysis (HbHNL variants), and ethyl acetate (CRL variants). The substrate was covalently attached to the serine and geometry was optimized with Maestro clean-up. The system was solvated with SPC (simple point charge) water model 10 Å beyond protein edge using orthorhombic system and neutralized with 5 Na+ and 0.05 M NaCl. The system was geometry optimized with Desmond (Desmond Molecular Dynamics System, version 3.1, D. E. Shaw Research, New York) using a convergence threshold of 1.0 kcal/mol/Å with maximum iterations of 2000. Molecular dynamics within Desmond was used to simulate 1.2 ns, recording energy every 1.2 ps and a trajectory every 4.8 ps. Temperature was set to 300 K and pressure to 1.01325 bar. Simulations were analyzed with Desmond’s Simulation Event Analysis to observe hydrogen bonds, which were defined as a distance between heavy atoms ≤3.2 Å and an angle of X-H-X > 120°. Images were created using PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC).
Supplementary Material
Acknowledgments
We thank the US National Science Foundation (CBET-0932762, CHE-1152804) and US National Institutes of Health (1R01GM102205-01) for financial support, the Minnesota Supercomputing Institute for use of computers and software, and Prof. Dr. Helmut Schwab (Technische Universität Graz, Austria) for the plasmid for expressing HbHNL, Nahomi Carrasquillo-Tirado (U. of Puerto Rico) and Reto Berchtold and Bryan J. Jones for performing some experiments and Prof. Dr. Uwe T. Bornscheuer (U. of Greifswald) for information from the α/β-hydrolase 3DM database about amino acid frequencies in esterases and lipases and Prof. Dr. Martina Pohl (Jülich Research Center) for information about Dr. A. Hickel’s Ph.D. thesis and Prof. Dr. Ulf Hanefeld (Technical U. Delft) for helpful discussions.
References
- 1.Padhi SK, Fujii R, Legatt GA, Fossum SL, Berchtold R, Kazlauskas RJ. Chem. Biol. 2010;17:863–871. doi: 10.1016/j.chembiol.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 2.Blomberg R, Kries H, Pinkas DM, Mittl PRE, Grütter MG, Privett HK, Mayo SL, Hilvert D. Nature. 2013;503:418–421. doi: 10.1038/nature12623. [DOI] [PubMed] [Google Scholar]
- 3.Syrén P-O, Hult K. ChemCatChem. 2011;3:853–860. [Google Scholar]
- 4.Jung S, Kim J, Park S. RSC Adv. 2013;3:2590–2594. [Google Scholar]
- 5.Pavlova M, Klvana M, Prokop Z, Chaloupkova R, Banas P, Otyepka M, Wade RC, Tsuda M, Nagata Y, Damborský J. Nat. Chem. Biol. 2009;5:727–733. doi: 10.1038/nchembio.205. [DOI] [PubMed] [Google Scholar]
- 6.Wittrup Larsen M, Zielinska DF, Martinelle M, Hidalgo A, Jensen LJ, Bornscheuer UT, Hult K. ChemBioChem. 2010;11:796–801. doi: 10.1002/cbic.200900743. [DOI] [PubMed] [Google Scholar]
- 7.Jiang Y, Morley KL, Schrag JD, Kazlauskas RJ. ChemBioChem. 2011;12:768–776. doi: 10.1002/cbic.201000693. [DOI] [PubMed] [Google Scholar]
- 8.Yin DLT, Kazlauskas RJ. Chem. Eur. J. 2012;18:8130–8139. doi: 10.1002/chem.201200052. [DOI] [PubMed] [Google Scholar]; Yin DLT, Purpero VM, Fujii R, Jing Q, Kazlauskas RJ. Chem. Eur. J. 2013;19:3037–3046. doi: 10.1002/chem.201202027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selmar D, Lieberei R, Biehl B, Conn EE. Physiol Plantarum. 1989;75:97–101. [Google Scholar]
- 10.Hasslacher M, Kratky C, Griengl H, Schwab H, Kohlwein SD. Prot. Struct. Func. Bioinform. 1997;27:438–449. [PubMed] [Google Scholar]; Gartler G, Kratky C, Gruber K. J. Biotechnol. 2007;129:87–97. doi: 10.1016/j.jbiotec.2006.12.009. [DOI] [PubMed] [Google Scholar]; Schmidt A, Gruber K, Kratky C, Lamzin VS. J. Biol. Chem. 2008;283:21827–21836. doi: 10.1074/jbc.M801056200. [DOI] [PubMed] [Google Scholar]
- 11.Forouhar F, Yang Y, Kumar D, Chen Y, Fridman E, Park SW, Chiang Y, Acton TB, Montelione GT, Pichersky E, Klessig DF, Tong L. Proc. Natl. Acad. Sci. U. S. A. 2005;102:1773–1778. doi: 10.1073/pnas.0409227102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Park SW, Kaimoyo E, Kumar D, Mosher S, Klessig DF. Science. 2007;318:113–116. doi: 10.1126/science.1147113. [DOI] [PubMed] [Google Scholar]
- 12.von Langermann J, Nedrud D, Kazlauskas RJ. ChemBioChem. 2014;15 doi: 10.1002/cbic.201402081. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin DLT, Bernhardt P, Morley KL, Jiang Y, Cheeseman JD, Purpero V, Schrag JD, Kazlauskas RJ. Biochemistry. 2010;49:1931–1942. doi: 10.1021/bi9021268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gruber K, Gartler G, Krammer B, Schwab H, Kratky C. J. Biol. Chem. 2004;279:20501–20510. doi: 10.1074/jbc.M401575200. [DOI] [PubMed] [Google Scholar]
- 15.Hickel A. Screening, kinetic and stabilization investigations of the enzyme hydroxynitrile lyase from Hevea brasiliensis. Ph.D. Thesis. Graz, Austria: Graz Technical University; 1996. pp. 90–91. [Google Scholar]
- 16.Kourist R, Jochens H, Bartsch S, Kuipers R, Padhi SK, Gall M, Böttcher D, Joosten H-J, Bornscheuer UT. ChemBioChem. 2010;11:1635–1643. doi: 10.1002/cbic.201000213. [DOI] [PubMed] [Google Scholar]
- 17.Krebsfanger N, Schierholz K, Bornscheuer UT. J. Biotechnol. 1998;60:105–111. doi: 10.1016/s0168-1656(97)00192-2. [DOI] [PubMed] [Google Scholar]
- 18.Hu CH, Brinck T, Hult K. Int. J. Quantum Chem. 1998;69:89–103. [Google Scholar]
- 19.Hur S, Bruice TC. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12015–12020. doi: 10.1073/pnas.1534873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forming an ion pair with a nearby glutamate would stabilize the protonated form of the catalytic histidine and therefore raise its thermodynamic (or equilibrium) pKa. However, measurement of the kinetic pKa showed no change. The kinetic pKa measures the pH dependence of catalysis, thus, the pKa of the catalytically active form. The distorted form, which can form the ion pair with Glu79, is not catalytically active, so it does not contribute to the kinetic pKa.
- 21.Li H, Robertson AD, Jensen JH. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]; Bas DC, Rogers DM, Jensen JH. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]; Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. J. Chem. Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]; Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH. J. Chem. Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 22.Grochulski P, Bouthillier F, Kazlauskas RJ, Serreqi AN, Schrag JD, Ziomek E, Cygler M. Biochemistry. 1994;33:3494–3500. doi: 10.1021/bi00178a005. [DOI] [PubMed] [Google Scholar]
- 23.Khersonsky O, Roodveldt C, Tawfik DS. Curr. Opin. Chem. Biol. 2006;10:498–508. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 24. Tokuriki N, Jackson CJ, Afriat-Jurnou L, Wyganowski KT, Tang R, Tawfik DS. Nat. Commun. 2012;3:1257–1259. doi: 10.1038/ncomms2246. see also Aharoni A, Gaidukov L, Khersonsky O, Gould SM, Roodveldt C, Tawfik DS. Nat. Genet. 2004;37:73–76. doi: 10.1038/ng1482.
- 25.Yasutake Y, Yao M, Sakai N, Kirita T, Tanaka I. J. Mol. Biol. 2004;344:325–333. doi: 10.1016/j.jmb.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 26.Sambrook J, Russell DW. Molecular Cloning: a Laboratory Manual 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. pp. 1.112–1.116. [Google Scholar]
- 27.Hasslacher M, Schwab H, Schall M, Hayn M, Griengl H, Kohlwein SD. J. Biol. Chem. 1996;271:5884–5891. doi: 10.1074/jbc.271.10.5884. [DOI] [PubMed] [Google Scholar]
- 28.Brosius J. DNA. 1989;8:759–777. doi: 10.1089/dna.1989.8.759. [DOI] [PubMed] [Google Scholar]
- 29.Kibbe WA. Nucleic Acids Res. 2007;35:W43–W46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kemmer G, Keller S. Nat. Protoc. 2010;5:267–281. doi: 10.1038/nprot.2009.182. [DOI] [PubMed] [Google Scholar]
- 31.Xu LL, Singh BK, Conn EE. Arch. Biochem. Biophys. 1986;250:322–328. doi: 10.1016/0003-9861(86)90733-2. [DOI] [PubMed] [Google Scholar]
- 32.Pocker Y, Stone JT. J. Am. Chem. Soc. 1965;87:5497–5498. doi: 10.1021/ja00951a049. [DOI] [PubMed] [Google Scholar]
- 33.Andexer J, Guterl J-K, Pohl M, Eggert T. Chem. Commun. 2006:4201–4203. doi: 10.1039/b607863j. [DOI] [PubMed] [Google Scholar]
- 34.König W. J. Prakt. Chem. 1904;69:105–137. [Google Scholar]
- 35.Fersht A, Freeman WH. New York: 1999. pp. 169–189. [Google Scholar]
- 36.Grochulski P, Li Y, Schrag JD, Bouthillier F, Smith P, Harrison D, Rubin B, Cygler M. J. Biol. Chem. 1993;268:12843–12847. [PubMed] [Google Scholar]
- 37.Jorgensen WL, Tirado-Rives J. J. Am. Chem. Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.