
Figure 5. An approach for the chemical optimization of allosteric modulators.
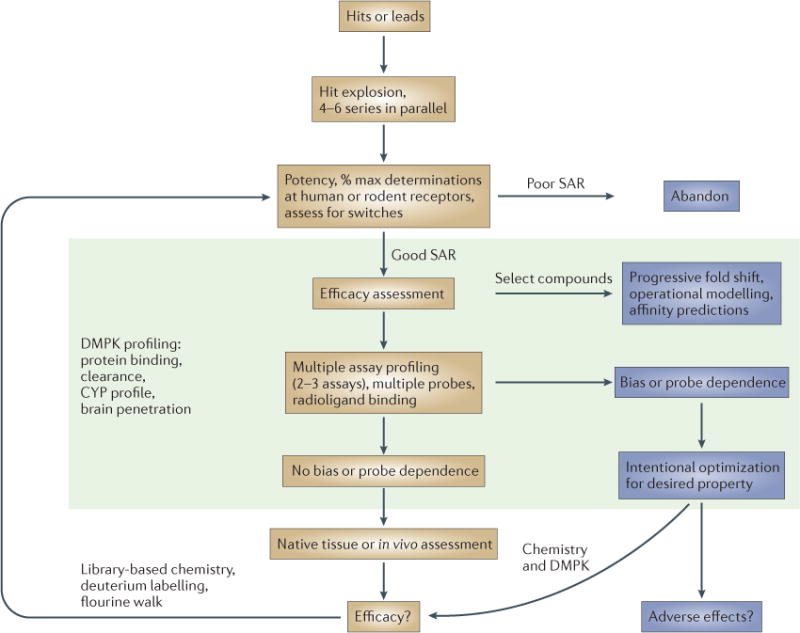
Our approach has been to drive an iterative chemistry effort primarily from the determination of the EC50 (effector concentration for half-maximum response) and maximal responses to an allosteric modulator. In the majority of cases, the most well-characterized endogenous ligand is used; in situations in which more than one endogenous ligand exists, profiling should include all relevant endogenous ligands, at least with selected compounds. Although the ‘% max’ values that are determined in this manner may be artificially capped (that is, the maximal response induced by the compound has reached the maximal response of the system), this capping can serve as an initial triage step to rank compounds as having ‘high’ versus ‘low’ efficacy. Such ranking can be validated with an efficacy experiment in which a constant amount of compound is incubated with increasing concentrations of orthosteric ligand (‘fold shift’). Selected compounds can then be progressed to produce concentration–response curves to examine the effects of multiple concentrations of allosteric modulator in the presence of a full orthosteric agonist. Application of the operational model of allosterism can then be used to predict compound affinity in the functional assay to be used for assessment. Additional profiling in other functional assays can be used to assess ligand bias; at this stage it is important to consider non-G-protein-dependent assays, such as β-arrestin recruitment. If an allosteric or orthosteric radioligand is available, compounds can be further profiled for displacement or effects on radioligand affinity. Intentional optimization of compounds with distinct in vitro profiles (signal bias, probe dependence) can then be pursued, with the goal of translating these properties into native or in vivo system models, such as for brain-slice electrophysiology or behavioural studies. Additionally, compounds with differential profiles can be tested for adverse effects to try to understand if a signalling profile that has been identified in vitro may predict efficacy and/or adverse effect profiles. Additional libraries of compounds can then be synthesized by rapidly identifying tractable structure–activity relationships (SARs) and using deuterium labelling or fluorine walk strategies to address the common finding that allosteric ligands often lose activity or switch pharmacology at the target with chemical modification. In parallel, drug metabolism and pharmacokinetics (DMPK) analyses (including the profiling of possible cytochrome-P450 (CYP) interactions and blood–brain barrier permeability) should be ongoing to identify compounds with the highest likelihood to progress.