

Abstract
Neuronal glucose transporter (GLUT) isoform 3 deficiency in null heterozygous mice led to abnormal spatial learning and working memory but normal acquisition and retrieval during contextual conditioning, abnormal cognitive flexibility with intact gross motor ability, electroencephalographic seizures, perturbed social behavior with reduced vocalization and stereotypies at low frequency. This phenotypic expression is unique as it combines the neurobehavioral with the epileptiform characteristics of autism spectrum disorders. This clinical presentation occurred despite metabolic adaptations consisting of an increase in microvascular/glial GLUT1, neuronal GLUT8 and monocarboxylate transporter (MCT) isoform 2 concentrations, with minimal to no change in brain glucose uptake but an increase in lactate uptake. Neuron-specific glucose deficiency has a negative impact on neurodevelopment interfering with functional competence. This is the first description of GLUT3 deficiency that forms a possible novel genetic mechanism for pervasive developmental disorders, such as the neuropsychiatric autism spectrum disorders, requiring further investigation in humans.
Keywords: Brain metabolism, seizures, cognition, monocarboxylate transporters, development
Autism spectrum disorders (ASD) constitute a chronic debilitating neuropsychiatric condition diagnosed in childhood and with long term consequences. At present there are no specific treatments targeted at complete reversal of symptoms towards achieving functional competence. ASDs are part of pervasive developmental disorders that present with abnormal social interactions, aberrant verbal and non-verbal communication skills, learning disabilities, mild mental retardation, stereotypic behaviors with a subset of individuals developing electroencephalographic seizure activity. These disorders are diagnosed in infancy/childhood and seen in adolescence and adults (1). While a genetic basis for this disorder has been put forth the underlying definitive cause has remained elusive. More recently proposed gene(s) for autism have been located on chromosome 16 which are responsible for only 1% of the clinical disorder (1). Mounting evidence has supported environmental factors such as perinatal events and nutrition to play an etiological role in ASD (2, 3). Positron emission tomography of human brain has revealed decreased brain glucose uptake in ASD (4, 5, 6). Abnormal neuronal energy metabolism due to aberrant fuel supply, particularly glucose, during development may form the common denominator for these disorders.
Glucose forms the essential substrate that fuels neuronal oxidative metabolism. This substrate is transported across the blood-brain barrier and into neurons and glia via a family of structurally related membrane-spanning glycoproteins, termed the facilitative glucose transporters (7). Of the multiple isoforms cloned to date, GLUT1 is the predominant isoform expressed in the blood-brain barrier and glial cells (8), and GLUT3 is the neuronal isoform (9). While other isoforms are expressed to a lesser extent in neurons, such as GLUT8 and GLUT4 (10, 11), GLUT3 is the predominant neuronal isoform that fuels ATP generation and thereby the energy metabolism (12). We therefore hypothesized that GLUT3 deficiency will alter brain metabolism and perturb neurobehavior through development resulting in a phenotype that may resemble clinical ASD. To test this hypothesis we created a glut3 null mouse and observed embryonic lethality in homozygotes (13) while the heterozygotes had no malformations and survived into adulthood. We examined these glut3 heterozygous null (glut3+/−) mice through development for neurobehavioral changes and observed features relevant to ASD.
Materials and Methods
Glut3 null mouse lines
The mouse glut3 (Slc2a3) knockout targeting construct was created by isolating and characterizing the entire glut3 gene in a P1 clone (Genome Systems, St. Louis, MO) and using the pKO Scrambler NTK 1903 vector (Stratagene, La Jolla, CA) to delete a 3.5 kb fragment consisting of exons 7, 8, 9 and the coding region of 10. (13). Glut3 (Slc2a3) knockout mouse lines were generated and back crossed to achieve a homogeneous C57/BL6 strain background, genotyped as described previously by us (13), housed in 12-h light and 12-h dark cycle and maintained in approved mouse housing areas. All mouse studies were approved by Animal Research Committee of the University of California, Los Angeles in accordance with guidelines set by the National Institutes of Health.
Glucose transporter protein studies
Whole brains at different postnatal stages (PN1, 7, 14, 21 and 60 days) were subjected to Western blot analysis as described previously (10, 11). The primary antibodies consisted of rabbit anti-mouse GLUT1 IgG (1:2000 dilution) (14), anti-mouse GLUT3 IgG (1:2000 dilution) (15), rabbit anti-mouse GLUT8 IgG, rabbit anti-rat MCT1 (1:200 dilution) and anti-rat MCT2 antibody (1:400 dilution) (the latter three antibodies were obtained from Alpha Diagnostic International, San Antonio, TX). The internal control was detected using a monoclonal anti-human vinculin antibody (1:4000 dilution) (Sigma Chemical Co., St. Louis, MO). The protein bands were quantified by densitometry and the arbitrary units expressed as a ratio to vinculin. The developmental expression of each protein was presented as a percent of PN60 WT value normalized to 100%.
Brain 2-deoxyglucose uptake
Cerebral 14C-2-deoxyglucose uptake in the PN60 WT and glut3+/− brains was measured using a modified Sokoloff method following the intraperitoneal administration of 0.5 μCi/gm of 2-deoxy-D-[14C]-glucose tracer. The specific activity of glucose in circulation and cerebral cortical tissue over a predetermined optimal duration allowed for the derivation of cerebral glucose uptake as described previously (16, 17).
Brain lactate uptake
Cerebral lactate uptake in the PN60 WT and glut3+/− brains was assessed using a modified Oldendorf method (18) in which a mixture of L[14C(U)]-lactate and [3H]-water (0.1 μCi each) was injected via the left ventricle (LV) with collection of whole brains 15–20 seconds later. The ratio of 14C to 3H in brain tissue relative to the ratio of 14C to 3H in the injectate determined the amount of lactate lost to brain tissue on a single passage through brain microcirculation.
Immunohistochemical studies
Paraformaldehyde pre-fixed vibratome floating coronal brain sections were subjected to immunofluorescence staining using the rabbit anti-mouse GLUT3, guinea-pig anti-GLUT1 or mouse anti-tubulin (1:500 dilution each) IgGs as primary antibodies with the fluorescent tag being FITC for GLUT1 and Texas Red for GLUT3 or tubulin. Slides with sections were examined under a Nikon E-600 fluorescence microscope (Nikon, Melville, NY) equipped with a cooled charged-coupled device (CCD) camera as previously described (11, 13). Neuronal nitric oxide synthase enzyme immunoreactivity was assessed by the primary rabbit anti-nNOS antibody (1:100 dilution) (BD Biosciences, San Jose, CA), biotinylated-peroxidase-DAB reaction and viewed under the light microscope and cells counted by the NIH image analysis system (10, 13).
Neuroligin-3 mRNA studies
Total RNA was isolated from snap frozen dissected hippocampi (RNeasy lipid tissue mini-kit, Qiagen, Valencia, CA) (19, 20). Real-time PCR primers used amplified a 75 bp isoform specific DNA fragment (forward:5′-ggagctggcagcattacagtt-3′, reverse:5′-aggcgaagtgtgtcatgtg-3′) of the mouse neuroligin-3 sequence (GenBank no. NM_172932). Taqman probe was the intervening sequence (5′-ccactcaccatgaatgtgaggccg-3′), which was synthesized and labeled with fluorescent dyes, 6-carboxyfluorescein (FAM) on the 5′ end and N,N,N,N-tetramethyl-6-carboxyrhodamine (TAMRA) on the 3′ end (Applied Biosystems, Foster City, CA). Quantitative real-time PCR (q-PCR) was performed in triplicate with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) serving as the internal control on the StepOnePlus™ Real-Time PCR system (Applied Biosystems, Foster City, CA). The amplification conditions consisted of initial 12-min activation at 95°C followed by 40 cycles of denaturation at 95°C for 30s, annealing at 58°C for 30s, and extension at 72°C for 30s. Relative quantification of the PCR amplification products was performed using the comparative critical threshold (CT) method as previously described (19, 20).
Continuous video-EEG monitoring
Following isoflurane anesthesia administration, the two leads of the EEG transmitter were operatively implanted on the dura through two drilled bilateral burr holes in the skull on each side of the fronto-parietal cranium, 1.4 mm in diameter, 3 mm posterior and 3 mm lateral to the Bregma. After surgery, the mice ranging in age from 4–6 to 8–12 months of age were transferred to the cage which was placed on top of the telemetry receiving platform (Data Sciences International, Arden Hills, MN) and continuous EEG monitoring was performed for 24–48 hours after three hours of recovery. Stellate software (Montreal, Quebec, Canada) was used to capture the data which was then manually reviewed offline for epileptiform events. Analysis was done based on amplitude, frequency and time with EEG evidence of seizures (21).
Neurobehavioral Studies
Different sets of naïve 4–6 month old mice were employed for SHIRPA, T-maze, Morris water maze and fear conditioning tests each. Another set of naïve mice of the same age was subjected to open field followed by the roto-rod testing after a minimal interval of 10 days between the two tests. This same set of mice underwent the radial arm maze test at 7–8 months of age. A different set of naïve mice at postnatal age of 7 days was subjected to the ultrasonic vocalization (USV) test and a separate set of 40 day old mice was engaged for social behavior testing and subsequently examined for stereotypic behaviors at 50 days of age. The same set along with a naïve set of mice at 60 days of age underwent the olfactory sensitivity testing.
SHIRPA primary screen
A primary observational screen based on a modified Irwin (22) profile was performed as described previously (23). Behavioral and functional profile screen included quantitative scoring to detect defects in gait or posture, motor control and coordination, changes in excitability and aggression, autonomic function such as salivation, lacrimation, piloerection, defecation and muscle tone.
Accelerating rotarod
Motor coordination was assessed with the accelerating rotarod (TSE-Systems, RotoRod 3375-5, Hamburg, Germany) as described previously (24) with slight modification. Each trial started either at a constant forward rotational speed of 5 rpm or at an accelerating speed beginning at 5 rpm up to 60 rpm. Trials were completed after experimental subjects fell of the rod or 300 s at a maximum speed of 60 rpm had elapsed, whichever came first. One daily trial was given for 4 consecutive days. Primary measure of interest was latency to fall.
Open Field Test
Spontaneous behavioral activity was recorded in an activity chamber (Med-Associates Inc, ENV-520, St. Albans, Vermont) as described previously (25). Exploratory activity was measured in a square Plexiglas enclosure for 20 min. Movement within the open field area was recorded by the computerized Med Associates Activity Monitor. Measured parameters included distance traveled, ratio of center distance traveled to total distance traveled, average velocity, ambulatory time, stereotypic behavior time, resting time and vertical time.
Sociability and social recognition
The social testing apparatus was a rectangular, three-chambered box with dimensions of each chamber being 16 in. in length × 9 in. in width × 9.25 in. in height. Dividing walls were made of clear Plexiglas containing small circular openings (3 inches in diameter) allowing access into each chamber. The procedure (Crawley social approach task) (26) involved 3 phases: habituation, tests of sociability and social recognition. The test mouse was first placed in the middle compartment and allowed to explore all 3 chambers for 10 min (apparatus did not contain any wire cages during habituation). After habituation, an unfamiliar mouse (stimulus mouse; age- and sex-matched to test mouse) was placed into one of the side compartments (constrained by a round wire cage that permitted nose contact between the bars but prevented extensive physical contact; stimulus mice were habituated to small wire cages for 5–10 minutes each for two days before testing), while the opposite side compartment only contained an empty wire cage. For evaluation of sociability, the test mouse was returned to the apparatus for 10 min. Active exploration (approach, sniffing, rearing) of the conspecific and the empty wire cage by the test mouse was recorded and scored offline. For the social recognition test, one side compartment of the apparatus contained a now familiar mouse (from the previous sociability phase), the other side an unfamiliar, novel mouse (both constrained by wire cages). Active exploration of familiar and novel unfamiliar mouse by the test mouse during the 10 min period was recorded for offline analysis and scored by an observer blinded to genotype.
Olfactory sensitivity test procedure
Two days prior to testing, an unfamiliar high carbohydrate containing food item (Froot loop) was placed in the home cages of the subject mice to ensure palatability. On the day of the test, each mouse was placed in a Plexiglas box (46 cm in length × 24 cm in width × 20 cm in height) containing 3 cm depth of bedding and allowed to explore for five minutes. The mouse was removed from the Plexiglas box and one Froot loop was buried in the bedding of the empty box. The mouse was returned to the box and allotted 15 min to locate the buried Froot loop. All activities were videotaped for offline analysis and the latency to find and consume the Froot loop was assessed (26).
Ultrasonic vocalization (USV) tests
Quantification of USVs at a frequency of 70 KHz was carried out at PN7 as described previously (27). Pups were removed from the breeding cages (contained dam and offspring in a temperature of 24–25°C) in groups of four and placed individually into glass containers (4″ × 3″ × 2.5″ in a temperature of 22–23°C) that were housed in sound attenuation chambers. A four-channel Noldus UltraVox system (Noldus Information Technology Inc, Leesburg, Virginia) was used to record ultrasonic emissions generated by each animal. Immediately after a six minute recording period, a tail biopsy was obtained from each animal to permit genotyping (13).
T maze acquisition and reversal learning
T maze test was performed as described previously (28). Briefly, mice were initially food-deprived until they had reached 85% of their ad libitum feeding body weight, habituated to the T-maze and shaped to consume 20 mg chow pellets (Test Diet Omnitreat Tab, 20 mg, Richmond, Indiana). Ten training trials per day were then initiated. For each mouse, one arm was designated as the correct arm that contained a food pellet during training trials (right and left side were counter balanced for both genotypes). At the beginning of each test session, mice were placed in the start box at the bottom of the T-maze stem. The start box door was opened, and the mice were given a choice of entering either arm. Upon making the correct choice, mice were given time to consume the food pellet before returning to the start box for the next trial. For each successive trial, the reward pellet was always placed in the same arm. Latency to enter an arm, number of errors, and number of days to criterion (80% correct responses on 3 consecutive days) was recorded by a human observer blind to genotype. Each mouse that met criterion for acquisition (66% of glut3+/− and 64% of WT) was subjected to reversal learning, during which the reinforcer location was switched to the opposite arm. Ten trials per day were administered for reversal learning, using the same methods and criterion.
Stereotypic behavior
Stereotypic behavior was assessed as described previously (29, 30). Mice (~50 days of age) were placed individually into a round Plexiglas cylinder (12 in. tall and 8 in. in diameter) and allowed to explore freely. Twenty-minute video recordings were made for each subject. Stereotypic behaviors including rotation, rears, sniffing, grooming and exploratory activity were subsequently scored by a human observer blinded to genotype.
Morris Water Maze Test
The hidden version of the Morris water maze (31) was performed to assess spatial learning. The task was carried out as described previously (32) with slight modification. Mice were initially trained to find an escape platform that was located in a constant location of the pool 1 cm below the water surface (pool diameter: 1 m; platform diameter: 12 cm; water temperature: 24°C). Animals were released into the pool from alternating starting positions. Training trials terminated when the animal remained on the platform for 1 s or 60 s had elapsed, whichever came first. Mice were allowed to remain on the platform for a 5 s post-trial interval. Two training trials per day were performed. The accuracy with which animals had learned the platform location was monitored with a single probe trial given after completion of training (probe trial duration: 60 s; platform was removed from the pool during probe trials). Visible platform training was given after the hidden version of the Morris water maze had been completed. Tracking information was processed by the HVS Water maze software package (HVS Image, Buckingham, United Kingdom). Primary measures of interest were escape latency for training trials and quadrant occupancy, target crossings and proximity for probe trials.
Radial Arm Maze
Spatial working memory was assessed using a win-shift version of the 8-arm radial maze (Lafayette Instrument Co, Lafayette, Indiana) as described previously (33). Mice were initially food-restricted (until they had reached 85% of their free-feeding body weight), shaped to consume 20 mg chow pellets (Noyes Precision Pellets, 20mg, P.J. Noyes Company, Inc, Lancaster, New Hampshire) and habituated to the maze (three 5 min sessions during which all arms were baited and accessible). Mice were given 1 daily session (consisting of 2 phases separated by 3 h) for 15 consecutive days on the radial arm maze. During phase A, animals were allowed to retrieve a 20 mg food pellet from 4 pseudo-randomly chosen arms of the 8-arm maze, while access to the remaining 4 arms was blocked. Phase A terminated after animals had retrieved all 4 food pellets or 5 min had elapsed, whichever came first. Mice were returned to the maze for phase B after a 3 h interval. During phase B, all arms were accessible but only the 4 previously blocked arms were baited. The baiting pattern of the maze was pseudo-random (no more than three adjacent arms were baited during either phase) and changed every session, as was the order in which the mice were run. Phase B terminated the same way as phase A after animals had retrieved all four food pellets or 5 min had elapsed, whichever came first. Three consecutive training sessions consisting of phases A and B were combined into one block session and a total of 5 block sessions were conducted. Phase B entries into arms that were baited only during phase A were scored as across-phase errors. Repeated entries into the same arm were scored as within-phase errors. In addition, total arm entries were scored and latencies to complete phase B recorded. As glut3+/− mice showed a reduced number of total arm entries, errors (across-phase errors, within-phase errors) were normalized to total arm entries. A minimum of 2 total arm entries during phase A was required for subjects to qualify for phase B error analysis.
Contextual Fear Conditioning
Mice were conditioned to the training context with 3×2 s, 0.75 mA shocks (placement to shock interval: 2 min; shocks were spaced by 1 min; training trial duration: 5 min) as described previously (34). Context fear was assessed after 24 h (5 min session). Freezing and activity levels were recorded and measured by an automated system (Med-Associates Inc, St. Albans, Vermont).
Statistical Analyses
All data are presented as mean±SEM. T-test or Fisher’s Exact test was employed for protein expression, metabolic and seizure studies. T-test with Bonferroni’s correction when indicated or two-way repeated-measures ANOVA was used for neurobehavioral studies. At a significance level of p < 0.05 for two-way repeated measures ANOVA, Tukey’s posthoc analysis was subsequently performed.
Results
Brain glucose and monocarboxylate transporters
The developmental profile of brain GLUT3 protein consisted of an age related increase from postnatal day (PN)1 and PN7 to that of PN14, 21 and 60 in both wild type (WT) and glut3+/− mice. The glut3+/− mice demonstrated lower GLUT3 protein concentrations throughout development (figure 1.I.a & b). Examination of intracellular localized neuronal GLUT8 (10) revealed no inter-genotype differences except at PN14 when glut3+/− mice exhibited higher concentrations (figure 1.I.a & c). The microvascular and glial GLUT1 (8) revealed an age-dependent increase in brain GLUT1 protein concentrations at PN21 and 60 in both WT and glut3+/− mice, however contrary to GLUT3, a higher concentration of GLUT1 was observed in glut3+/− mice versus WT at these two ages (figure 1.I.a & d). Investigation of the monocarboxylate transporters demonstrated no change in brain (microvascular and glial) MCT1 (35) through development and between the genotypes (figure 1.I.a & e). In contrast, neuronal MCT2 (35) increased with development at PN60 in WT mice, with the glut3+/− mice demonstrating an earlier increase from PN14 through PN60 when compared to the age-matched WT mice (figure 1.I.a & f).
Figure 1. I. Brain GLUT and MCT proteins.


a. Western blots show GLUT3, GLUT8, GLUT1, MCT1 and MCT2 with respective vinculin protein bands in PN 1–60 WT and glut3+/− mice. b–f. Densitometric quantification as a percent of PN60 value is shown (n = 7/genotype) b. GLUT3, *p < 0.05 vs. age-matched WT, #p < 0.05 vs. PN1 glut3+/−, **p < 0.01 vs. PN1 WT, c. GLUT8, *p < 0.05 vs. PN7 glut3+/−, #p < 0.05 vs PN14 WT, d. GLUT1, *p < 0.05 vs. age-matched WT, #p < 0.05 vs. PN1 WT, **p < 0.001 vs. PN1 glut3+/−, e. MCT1 and f. MCT2, *p < 0.05 vs. age-matched WT, **p < 0.01 vs PN1 WT, ***p < 0.001 vs PN1 glut3+/−. II. GLUT3 and GLUT1 immunofluorescence a, b. Sub-fornical organ (SFO) with GLUT3 in neuronal processes (red; arrows), GLUT1 in microvessels (green; arrowhead) and DAPI nuclear staining (blue) in PN60 WT (n = 10) and glut3+/− (n = 10) brain sections. c–d. Tubulin in cellular processes (arrows) is observed in PN60 WT (n = 4) and glut3+/− (n = 4) cerebral cortical regions. Scale bar = 100μm.
Brain 2-deoxyglucose and lactate uptake
These age-related changes in other GLUT and MCT isoforms led to a lower trend in glut3+/− versus WT in total brain 14C-2-deoxy-glucose uptake (WT = 1113±187 vs. glut3+/− = 887±91 μmol/mg/min; n = 12–13 each). In contrast, brain lactate uptake increased in glut3+/− versus WT mice (WT = 288±9 vs. glut3+/− = 324±14 μmol/mg/min, n = 6 each, unpaired t-test p < 0.05). Gross body weights (WT = 32.8±5 vs. glut3+/− = 34±4 gm, n = 20 each), and brain weights (WT = 0.33±0.02 vs. glut3+/− = 0.32±0.03 gm, n = 20 each) were no different between the PN60 glut3+/− and WT mice.
Glucose transporter and other marker proteins localization and expression
Brain histology was no different between the PN60 glut3+/− and WT mice. Neuronal nitric oxide synthase immunoreactivity, a surrogate marker for neuronal oxidative injury due to glucose deprivation, was no different between the two genotypes (n = 10/genotype, data not shown). No ectopic expression of GLUT1 in neurons despite GLUT3 deficient neurons was noted in any brain structures; instead GLUT1 immunolocalization was limited to microvasculature in brain sections of glut3+/− and WT (figure 1.II.a & b) mice. Tubulin immunoreactivity, a surrogate for intact cellular processes, was preserved (figure 1.II.c & d). In contrast, the expression of hippocampus neuroligin-3, a marker of mature functional GABA and glutaminergic synapses, was slightly lower in glut3+/− versus WT mice (WT = 1.01±0.03 vs. glut3+/− = 0.93±0.02* arbitrary units, n = 11 to 12 each, unpaired t-test, *p < 0.05).
Continuous Video-Electroencephalographic monitoring
Functional assessment by continuous electroencephalographic (EEG) telemetry recording demonstrated seizure activity in some of the glut3+/− versus age matched WT mice (WT = 0.034±0.017 and glut3+/− = 0.55±0.24 average seizure frequency/hr, unpaired t-test, *p < 0.05) with a trend towards increased spike activity in the former versus the latter (WT = 6.90±3.97 and glut3+/− = 19.35±7.10 average spike frequency/hr) (figure 2.I. d & e). The incidence of EEG seizures was also higher in glut3+/− versus WT mice (Fisher Exact test, p < 0.05) as was the increasing trend in spike activity (figure 2.I. a–c). As described previously in sham rats, idiopathic seizures were observed in C57BL/6 WT mice as well, albeit at a 16-fold lower rate than the glut3+/− mice, reinforcing the importance of simultaneous systematic monitoring of appropriate strain-specific controls (36). Rarely glut3+/− mice were observed to demonstrate tonic posturing (figure 2.I. a) or a behavioral arrest but most glut3+/− mice were asymptomatic when seizures were observed on EEG (figure 2.I. b) unlike the EEG tracing seen in most of the WT mice (figure 2.I. c). All behavioral and cognitive analyses were performed in mice lacking spontaneous seizures, which constituted most of them.
Figure 2. I. Extended video-EEG monitoring.

a–c. Epileptiform activity accompanied by distinct behavioral phenotypes in glut3+/− mice is shown. a. Periodic seizures were seen with exploratory behavior that progressed into tonic extension of forelimb and tail (inset) followed by post-ictal depression (arrowhead). b. Prolonged recordings reveal distinct epileptiform activity with no clear motor component. The dashed lines encompass the segment of recording that corresponds to the behavior shown. The horizontal scale bar is 1 s for the tracings, and 4 cm for the insets. c. A representative baseline tracing consisting of alpha slow waves in WT mice is shown. d–e. EEG seizures (>6 s) (d) and spikes (<6 s) (e) per hour were quantified in WT (n = 19) and glut3+/− (n = 16) mice; unpaired t-test, *p<0.05. II. a–d. Glut3 deficiency led to impaired social interaction and reduced separation calls. a. WT mice (n = 18) spent significantly more time exploring a conspecific than an empty cage in a social choice paradigm (two-way repeated-measures ANOVA with genotype as between-subjects factor and compartment as within-subjects factor, genotype x compartment interaction: F (1, 40) = 30.312, p < 0.001; Tukey post-hoc analysis: WT-conspecific vs. WT-empty cage: ***p < 0.001). Glut3+/− mice (n = 24), in contrast, spent more time exploring the empty cage than they spent interacting with the conspecific (**Tukey post-hoc analysis, p = 0.004). Moreover, glut3+/− mice spent significantly less time exploring the conspecific than WT controls (Tukey post-hoc analysis, ***p < 0.001), and therefore lacked normal levels of social exploration. b. Glut3+/− mice failed to show preference for the novel mouse in a social recognition task (two-way repeated-measures ANOVA, genotype x compartment interaction: F (1, 40) = 45.41, p < 0.001; Tukey post-hoc analyses: time spent exploring familiar mouse vs. novel mouse; WT: n = 18, ***p < 0.001; glut3+/−: n = 24, p = 0.106; time spent exploring unfamiliar mouse, WT vs. glut3+/−: ***p < 0.001). c–d. Ultrasonic vocalization (UV) testing (WT n = 38 mice, glut3+/− n = 35 mice) revealed diminished time spent in vocalizations (c) (two-way repeated-measures ANOVA with genotype as between-subjects factor and time point as within-subjects factor, effect of genotype: F(1, 71) = 4.88, *p = 0.03) and a diminished number of vocalizations/min (d) (two-way repeated-measures ANOVA with genotype as between-subjects factor and time point as within-subjects factor, effect of genotype: F(1, 71) = 7.558, **p = 0.008) in the glut3+/− vs. WT mice.
Neurobehavior testing: SHIRPA primary screen, motor skills and locomotor activity
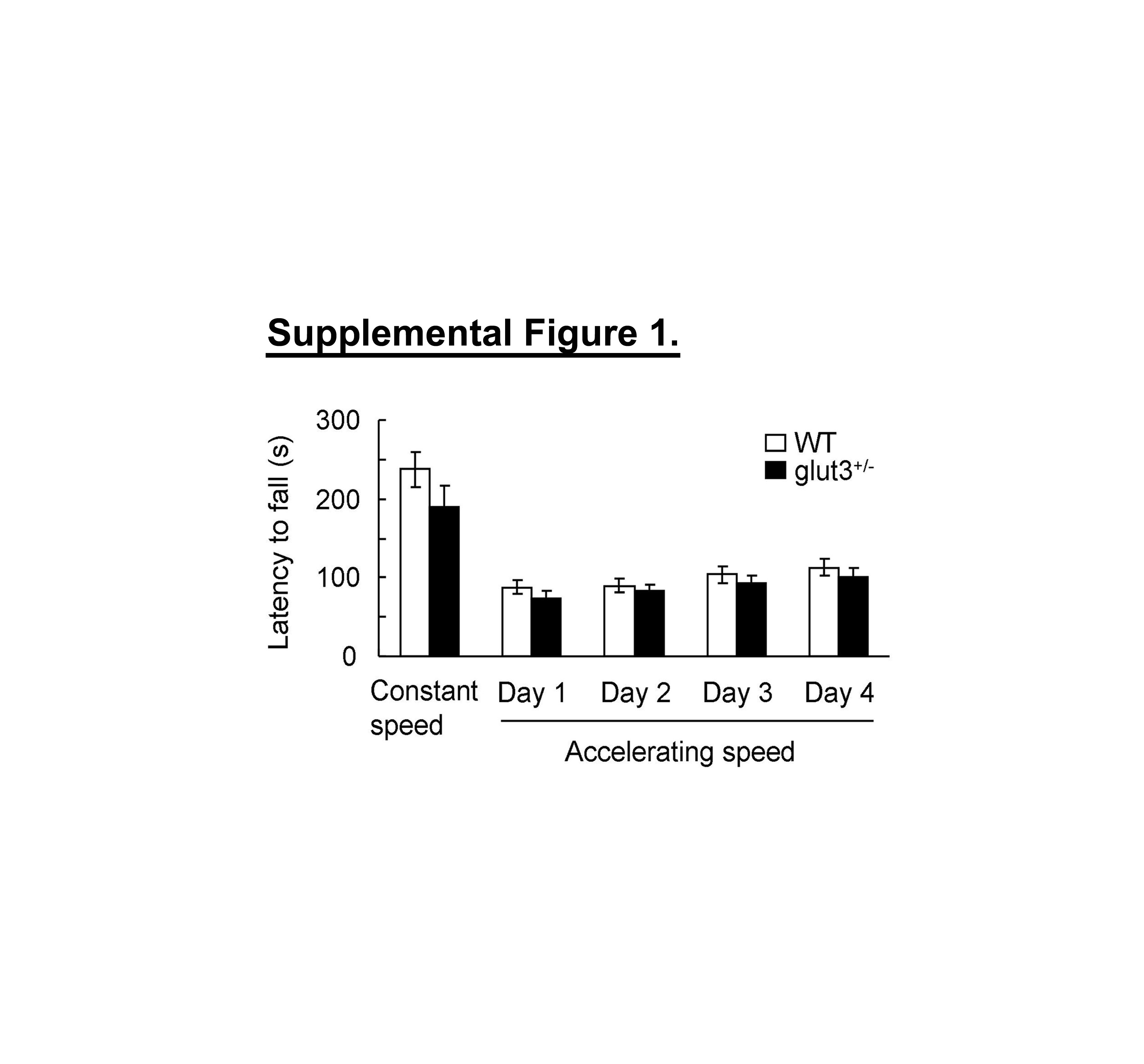
Quantitative measures of SHIRPA primary screen (23) are presented in table 1. No differences were initially observed between WT controls (n = 27 mice) and glut3+/− mice (n = 28 mice) The latency to fall in the rotorod test (supplemental figure 1), the total distance traveled, the ratio of center distance to total distance traveled, average velocity, time spent in center versus periphery and time spent in different activities in the open field test (supplemental figure 2) and the path length and speed of travel in the water maze swim test (supplemental figure 3) were no different between the two mouse genotypes.
Table 1.
SHIRPA primary screen score in glut3+/− mice (n = 28) and WT control mice (n = 27).
| WT littermates | glut3+/− | p-value | |
|---|---|---|---|
| Viewing jar | |||
| Body position | 3.30±0.09 | 3.04±0.04 | 0.097 |
| Spontaneous activity | 1.82±0.14 | 1.89±0.09 | 0.595 |
| Respiration rate | 2.00±0.00 | 2.00±0.00 | 1.000 |
| Tremor | 0.67±0.13 | 0.68±0.10 | 0.826 |
| Urination | 0.07±0.05 | 0.07±0.05 | 0.993 |
| Defecation | 3.63±0.27 | 4.11±0.48 | 0.539 |
| Arena | |||
| Transfer arousal | 3.41±0.23 | 3.79±0.17 | 0.288 |
| Locomotor activity | 8.82±0.77 | 8.50±0.72 | 0.686 |
| Palpebral closure | 0.00±0.00 | 0.00±0.00 | 1.000 |
| Piloerection | 0.00±0.00 | 0.00±0.00 | 1.000 |
| Startle response | 0.96±0.04 | 1.00±0.00 | 0.819 |
| Gait | 0.00±0.00 | 0.00±0.00 | 1.000 |
| Pelvic elevation | 2.22±0.08 | 2.25±0.08 | 0.866 |
| Tail elevation | 1.41±0.10 | 1.32±0.09 | 0.589 |
| Touch escape | 2.04±0.08 | 2.07±0.10 | 0.839 |
| Positional Passivity | 1.00±0.00 | 0.96±0.04 | 0.825 |
| Above Arena | |||
| Abnormal behavior | 0.00±0.00 | 0.00±0.00 | 1.000 |
| Visual placing | 3.00±0.00 | 3.00±0.00 | 1.000 |
| Grip strength | 2.04±0.10 | 1.50±0.15 | 0.006 |
| Body tone | 1.00±0.00 | 1.00±0.00 | 1.000 |
| Pinna reflex | 1.00±0.00 | 1.00±0.00 | 1.000 |
| Corneal reflex | 1.00±0.00 | 1.00±0.00 | 1.000 |
| Toe Pinch | 2.52±0.15 | 2.75±0.13 | 0.316 |
| Wire manoeuvre | 0.11±0.08 | 0.04±0.04 | 0.806 |
| Supine Restraint | |||
| Skin color | 0.96±0.04 | 0.89±0.06 | 0.659 |
| Heart rate | 1.48±0.10 | 1.32±0.09 | 0.311 |
| Limb tone | 1.15±0.07 | 1.11±0.06 | 0.799 |
| Abdominal tone | 1.00±0.00 | 1.00±0.00 | 1.000 |
| Lacrimation | 0.00±0.00 | 0.00±0.00 | 1.000 |
| Salivation | 0.37±0.10 | 0.43±0.11 | 0.826 |
| Provoked biting | 0.67±0.09 | 0.68±0.09 | 0.946 |
| Righting reflex | 0.00±0.00 | 0.00±0.00 | 1.000 |
| Contact righting reflex | 1.00±0.00 | 1.00±0.00 | 1.000 |
| Negative geotaxis | 0.11±0.06 | 0.32±0.12 | 0.352 |
| Fear | 0.59±0.10 | 0.54±0.10 | 0.723 |
| Irritability | 0.41±0.10 | 0.36±0.09 | 0.755 |
| Aggression | 0.59±0.10 | 0.39±0.09 | 0.206 |
| Vocalization | 0.19±0.08 | 0.25±0.08 | 0.685 |
Unpaired t-test with Bonferroni’s correction, significance is achieved at a p < 0.001.
Sociability and Social Recognition
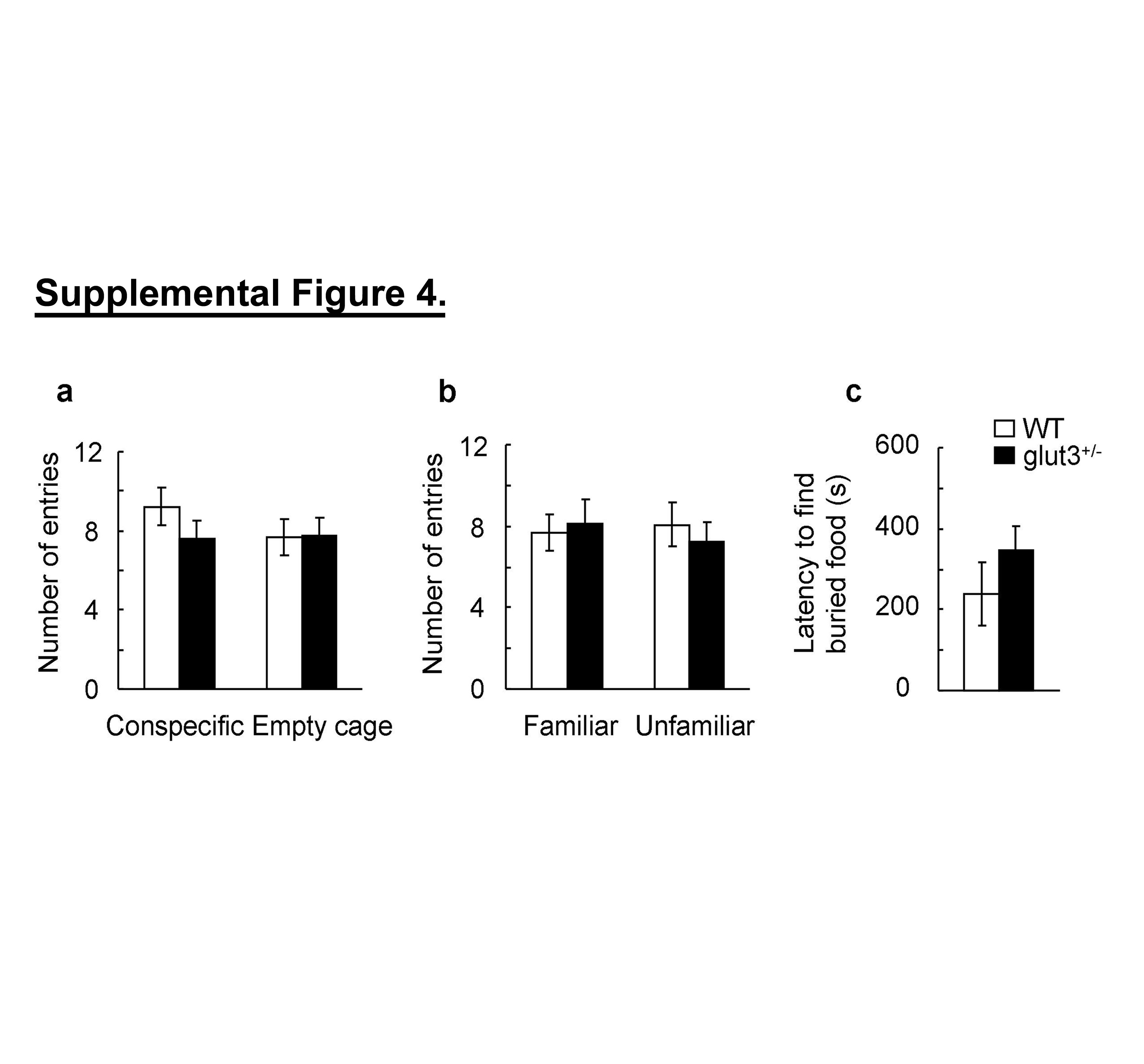
Impaired social behaviors, including lack of social interest, are an important feature of ASD. Accordingly, social interaction of glut3+/− mice and WT controls was examined using a social choice paradigm (26). After a brief habituation phase, mice were allowed to freely explore a 3-compartment apparatus. One side compartment contained a stimulus mouse constrained to this location by a round wire cage, while the other side compartment contained an empty wire cage. Consistent with previous reports (26), WT mice spent more time actively exploring the cage containing the conspecific than the empty cage; this pattern was reversed in glut3+/− mice which spent more time exploring the empty cage than the cage containing the stimulus mouse (figure 2.II. a). Moreover, glut3+/− mice spent significantly less time exploring the conspecific than WT controls (figure 2.II. a) and therefore lacked normal levels of social exploration. Further, these glut3+/− mice when subjected to a social recognition test spent less time with the unfamiliar than the familiar mouse, supporting diminished aptitude for social novelty (figure 2.II. b). Sex-specific analysis revealed no differences in either sociability or social recognition. These data demonstrate that heterozygous glut3 deletion results in abnormal social interaction, including diminished social approach behavior and a lack of preference for social novelty. Other factors such as locomotor activity that could influence the transition between chambers (supplemental figure 4. a–b) and olfactory sensitivity that could affect sociability (supplemental figure 4. c) were no different between the two mouse genotypes.
Communication deficits by ultrasonic vocalization (USV) tests
Human subjects with ASD frequently show language and communication deficits. It is unclear to what extent vocalizations in mice serve communicative functions and how impairments in mice relate to autism-associated communication deficits. Nevertheless, at least limited forms of vocal communication appear to exist in mice: Upon separation from the dam, mouse pups emit vocalizations in the ultrasonic range that induce retrieval by the dam (27). To assess whether GLUT3 deficiency impacts pup vocalizations, these separation calls were quantified in glut3+/− mice and WT controls. Glut3+/− mice in the absence of any thermal regulatory differences showed a significantly lower duration (figure 2.II. c) and significantly lower rates of vocalizations (figure 2.II. d) than WT controls, demonstrating that GLUT3 deficiency impairs vocalizations in mice.
Cognitive flexibility by T maze acquisition and reversal learning
The range of behavioral repertoires in subjects with ASD is often restricted and not flexibly applied to environmental demands. We examined cognitive flexibility in glut3+/− mice and WT controls using a T-maze task (28). During the acquisition phase, food-deprived mice were trained to retrieve a food reward from one constantly baited arm of a T-maze, while the other arm remained unbaited. After the animal had reached a performance criterion (visiting the correct arm in at least 80% of cases for 3 consecutive days; 65% of mice), the contingency of the task was changed such that the food reward was located exclusively in the previously unbaited arm (reversal learning). While WT mice and glut3+/− mutants learned to correctly choose the baited arm at comparable rates during acquisition (figure 3. a), reversal learning was significantly slower in glut3+/− mutant mice (figure 3. a), suggesting impaired cognitive flexibility in glut3+/− mutant mice.
Figure 3. Glut3+/− mice showed impaired cognitive flexibility and motor stereotypies.
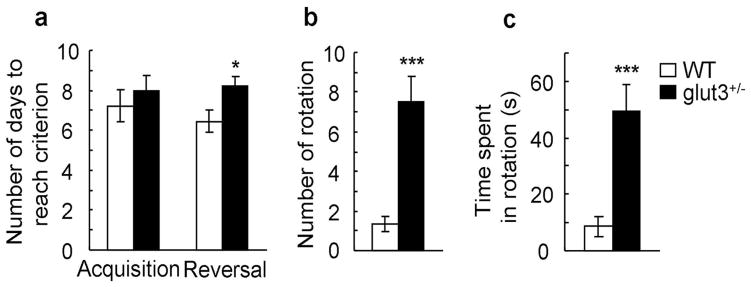
a. The time to reach criterion during acquisition training and reversal learning on a T-maze task is shown (WT: n = 9 mice, glut3+/−: n = 16 mice). No genotype difference was noted on acquisition (unpaired t-test, p = 0.518) but glut3+/− mice required more training than WT mice to reach criterion during the reversal phase (unpaired t-test, *p = 0.033). b–c. Stereotypic behavior tests: The number (b) (unpaired t-test, ***p < 0.001) and time (c) (unpaired t-test, ***p < 0.001) spent in rotations were increased in glut3+/− (n = 25) vs. WT (n = 22) mice.
Stereotypic behaviors
Repetitive behaviors in subjects with ASD may manifest as motor stereotypies, such as hand flapping, body rocking or head rolling. Stereotypical movements were quantified in freely behaving glut3+/− mice and WT controls (29, 30). While there was no significant difference between genotypes in rearing, sniffing, exploring and grooming (supplemental figure 5. a–e), glut3+/− mice spent substantially more time in rotational movements than WT controls (figure 3. b & c), demonstrating GLUT3 deficiency resulted in abnormal motor stereotypies. Interestingly, excessive motor stereotypies in glut3+/− mice were not seen in the social interaction tasks (data not shown) and open field test (data not shown), suggesting that these behaviors may be present in certain environmental conditions only (e.g. cylindric environment for the study of stereotypical behaviors as opposed to larger square-shaped environments in the open field and social interaction task). Accordingly, it can be ruled out that repetitive rotational behaviors may have confounded the assessment of social behaviors in our studies.
Spatial cognition by Morris water maze test
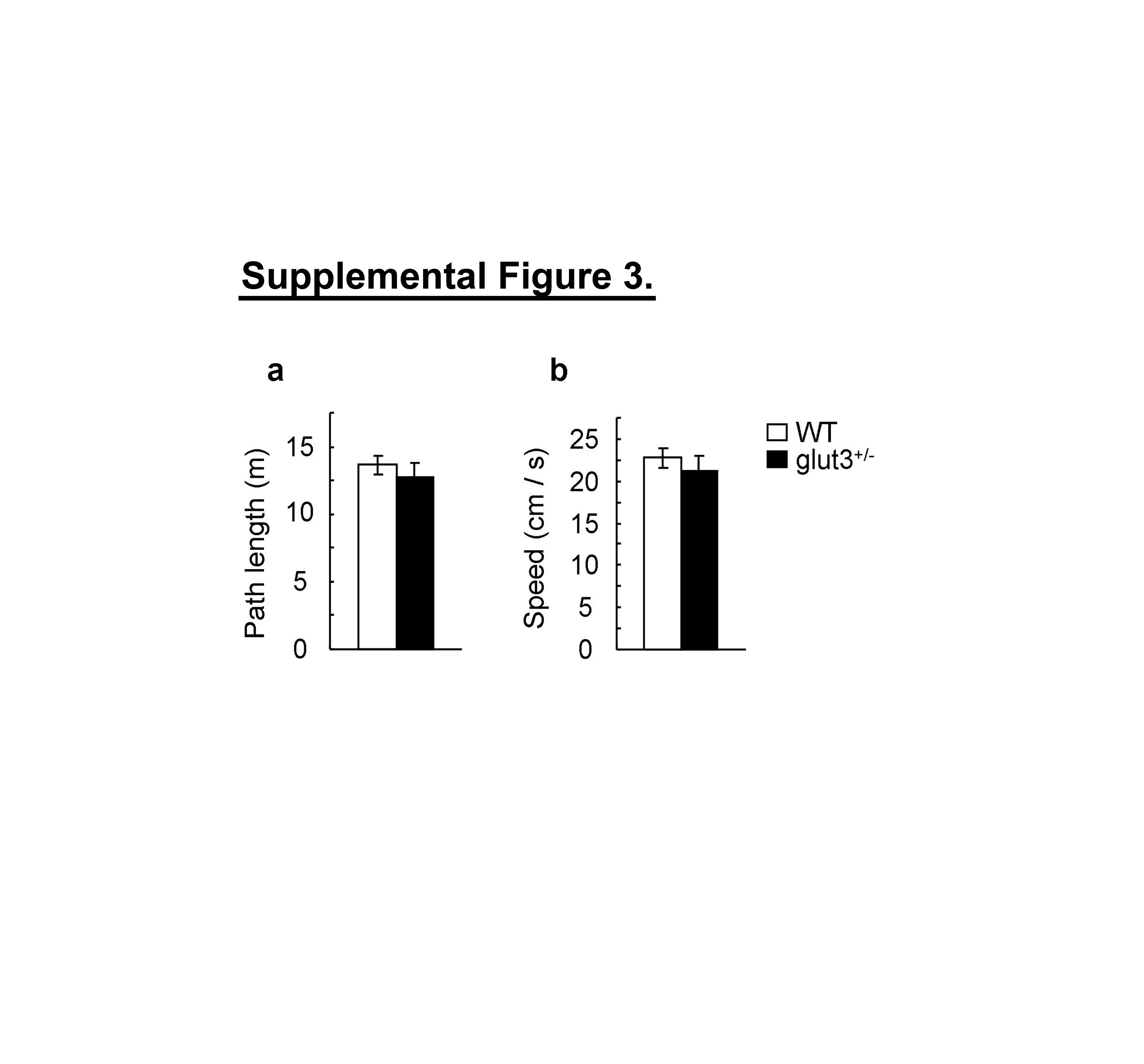
Mental retardation and specific learning disabilities constitute important co-morbidities of ASD. To examine whether GLUT3 deficiency adversely affects spatial cognition, mutant mice and WT controls were trained on the hidden version of the Morris water maze (31, 32). Analysis of escape latencies showed a non-significant trend towards higher escape latencies in glut3+/− mice (figure 4. d). As escape latencies generally are a poor indicator of spatial learning (37), we tested the accuracy with which the platform location was learned by delivering a probe trial (during which the platform was removed from the pool) after completion of acquisition training. Probe trial data indicated that WT mice had a trend towards higher target quadrant occupancy scores (figure 4. a), searched closer to the previous platform location (figure 4. c) and crossed the exact target position more frequently (figure 4. b) than glut3+/− mice. Total path length covered and swim speed during the probe trial did not differ between genotypes (supplemental figure 3. a-b). Moreover, training in the visible version of the Morris water maze revealed normal escape latencies in glut3+/− mice (figure 4. d). These data suggest that perceptive, motivational and motor skills were normal in glut3 mutant mice and that the observed deficit in the hidden version of the Morris water maze reflects impaired spatial cognition in glut3+/− mice.
Figure 4.

Glut3+/− mice showed impaired spatial learning in the Morris water maze and spatial working memory deficits in a win-shift version of the 8-arm radial maze. a–c. Quadrant occupancy, target crossings and proximity during the probe trial (WT: n = 13 mice; glut3+/−: n = 15 mice; pool quadrant: target (T), right (R), opposite (O) and left (L) quadrants). a. WT mice showed a trend towards higher target quadrant occupancy scores in comparison with mutant mice (two-way repeated-measures ANOVA, genotype x quadrant interaction: F (3, 78) = 5.013, p = 0.003; Tukey post-hoc analysis, target quadrant occupancy in WT vs. mutant mice, p = 0.129). b. WT mice crossed the exact target position more frequently than glut3+/− mice (two-way repeated-measures ANOVA, genotype x quadrant interaction: F (3, 78) = 3.116, p = 0.031; Tukey post-hoc analysis, target crossing in WT vs. mutant mice, *p = 0.013). c. Proximity to the target location was closer in WT mice than in mutant mice (two-way repeated-measures ANOVA, genotype x quadrant interaction: F (3, 78) = 5.065, p = 0.003; Tukey post-hoc analysis, proximity to target quadrant in WT vs. mutant mice, ***p = 0.001). d. Escape latency: A non-significant trend towards higher escape latencies was observed in glut3+/− mice (two-way repeated-measures ANOVA with genotype as between-subjects factor and session as within-subjects factor, effect of genotype: F (1, 26) = 4.121, p = 0.053; data from 4–6 training sessions were averaged and are shown as one block session). In the visible platform task (VP), the escape latency of glut3+/− mice was similar to that of WT controls (unpaired t-test, p = 0.963). e. Radial arm maze: The number of across-phase errors normalized to total arm entries was higher in glut3+/− mice than WT controls (WT, n = 18 mice, glut3+/−, n = 12 mice, two-way repeated-measures ANOVA with genotype as between-subjects factor and session as within-subjects factor, effect of genotype: F (1, 28) = 9.16, **p = 0.005).
Spatial working memory by radial arm maze test
Next, we tested spatial working memory in glut3+/− mice and WT controls in a delayed-non-match-to-place version of the 8-arm radial maze (33). During phase A of the task, food-deprived mice were allowed to retrieve reward pellets from four pseudo-randomly baited arms, while access to the other four arms was blocked. After a 3 h delay, mice were returned to the maze and were allowed to enter any of the 8 arms. However, only the previously not accessible arms were baited during this phase of the task (phase B). Mice can commit different types of errors in this task: they can fail to visit a baited arm (omission error), visit an arm that was only baited during phase A (across-phase error) or repeatedly enter an arm during phase B (within-phase error). Glut3+/− mice made significantly more omission errors than WT controls (two-way repeated-measures ANOVA with genotype as between-subjects factor and session as within-subjects factor, effect of genotype: F (1, 28) = 14.757, p < 0.001, data not shown) and showed a lower number of total arm entries (supplemental figure 6. a). Glut3+/− mice committed more across-phase errors relative to total arm entries than WT mice (figure 4. e), while within-phase errors/total arm entries were not affected in glut3 mutants (two-way repeated-measures ANOVA with genotype as between-subjects factor and session as within-subjects factor, effect of genotype: F (1, 28) = 0.114, p = 0.738, data not shown). These observations suggest that GLUT3 deficiency led to a cognitive impairment suggestive of hippocampal dysfunction, while also affecting overall arm entries.
Altogether, the data show that GLUT3 deficiency caused cognitive deficits in two hippocampus-dependent tasks, namely the water and radial arm maze tasks.
Emotional working memory by contextual fear conditioning test
No deficits in one other emotion based measure of context learning in a fear conditioned task were observed (supplemental figure 7). The acquisition of contextual conditioning can be hippocampus-dependent. However, animals can use non-hippocampal strategies to recognize the training context (38). Thus, no difference between glut3+/− and WT mice supports non-hippocampal mediated learning and working memory to be intact.
Discussion
We have characterized the neurodevelopmental phenotype of glut3 heterozygous null mice. These mice demonstrate compensatory brain metabolic changes, increased electroencephalographic seizure activity, abnormal spatial learning and working memory, perturbed cognitive flexibility with intact gross motor ability, altered social behavior, reduced vocalization with low frequency stereotypies.
Our observations support an association between the decline in neuronal GLUT3 and an increase in brain GLUT1 and MCT2 concentrations during the suckling and early post-suckling phases of development. Since GLUT1 mediates glucose transport across the blood brain barrier and into glial cells, compensatory increase in GLUT1 noted in glut3+/− mice was associated with none to minimal diminution in total brain 2-deoxy-glucose uptake. This finding may reflect the net effect of the increase in endothelial and glial GLUT1 but decrease in neuronal GLUT3 mediated glucose uptake. The compensatory increase in neuronal MCT2 but not endothelial/glial MCT1 paralleled the enhanced brain lactate uptake. Thus, neuronal glucose deficiency was compensated by increased lactate supply suggesting an exclusive increase in neuronal lactate uptake.
To ascertain this phenomenon isolation of specific brain cell types is required. However, in-vitro isolation and culturing of neurons induces GLUT1 (39, 40), while culturing of astrocytes induces GLUT3 expression (41) thereby no longer reflecting the in-vivo state. Accurate in-vivo ascertainment of neuronal glucose uptake separate from that of glial cells without disrupting the inter-cellular communications is difficult to accomplish. In keeping with these experimental barriers, previous investigations employing isolated and cultured astrocytes from newborn glut3+/− and WT mice failed to detect differences in either 2-deoxyglucose uptake or lactate release into the culture medium (42). If these in-vitro results are taken at face value, with no information regarding cellular GLUT1, GLUT3, MCT1 and MCT2 expression, the lack of any inter-genotype difference supports no protective role for the astrocyte in the presence of neuronal glucose deficiency. This observation goes against the “astrocyte-neuron lactate shuttle” concept put forth by other investigator groups (43). In contrast, our present in-vivo observations support a compensatory increase in blood-brain barrier GLUT1 mediated 2-deoxy-glucose transport from the circulation into the brain interstitium/glial cells, thereby minimizing the decrease in glut3+/− brain glucose uptake. Since neuronal glut3+/− deficiency sets the stage for a neuronal energy deficit, an increased neuronal MCT2 dependent lactate uptake based on enhanced blood-brain barrier transfer of lactate is plausible. Despite these metabolic compensations, disturbances in neuronal function were observed. These disturbances consisted of electroencephalographic seizure activity and certain neurobehavioral abnormalities.
In contrast, glut1 mutation created by anti-sense technology has serious structural effects causing major brain malformations in zebra fish and mouse (44, 45). On the other hand, glut1 haplo-deficient mice demonstrated a normal body size but a small sized brain, diminished brain glucose uptake on positron emission tomography, with abnormal motor activity and EEG seizures (46). However, neurobehavior related to learning, memory or anxiety was not assessed. Paralleling findings in mice, the autosomal dominant GLUT1 deficiency syndrome due to Glut1 mutations in human infants is noteworthy for developmental delay, a spastic ataxic gait and seizure disorder (47–49). This aberrant neurodevelopment is partially ameliorated by a ketogenic diet (50). GLUT1 deficiency has the propensity to globally affect both neurons and glia by curbing glucose entry across the blood-brain barrier (49). GLUT1 expressing astroglia that are thought to serve as a local supplier of an alternate substrate, i.e. lactate, to glucose deficient neurons may also be negatively affected (7, 43). Thus GLUT1 deficiency that leads to low cerebrospinal fluid glucose and lactate concentrations in the presence of normal plasma glucose concentrations reflects diminished glucose transport across the blood-brain barrier (47, 49). GLUT3 deficiency on the other hand selectively affects the glucose supply to GLUT3 expressing neurons compromising their energy metabolism.
Although GLUT4 and GLUT8 are also expressed in neurons within certain brain structures (10, 11), neurobehavior has not been examined in glut4 null homozygous or heterozygous mutant mice (51, 52). In contrast, glut8 null homozygous mutation caused minimal increase in hippocampus volume (53), with hyperactivity, reduced risk assessment and increased emotional reactivity to a novel environment (54). This presentation consistent with an anxiety disorder was not replicated in glut3+/− mice previously (42). Instead we presently observed a phenotypic presentation in glut3+/− mice that resembled clinical features of ASD in humans.
ASDs affect an estimated 5 to 6 per 1000 children at 3–10 years of age. Diagnostic screens may detect this disorder as early as 12 to 36 months of age. The presenting features consist of deficiencies in sociability, communication difficulties with problems in learning and utilizing language appropriately, difficulties in executive functioning, and repetitive behaviors although the individuals appear physically normal and exhibit good muscle control (55). Akin to this clinical presentation the glut3 haplo-insufficient mice demonstrated stereotypic behaviors consisting of rotational activity at low frequency and expressed perturbed vocalizations and social behaviors.
The stereotypic behavior observed was seen in more restrictive environments and did not affect grooming behavior. In contrast, another in-bred mouse strain with features relevant to autism (BTBR T+tf/J) demonstrated high levels of repetitive self-grooming (29). The reduction in USVs may reflect the early signs of disturbed communication in ASD. Maternal and sibling separation lends to social isolation resulting in vocalizations in response to maternal potentiation (56). C57BL/6 mice in general demonstrate a low rate of vocalization reflective of a higher maternal responsiveness (56). Nevertheless, in this strain we observed a genotype specific difference in vocalization. Previously, hypothermia incurred on separation of pups from the maternal nest (30°C to 19°C) stimulated vocalization abnormalities (57). In our present investigation, we avoided hypothermia and have previously observed no difference in body temperature regulation between the glut3+/− and WT mice (58). Others have examined the role of larger lung capacity or abdominal pressure induced increased venous return on perturbing postnatal USVs in mice (56). Our present investigations examined WT age-matched littermates and glut3+/− mice under similar conditions and detected a reduction in USVs reflective of a communication disability in the mutant mice. This reduction in USVs has been observed in other mutant mice that have neurodevelopmental disorders related to abnormal synapses, examples include the Reeler mice, neuroligin-3 and neuroligin-4 null mice (56, 59, 60). A reduction in USVs is also observed in Foxp2 mutant mice, this gene mutation being responsible for human verbal dyspraxia, dysphagia and other severe language and speech disorders (56). These previous observations are of particular significance, since GLUT3 is localized to synapses (7, 9, 12).
In contrast to our findings in glut3+/− mice, the Mecp2 mutant mouse model of Rett’s syndrome demonstrated a dramatic increase in USVs perhaps related to associated alterations of respiratory function and cholinergic deficits (56). Similarly, in another inbred strain of mice, BTBR T+tf/J, several traits relevant to autism were detected including emission of a higher number albeit restricted categories of calls (27). In the human disorder of ASD the difficulty in communication spans a spectrum ranging from no response to external social stimuli to inappropriate outbursts (1, 61). Earlier during infancy, the normal cooing and babbling may not exist or is replaced by humming or grunting (1, 61). The impaired social behavior detected in glut3+/− mice is similar to that reported in the BTBR T+tf/J mouse relevant to autism (26, 27).
Other problems that may accompany the clinical picture of ASD are sensory problems, mental retardation and specific learning disabilities. We observed impaired working memory and spatial cognition that relied on environmental cues and cognitive inflexibility in glut3 haplo-insufficient mice. These features were previously either not examined at all, investigated with incomplete testing (e.g. lack of probe trial in the water maze test) or inadequate power to detect differences or were masked by a battery of neurobehavioral tests imposed in succession on the same set of glut3+/− mice (42). However, in our present investigation emotional working memory which may be non-hippocampus mediated (38) was not altered under fear contextual conditions.
One in four children with ASD develops seizures that may start during childhood or adolescence. While occasionally these seizures are clinically apparent, frequently sub-clinical epileptiform activity can only be detected by overnight EEG monitoring. These EEG changes may contribute to abnormal neurobehavior and cognition which are diagnostic of ASD (55). Wide variations in prevalence rates of epileptiform activity in ASD patients have been reported. Typically, antiepileptic therapy is used in ASD patients with epilepsy, contributing towards some of the variability reported. Our glut3+/− mice demonstrated ~62% incidence of seizures akin to the higher end reported in human ASD. The glut3 mutant mice displayed a mixture of seizure phenotypes; some mice demonstrated overt tonic seizures while others had subtle clinical manifestations such as behavioral arrest or were asymptomatic, making detection difficult without extensive video-EEG recording. Recent neuro-imaging investigations demonstrate decreased brain glucose uptake and aberrant intra-cortical connectivity in this disorder (4). Whether decreased brain glucose uptake and aberrant inter-neuronal connectivity form the cause or effect of ASD is unclear from such studies. Aberrant neuronal GLUT3 mediated glucose uptake as the common mechanistic denominator of ASD has never been explored previously.
GLUT3 is mainly noted on neuronal processes that mediate inter-neuronal connectivity particularly in the cortical gray matter thereby ensuring adequate glucose supply at synapses that require high energy (7, 9, 12). Neuroligin-3 (NLGN3), an extra-cellular matrix protein that interacts with neurexin (NRXN) receptors in the synaptic cleft to mediate synaptic maturation and neurotransmission is dysfunctional in ASD (62, 63). Polymorphisms or mutations of NLGN3 that cause protein unfolding and aggregation in the endoplasmic reticulum are seen in ASD (64). Due to redundancy of neuroligins and neurexins, NLGN3 mutant mice did not while NLGN4 mutant mice displayed apparent autism-like phenotype (59, 60). More recent evidence in human autism however demonstrates deletion of genes (NLGN3, NLGN4, NRXN1, PCDH10 and CNTN3) responsible for activity-regulated synaptic development mediating postnatal experience-dependent neural activity which in turn forms the basis for learning and memory (65). Our present glut3 heterozygous null mice show neurobehavioral features relevant to ASD, a diminution of the GLUT3 protein and a mild but significant decline in NLGN3 expression on inter-neuronal connecting processes. Our present novel observations set the stage for investigating the association of Glut3 mutations or polymorphisms in similar human neuropsychiatric disorders with EEG seizures or EEG seizures that result in neurobehavioral consequences.
In conclusion, neuronal GLUT3 deficiency causes increased EEG seizures which may not be clinically detected, significant deficiencies in spatial learning and working memory reliant on environmental cues, cognitive inflexibility, stereotypic behaviors of low frequency and abnormal social behaviors and vocalizations, features not unlike those encountered in ASD. This novel observation lends credence to future investigations of dietary or pharmacological interventions targeted at potentially rescuing and/or reversing the progression of this devastating condition.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by grants from the NIH HD33997 and HD46979 (SUD), while the EEG studies were partly supported by NS 046516 (RS). Camille Fung was supported by a NIH T32 HD07549 training grant. We thank Lynn Talton, Ph.D. in the UCLA Behavioral Testing Core Laboratory, Brian Wiltgen and Brett Abrahams for their guidance with certain neurobehavioral studies.
References
- 1.Weiss LA, et al. Association between microdeletion and microduplication at 16p11. 2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 2.Kolevzon A, Gross R, Reichenberg A. Prenatal and perinatal risk factors for autism: a review and integration of findings. Arch Pediatr Adolesc Med. 2007;161:326–333. doi: 10.1001/archpedi.161.4.326. [DOI] [PubMed] [Google Scholar]
- 3.Limperopoulos C, Bassan H, Sullivan NR, Soul JS, Robertson RL, Jr, Moore M, et al. Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics. 2008;121:758–765. doi: 10.1542/peds.2007-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toal F, Murphy DG, Murphy KC. Autistic-spectrum disorders:lesson from neuroimaging. Br J Psychiatry. 2005;187:395–397. doi: 10.1192/bjp.187.5.395. [DOI] [PubMed] [Google Scholar]
- 5.Haznedar MM, Buchsbaum MS, Wei TC, Hof PR, Cartwright C, Bienstock CA, Hollander E. Limbic circuitry in patients with autism spectrum disorders studied with positron emission tomography and magnetic resonance imaging. Am J Psychiatry. 2000;157:1994–2001. doi: 10.1176/appi.ajp.157.12.1994. [DOI] [PubMed] [Google Scholar]
- 6.Haznedar MM, Buchsbaum MS, Hazlett EA, LiCalzi EM, Cartwright C, Hollander E. Volumetric analysis and three-dimensional glucose metabolic mapping of the striatum and thalamus in patients with autism spectrum disorders. Am J Psychiatry. 2006;163:1252–63. doi: 10.1176/ajp.2006.163.7.1252. [DOI] [PubMed] [Google Scholar]
- 7.Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow & Metab. 2007;27:1766–1791. doi: 10.1038/sj.jcbfm.9600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devaskar S, Zahm DS, Holtzclaw L, Chundu K, Wadzinski BE. Developmental regulation of the distribution of rat brain insulin-insensitive (Glut 1) glucose transporter. Endocrinology. 1991;129:1530–1540. doi: 10.1210/endo-129-3-1530. [DOI] [PubMed] [Google Scholar]
- 9.Mantych GJ, James DE, Chung HD, Devaskar SU. Cellular localization and characterization of Glut 3 glucose transporter isoform in human brain. Endocrinology. 1992;131:1270–1278. doi: 10.1210/endo.131.3.1505464. [DOI] [PubMed] [Google Scholar]
- 10.Sankar R, Thamotharan S, Shin D, Moley KH, Devaskar SU. Insulin-responsive glucose transporters-GLUT8 and GLUT4 are expressed in the developing brain. Brain Res Mol Brain Res. 2002;107:157–165. doi: 10.1016/s0169-328x(02)00487-4. [DOI] [PubMed] [Google Scholar]
- 11.Shin BC, McKnight RA, Devaskar SU. Glucose transporter GLUT8 translocation in neurons is not insulin responsive. J Neurosci Res. 2004;75:835–844. doi: 10.1002/jnr.20054. [DOI] [PubMed] [Google Scholar]
- 12.Maher F, Davies-Hill TM, Simpson IA. Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem J. 1995;315 (Pt 3):827–831. doi: 10.1042/bj3150827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganguly A, McKnight RA, Raychaudhuri S, Shin BC, Ma Z, Moley K, et al. Glucose transporter isoform-3 mutations cause early pregnancy loss and fetal growth restriction. Am J Physiol Endocrinol Metab. 2007;292:E1241–1255. doi: 10.1152/ajpendo.00344.2006. [DOI] [PubMed] [Google Scholar]
- 14.Khan JY, Rajakumar RA, McKnight RA, Devaskar UP, Devaskar SU. Developmental regulation of genes mediating murine brain glucose uptake. Am J Physiol. 1999;276:R892–900. doi: 10.1152/ajpregu.1999.276.3.R892. [DOI] [PubMed] [Google Scholar]
- 15.Rajakumar A, Thamotharan S, Raychaudhuri N, Menon RK, Devaskar SU. Trans-activators regulating neuronal glucose transporter isoform-3 gene expression in mammalian neurons. J Biol Chem. 2004;279:26768–26779. doi: 10.1074/jbc.M402735200. [DOI] [PubMed] [Google Scholar]
- 16.Ito K, Sawada Y, Ishizuka H, Sugiyama Y, Suzuki H, Iga T, et al. Measurement of cerebral glucose-utilization from brain uptake of {C-14} 2-deoxyglucose and {H-3} 3-O-methylglucose in the mouse. J Pharmacol Methods. 1990;23:129–140. doi: 10.1016/0160-5402(90)90040-r. [DOI] [PubMed] [Google Scholar]
- 17.Zovein A, Flowers-Ziegler J, Thamotharan S, Shin D, Sankar R, Nguyen K, et al. Postnatal hypoxic-ischemic brain injury alters mechanisms mediating neuronal glucose transport. Am J Physiol Regul Integr Comp Physiol. 2004;286:R273–282. doi: 10.1152/ajpregu.00160.2003. [DOI] [PubMed] [Google Scholar]
- 18.Oldendorf WH. Measurement of brain uptake of radio labeled substances using a tritiated water internal standard. Brain Res. 1970;24:372–376. doi: 10.1016/0006-8993(70)90123-x. [DOI] [PubMed] [Google Scholar]
- 19.Choeiri C, Staines WA, Miki T, Seino S, Renaud JM, Teutenberg K, et al. Cerebral glucose transporters expression and spatial learning in the K-ATP Kir6. 2(−/−) knockout mice. Behav Brain Res. 2006;172:233–239. doi: 10.1016/j.bbr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Thamotharan M, Shin BC, Suddirikku DT, Thamotharan S, Garg M, Devaskar SU. GLUT4 expression and subcellular localization in the intrauterine growth-restricted adult rat female offspring. Am J Physiol Endocrinol Metab. 2005;288:E935–947. doi: 10.1152/ajpendo.00342.2004. [DOI] [PubMed] [Google Scholar]
- 21.Dube C, Richichi C, Bender RA, Chung G, Litt B, Baram TZ. Temporal lobe epilepsy after experimental prolonged febrile seizures: prospective analysis. Brain. 2006;129:911–922. doi: 10.1093/brain/awl018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irwin S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia. 1968;13:222–257. doi: 10.1007/BF00401402. [DOI] [PubMed] [Google Scholar]
- 23.Rafael JA, Nitta Y, Peters J, Davies KE. Testing of SHIRPA, a mouse phenotypic assessment protocol, on Dmd(mdx) and Dmd(mdx3cv) dystrophin-deficient mice. Mamm Genome. 2000;11:725–728. doi: 10.1007/s003350010149. [DOI] [PubMed] [Google Scholar]
- 24.Lawhorn C, Smith DM, Brown LL. Striosome-matrix pathology and motor deficits in the YAC128 mouse model of Huntington’s disease. Neurobiol Dis. 2008;32:471–478. doi: 10.1016/j.nbd.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Avale ME, Falzone TL, Gelman DM, Low MJ, Grandy DK, Rubinstein M. The dopamine D4 receptor is essential for hyperactivity and impaired behavioral inhibition in a mouse model of attention deficit/hyperactivity disorder. Mol Psychiatry. 2004;9:718–726. doi: 10.1038/sj.mp.4001474. [DOI] [PubMed] [Google Scholar]
- 26.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 27.Scattoni ML, Gandhy SU, Ricceri L, Crawley JN. Unusual repertoire of vocalizations in the BTBR T+tf/J mouse model of autism. PLoS ONE. 2008;3:e3067. doi: 10.1371/journal.pone.0003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, et al. Mouse behavioral tasks relevant to autism: Phenotypes of 10 inbred strains. Behav Brain Res. 2007;176:4–20. doi: 10.1016/j.bbr.2006.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McFarlane HG, Kusek GK, Yang M, Phoenix JL, Bolivar VJ, Crawley JN. Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav. 2008;7:152–163. doi: 10.1111/j.1601-183X.2007.00330.x. [DOI] [PubMed] [Google Scholar]
- 30.Kelley AE. Measurement of rodent stereotyped behavior. Curr Protoc Neurosci. 2001;Chapter 8(Unit 8.8) doi: 10.1002/0471142301.ns0808s04. [DOI] [PubMed] [Google Scholar]
- 31.Morris R. Development of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 32.Nakajo Y, Miyamoto S, Nakano Y, Xue JH, Hori T, Yanamoto H. Genetic increase in brain-derived neurotrophic factor levels enhances learning and memory. Brain Res. 2008;1241:103–109. doi: 10.1016/j.brainres.2008.08.080. [DOI] [PubMed] [Google Scholar]
- 33.Mizuno M, Yamada K, Takei N, Tran MH, He J, Nakajima A, et al. Phosphatidylinositol 3-kinase: a molecule mediating BDNF-dependent spatial memory formation. Mol Psychiatry. 2003;8:217–224. doi: 10.1038/sj.mp.4001215. [DOI] [PubMed] [Google Scholar]
- 34.Blaeser F, Sanders MJ, Truong N, Ko S, Wu LJ, Wozniak DF, et al. Long-term memory deficits in Pavlovian fear conditioning in Ca2+/calmodulin kinase kinase alpha-deficient mice. Mol Cell Biol. 2006;26:9105–9115. doi: 10.1128/MCB.01452-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94:1–14. doi: 10.1111/j.1471-4159.2005.03168.x. [DOI] [PubMed] [Google Scholar]
- 36.D’Ambrosio R, Fender JS, Fairbanks JP, Simon EA, Born DE, Doyle DL, et al. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain. 2005;128:174–188. doi: 10.1093/brain/awh337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolfer DP, Stagljar-Bozicevic M, Errington ML, Lipp HP. Spatial memory and learning in transgenic mice: fact or artifact? News Physiol Sci. 1998;13:118–123. doi: 10.1152/physiologyonline.1998.13.3.118. [DOI] [PubMed] [Google Scholar]
- 38.Matus-Amat P, Higgins EA, Barrientos RM, Rudy JW. The role of the dorsal hippocampus in the acquisition and retrieval of context memory representations. J Neurosci. 2004;24:2431–9. doi: 10.1523/JNEUROSCI.1598-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu S, Zhao T, Guo M, Fang H, Ma J, Ding A, et al. Hypoxic preconditioning up-regulates glucose transport activity and glucose transporter (GLUT1 and GLUT3) gene expression after acute anoxic exposure in the cultured rat hippocampal neurons and astrocytes. Brain Res. 2008;1211:22–29. doi: 10.1016/j.brainres.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 40.Bruckner BA, Ammini CV, Otal MP, Raizada MK, Stacpoole PW. Regulation of brain glucose transporters by glucose and oxygen deprivation. Metabolism. 1999;48:422–31. doi: 10.1016/s0026-0495(99)90098-7. [DOI] [PubMed] [Google Scholar]
- 41.Sadiq F, Holtzclaw L, Chundu K, Muzzafar A, Devaskar S. The ontogeny of the rabbit brain glucose transporters. Endocrinology. 1990;126:2417–24. doi: 10.1210/endo-126-5-2417. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt S, Richter M, Montag D, Sartorius T, Gawlik V, Hennige AM, et al. Neuronal functions, feeding behavior, and energy balance in Slc2a3+/− mice. Am J Physiol Endocrinol Metab. 2008;295:E1084–E1094. doi: 10.1152/ajpendo.90491.2008. [DOI] [PubMed] [Google Scholar]
- 43.Pellerin L. How astrocytes feed hungry neurons. Mol Neurobiol. 2005;32:59–72. doi: 10.1385/MN:32:1:059. [DOI] [PubMed] [Google Scholar]
- 44.Heilig CW, Saunders T, Brosius FC, 3rd, Moley K, Heilig K, Baggs R, et al. Glucose transporter-1-deficient mice exhibit impaired development and deformities that are similar to diabetic embryopathy. Proc Natl Acad Sci U S A. 2003;100:15613–15618. doi: 10.1073/pnas.2536196100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen PJ, Gitlin JD, Carayannopoulos MO. GLUT1 deficiency links nutrient availability and apoptosis during embryonic development. J Biol Chem. 2006;281:13382–13387. doi: 10.1074/jbc.M601881200. [DOI] [PubMed] [Google Scholar]
- 46.Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, et al. A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet. 2006;15:1169–1179. doi: 10.1093/hmg/ddl032. [DOI] [PubMed] [Google Scholar]
- 47.De Vivo DC, Trifiletti R, Jacobson RI, Ronen GM, Behmand RA, Harik SI. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. New Engl J Med. 1991;325:703–709. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- 48.Seidner G, Alvarez MG, Yeh J-I, Driscoll KR, Klepper J, Stump TS, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nature Genetics. 1998;18:188–191. doi: 10.1038/ng0298-188. [DOI] [PubMed] [Google Scholar]
- 49.Klepper J, Wang D, Fischbarg J, Vera JC, Jarjour IT, O’Driscoll KR, et al. Defective glucose transport across brain tissue barriers: a newly recognized neurological syndrome. Neurochem Res. 1998;24:587–594. doi: 10.1023/a:1022544131826. [DOI] [PubMed] [Google Scholar]
- 50.Wang D, Pascual JM, Yang H, Engelstad K, Jhung S, Sun RP, et al. Glut-1 deficiency syndrome: Clinical, genetic, and therapeutic aspects. Ann Neurol. 2005;57:111–118. doi: 10.1002/ana.20331. [DOI] [PubMed] [Google Scholar]
- 51.Katz EB, Stenbit AE, Hatton K, DePinho R, Charron MJ. Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature. 1995;377:151–155. doi: 10.1038/377151a0. [DOI] [PubMed] [Google Scholar]
- 52.Kotani K, Peroni OD, Minokoshi Y, Boss O, Kahn BB. GLUT4 glucose transporter deficiency increases hepatic lipid production and peripheral lipid utilization. J Clin Invest. 2004;114:1666–1675. doi: 10.1172/JCI21341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Membrez M, Hummler E, Beermann F, Haefliger JA, Savioz R, Pedrazzini T, et al. GLUT8 is dispensable for embryonic development but influences hippocampal neurogenesis and heart function. Mol Cell Biol. 2006;26:4268–4276. doi: 10.1128/MCB.00081-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt S, Gawlik V, Holter SM, Augustin R, Scheepers A, Behrens M, et al. Deletion of glucose transporter GLUT8 in mice increases locomotor activity. Behav Genet. 2008;38:396–406. doi: 10.1007/s10519-008-9208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Myers SM, Johnson CP. Management of children with autism spectrum disorders. Pediatrics. 2008;120:1162–1182. doi: 10.1542/peds.2007-2362. [DOI] [PubMed] [Google Scholar]
- 56.Scattoni ML, Crawley J, Ricceri L. Ultrasonic vocalizations: A tool for behavioral phenotyping of mouse models of neurodevelopmental disorders. Neuroscience and Behavioral Reviews. 2009;33:508–515. doi: 10.1016/j.neubiorev.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dirks A, Fish EW, Kikusui T, van der Gugten J, Groenink L, Olivier B, Miczek KA. Effects of corticotropin-releasing hormone on distress vocalizations and locomotion in maternally separated mouse pups. Pharmacol Biochem Behav. 2002;72:993–9. doi: 10.1016/s0091-3057(02)00809-2. [DOI] [PubMed] [Google Scholar]
- 58.Ganguly A, Devaskar SU. Glucose transporter-3 null heterozygous mutation causes sexually dimorphic adiposity with insulin resistance. Am J Physiol Endocrinol Metab. 2008;294:E1144–51. doi: 10.1152/ajpendo.90251.2008. [DOI] [PubMed] [Google Scholar]
- 59.Chadman KK, Gong S, Scattoni ML, Boltuck SE, Gandhy SU, Heintz N, Crawley JN. Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Research. 2008;1:147–158. doi: 10.1002/aur.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F, et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proceedings of the National Academy of Sciences USA. 2008;105:1710–1715. doi: 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheinkopf SJ, Mundy P, Oiler DK, Steffens M. Vocal atypicalities of preverbal autistic children. Autism Dev Disord. 2000;30:345–54. doi: 10.1023/a:1005531501155. [DOI] [PubMed] [Google Scholar]
- 62.Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann L, et al. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 64.De Jaco A, Comoletti D, King CC, Taylor P. Trafficking of cholinesterases and neuroligins mutant proteins: An association with autism. Chem Biol Interact. 2006;175:349–351. doi: 10.1016/j.cbi.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morrow EM, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.