

Significance
Protein–protein interactions typically involve some degree of induced fit, producing complementary surfaces that account for high affinity and specificity. However, there are increasingly more examples of intrinsically disordered regions (IDRs) that exert important biologic effects despite never attaining a rigid structure. Here we show how a particularly disordered region of cardiac troponin I impacts the overall global conformation and function of its binding partner, cardiac troponin C. This newly described role for an IDR is accomplished through electrostatic interactions, which are particularly suited to IDRs. The regulation of electrostatic interactions in IDRs through phosphorylation is an emerging concept in cellular signaling, and troponin I is now another important example, one known by cardiac physiologists for 40 y.
Keywords: heart failure, dilated cardiomyopathy, hypertrophic cardiomyopathy, post-translational modification, fuzzy complex
Abstract
The cardiac isoform of troponin I (cTnI) has a unique 31-residue N-terminal region that binds cardiac troponin C (cTnC) to increase the calcium sensitivity of the sarcomere. The interaction can be abolished by cTnI phosphorylation at Ser22 and Ser23, an important mechanism for regulating cardiac contractility. cTnC contains two EF–hand domains (the N and C domain of cTnC, cNTnC and cCTnC) connected by a flexible linker. Calcium binding to either domain favors an “open” conformation, exposing a large hydrophobic surface that is stabilized by target binding, cTnI[148–158] for cNTnC and cTnI[39–60] for cCTnC. We used multinuclear multidimensional solution NMR spectroscopy to study cTnI[1–73] in complex with cTnC. cTnI[39–60] binds to the hydrophobic face of cCTnC, stabilizing an alpha helix in cTnI[41–67] and a type VIII turn in cTnI[38–41]. In contrast, cTnI[1–37] remains disordered, although cTnI[19–37] is electrostatically tethered to the negatively charged surface of cNTnC (opposite its hydrophobic surface). The interaction does not directly affect the calcium binding affinity of cNTnC. However, it does fix the positioning of cNTnC relative to the rest of the troponin complex, similar to what was previously observed in an X-ray structure [Takeda S, et al. (2003) Nature 424(6944):35–41]. Domain positioning impacts the effective concentration of cTnI[148–158] presented to cNTnC, and this is how cTnI[19–37] indirectly modulates the calcium affinity of cNTnC within the context of the cardiac thin filament. Phosphorylation of cTnI at Ser22/23 disrupts domain positioning, explaining how it impacts many other cardiac regulatory mechanisms, like the Frank–Starling law of the heart.
The balance between contraction and relaxation must be carefully regulated in the heart. Impaired relaxation can lead to diastolic heart failure, whereas systolic failure is characterized by insufficient contractility. Despite having different etiologies, both forms of heart failure are similar in terms of prevalence, symptoms, and mortality (1). Of all of the signaling pathways that regulate contractile function, the best studied is sympathetic β1-adrenergic stimulation (2), which leads to cardiomyocyte cAMP production and activation of protein kinase A (PKA). Downstream phosphorylation of L-type calcium channels and phospholamban increases calcium fluxes, whereas phosphorylation of sarcomeric proteins, cardiac troponin I (cTnI), cardiac myosin binding protein-C, and titin (3) regulates the calcium-induced mechanical response.
In human cTnI, Ser22 and Ser23 are the residues most consistently phosphorylated (4, 5). (There are some numbering inconsistencies in the literature, and we will refer to Ser22/23 instead of Ser23/24 to account for physiologic removal of the N-terminal methionine residue.) Originally identified as PKA targets, Ser22/23 are now known to be phosphorylated by other kinases, including PKG, PKCβ, PKCδ, and PKD1 (6), showing it to be an important locus at which multiple signaling pathways converge. Phosphorylation at cTnI Ser22/23 decreases the calcium sensitivity of the cardiac sarcomere (7). High levels of phosphorylation are seen in healthy individuals, but decreased phosphorylation levels occur in a number of pathologic states, including heart failure with reduced ejection fraction, heart failure with preserved ejection fraction, dilated cardiomyopathy, and hypertrophic cardiomyopathy (5, 8). Although dephosphorylation is likely a compensatory mechanism in many cases, it may be a disease-driving dysregulation in others.
Other regulatory mechanisms are strongly influenced by the phosphorylation state of Ser22/23. The Frank–Starling law of the heart, also known as length-dependent activation or stretch activation, is more pronounced when Ser22/23 are phosphorylated (9, 10). In contrast, Ser5 (11) or Ser41/43 (12, 13) phosphorylation has more of an impact when Ser22/23 are unphosphorylated. Finally, some mutations that cause familial dilated cardiomyopathy have been shown to mitigate the effect of Ser22/23 phosphorylation (14). Despite the physiologic importance of Ser22/23 phosphorylation in regulating cardiac calcium sensitivity, the extent of its modulatory capacity has remained elusive.
Ser22/23 lie within the cardiac-specific N-terminal region, cTnI[1–31], not present in the skeletal muscle isoforms. cTnI[1–209] forms long stretches of helical structure along a winding course that binds to troponin C, troponin T, and actin–tropomyosin. The X-ray structure of the cardiac troponin complex (15) did not include cTnI[1–34], so the structure of this region has not been determined, although there have been some preliminary investigations (16, 17). It is known that cTnI[1–31] interacts with cTnC in its unphosphorylated state (18), but phosphorylation abolishes this interaction, having an effect similar to truncation or removal of cTnI[1–31] (19). Our present study provides a detailed analysis of the structure and dynamics of cTnI[1–73] in complex with cTnC using solution NMR spectroscopy, highlighting its unique mechanism of action and physiologic implications.
Results
Chemical Shift Assignment and Secondary Structure of cTnI[1–73] Free and in Complex with cTnC.
NMR chemical shift assignment of protein backbone atoms 1Hα, 13Cα, 13Cβ, 13CO, 1HN, and 15N provides information about secondary structure on a per-residue basis. For example, α-helical residues have downfield 13Cα chemical shifts compared with random coil values, whereas residues with extended structure appear upfield. In free cTnI[1–73], residues 42–67 possess downfield 13Cα shifts indicative of a nascent helical structure (Fig. 1). The chemical shift analysis program δ2D (20) indicates overall <25% helical content for this region and random coil for the rest of the protein (Fig. S1A). There is no suggestion of a stable helix from residues 19–31, as had been suggested by a previous NMR analysis (16) that did not have the benefit of 13C chemical shift data.
Fig. 1.
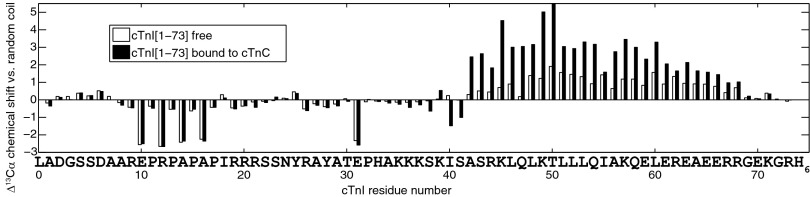
Deviation from random coil values for backbone 13Cα chemical shifts in free cTnI[1–73] (open bars) and cTnI[1–73] in complex with cTnC (filled bars).
Large deviations from random coil chemical shifts are observed in residues preceding proline (Fig. 1), which are almost always in an extended conformation. (Prolines themselves can adopt either turn-like or extended conformations.) The chemical shift analysis program DANGLE analyzes glycine, proline, and preproline residues separately from other amino acid residues because of their unique conformational preferences (21). DANGLE predicted an extended conformation for the proline-rich region spanning residues 10–18 (containing P11, P13, P15, and P17), although it does not specify a percentage, like δ2D, which predicted a random coil conformation with a small degree of extended conformation (Fig. S1). NOE assignments showed that all prolines in cTnI[1–73] preferred the trans conformation, although there is evidence of some minor peaks corresponding to cis forms.
Addition of wild type– or C35S,C84S–cTnC to cTnI[1–73] causes pronounced chemical shift changes in cTnI residues 42–68, suggestive of increased helical content (Fig. 1 and Fig. S1B), with Ser41 acting as the N-terminal helix cap residue (22). Helical structure is induced by binding of cTnI[39–60] to the large hydrophobic patch of the cTnC C-domain (cCTnC), forming a very tight complex with a nanomolar range dissociation constant (18) that effectively anchors cCTnC to the rest of troponin at all physiologic calcium concentrations.
Negligible chemical shift changes occur in cTnI residues 1–34 upon addition of cTnC (Fig. 1), despite a wealth of biochemical data documenting an interaction between this region and the N domain of cTnC (cNTnC) (for example, see refs. 23, 24). Using chemical shift data alone, there is no evidence of helix formation or other induced conformational preference anywhere along cTnI[1–34] as a result of the interaction (Fig. S1). There are examples in the literature of “fuzzy complexes” (25), some of which involve intrinsically disordered regions (IDRs) that show minimal chemical shift changes upon binding, like the phosphorylated Sic1–Cdc4 protein complex (26) and cystic fibrosis transmembrane conductance regulator (CFTR) R domain complexes (27). Evidence of an interaction can still be found through 1H-1H NOEs, signal broadening, or relaxation measurements, as turns out to be the case for cTnI[19–37]–cNTnC.
Reciprocal chemical shift changes in cTnC are also observed when cTnI[1–73] is added. The hydrophobic binding of cTnI[39–60] induces large chemical shift changes in cCTnC, but the interaction with cNTnC produced only minimal chemical shift changes (Fig. 2). It has previously been suggested that the interaction between cTnI[1–31] and cNTnC shifts the equilibrium of cNTnC toward the “open” state (28). The dominant conformation of calcium-bound cNTnC is in fact closed (29), with the open conformation sampled less than 20% of the time (30). The equilibrium is shifted to the open conformation when cNTnC binds hydrophobically to the helical switch region cTnI[148–158] (31), the critical step that releases the flanking inhibitory segments of cTnI and triggers muscle contraction. The shift to the open conformation manifests in large NMR chemical shift changes, particularly in loops I and II (Fig. 2). Stabilization of the calcium-bound open conformation also markedly increases the calcium affinity of cNTnC (32). However, because minimal chemical shift changes are observed when cNTnC interacts with cTnI[1–73], the cardiac-specific cTnI[1–31] must exert its calcium sensitizing effect through a different mechanism than stabilization of the open conformation.
Fig. 2.
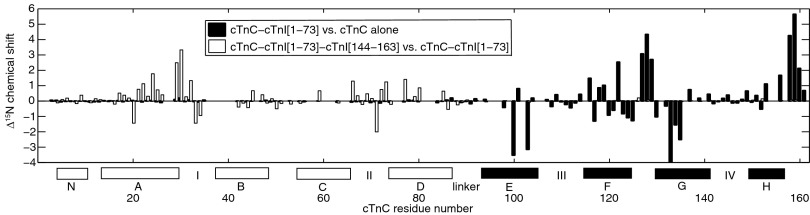
Binding-induced structural changes in C35S,C84S–cTnC are highlighted by backbone amide 15N chemical shift changes. The filled bars denote changes that occur when cTnC binds to cTnI[1–73]. The largest changes occur in cCTnC (see helices E–H along the x axis), whereas negligible changes are observed in cNTnC (helices N, A–D). This is because of the extensive hydrophobic interface between cTnI[39–60] and cCTnC, as opposed to the more superficial electrostatic interactions between cTnI[19–37] and cNTnC. In contrast, the open bars highlight the structural changes that occur when cTnI[144–163] binds cNTnC hydrophobically, driving a closed-to-open conformational transition in cNTnC. In this case, the predominant structural changes occur in cNTnC and not cCTnC. Stretches of missing assignments due to conformational exchange include residues 36–41, 60–62, 81–83, and 95–97.
In summary, the interaction between cTnI[1–31] and cNTnC has no observable effect on the secondary structure of cTnI[1–31] or the closed–open equilibrium of cNTnC.
1H-1H NOE Analysis of the cTnC–cTnI[1–73] Complex.
Homonuclear 1H-1H NOEs can be observed between any two 1H atoms within ∼6 Å, critical for any NMR-based structure determination. The vast majority of intermolecular 1H-1H NOEs occur between cTnI[39–60] and cCTnC, consistent with extensive hydrophobic binding. There are few intermolecular NOEs observed between cNTnC and cTnI[1–73], but the most critical NOEs are between cNTnC Ala7 and cTnI Ala42/Ser43. The importance of this contact is underscored by the fact that a double Ser41/43 mutation to aspartate (mimicking phosphorylation) markedly decreases troponin calcium affinity (33). Because cTnI Ala42/Ser43 are part of a rigid alpha helix bound to cCTnC and Ala7 is part of cNTnC helix N, the interaction fixes the position of cNTnC relative to cCTnC. This contact is actually present in the X-ray crystal structure (Fig. 3 A and B), suggesting that the true interdomain orientation in solution is in fact very similar. This single contact point is alone insufficient to stably fix the domains in solution, but it is stabilized by crystal packing contacts in the X-ray structure. In solution, the orientation is bolstered by additional electrostatic interactions from the disordered N-terminal tail of cTnI.
Fig. 3.

Ribbon diagrams of the troponin complex drawn by PyMol and derived from ref. 15. cNTnC is shown in magenta, and cCTnC is shown in cyan. Calcium ions are shown as green spheres. Troponin T, residues 226–271, is shown in yellow. cTnI, residues 34–135 and 147–161, is shown in orange. (A and B) The key contact that fixes the position of cNTnC relative to cCTnC is between A7 of cNTnC and A42/S43 of cTnI, shown as space-filling spheres. The side chains of cTnI[35–39] are also shown in stick figures, with K39 of cTnI contacting D131 and E135 of cCTnC (shown in sticks) and K35-S38 hovering over cNTnC. (C) Electrostatic surface representation of cNTnC showing the negatively charged surface that interacts with cTnI[19–37].
cTnI Ser41 is the N-terminal helix cap residue, but it is also part of a type VIII turn based on Ser38–Lys39–Ile40–Ser41 backbone chemical shifts, according to the Motif Identification from Chemical Shifts (MICS) program (22). Lys39 shows NOE contacts with Asp131 and Glu135 of helix G in cCTnC, whereas Ser38 has intermolecular NOEs to a lysine residue consistent with cNTnC Lys6 in the crystal structure (Fig. 3C). This brings Lys35–Lys36–Lys37 into contact with a prominent negatively charged surface that includes EF–hand loop II, the all-important calcium-binding loop (Fig. 3C). cTnI Lys35–37 display NOEs to residues with Asp- and Glu-like chemical shifts, but these could not be unambiguously separated from each other. In the crystal structure, there are two troponin complexes per asymmetric unit. In one complex, Lys35–37 are invisible, but in the other, they are modeled hovering over EF–hand loop II, with Lys37 closest to Asp75 of cNTnC. Finally, cTnI Arg21 and Arg26 also make weak NOEs to cTnC residues with Glu-like chemical shifts (Fig. S2), but these could not be unambiguously assigned either.
The considerable chemical shift overlap for charged Lys, Arg, Asp, and Glu side chains makes unambiguous intermolecular NOE assignment near impossible. Nevertheless, these NOEs demonstrate specific and stable electrostatic interactions involving cTnI Arg21, Arg26, and part of Lys35–37. This is in marked contrast to the lack of NOEs involving hydrophobic residues, which are usually critical for defining intermolecular contacts. cTnI Pro17, Ile18, Tyr25, Ala27, Tyr28, and Ala29 showed no intermolecular NOEs (see Fig. S2 for an example).
Classically, protein–protein interactions are dominated by hydrophobic interactions, which require close packing, exclusion of water, and a rigid structuring of the backbone, as exemplified by the interaction between cTnI[39–60] and cCTnC. In contrast, the electrostatic interactions involving cTnI[19–37] occur in a solvated environment that does not require as much rigidification of the backbone. It does require many positive charges, with as many as eight potentially involved (if His33 is included). Notably, Lys35 has been strongly implicated in autosomal dominant dilated cardiomyopathy (K36Q by the alternate numbering scheme), decreasing the calcium sensitivity of reconstituted thin filaments by 0.3–0.6 pCa units (14, 34).
15N Relaxation of cTnI[1–73] Free and in Complex with cTnC.
15N relaxation rates provide a window into nanosecond to picosecond (10−9 to 10−12 s) timescale conformational fluctuations in a protein. Structured proteins tumble with a global rotational correlation time, τC, on the order of several nanoseconds for a small domain, with τC scaling roughly proportionally to molecular weight. In rigidly structured regions, the overall motion is approximated by the global correlation time. As structural flexibility increases, for example, toward the N or C terminus, faster internal motions begin to dominate NMR relaxation behavior. Binding causes a decrease in internal motions that can be detected via changes in relaxation.
15N backbone relaxation studies were obtained for cTnI[1–73] both free and bound to C35S,C84S–cTnC (Fig. 4). The transverse relaxation rate, R2, is roughly proportional to the weighted average correlation time (including global tumbling and internal motions) at each backbone amide site. R1 relaxation is most effectively induced by motions with a timescale near 2–3 ns (15N nucleus on a 600-MHz spectrometer). A negative 1H-15N heteronuclear NOE is most effectively induced by motions with a timescale near 0.2–0.3 ns.
Fig. 4.

15N backbone relaxation data for cTnI[1–73]; “x” denotes free, and “o” denotes bound to C35S,C84S–cTnC.
Looking at the plots of R1, R2, and heteronuclear NOEs in free cTnI[1–73], it becomes evident that residues 45–67 are the most rigid in all of cTnI[1–73], with the highest R1, R2, and least negative heteronuclear NOE values, consistent with nascent helix formation. When this region binds to cTnC, it appears more massive and tumbles with a correlation time of about 15 ns (calculated using a model-free analysis: R2, ∼29 s−1; R1, ∼0.9 s−1; NOE, ∼0.8). The 15-ns correlation time is consistent with a 28-kDa protein. This suggests that not only is cTnI[1–73] tightly bound to cCTnC, but cNTnC is immobilized as well, with the whole complex tumbling as a single 28-kDa unit.
R2 values for cTnI[1–73] are consistent with those measured by Rosevear and coworkers for cTnC bound to cTnI[1–80] (35). In the absence of cTnI[1–80], the two domains of cTnC tumbled independently (like two smaller proteins) (mean R2, 13 s−1). Upon addition of cTnI[1–80], the two domains became rigidly fixed and tumbled as a single unit (mean R2, 31 s−1). Pseudophosphorylation of cTnI Ser22/Ser23 by mutation to aspartate caused the two domains of cTnC to tumble as two separate proteins (mean R2, 15 s−1). This work, along with the present study, shows that interaction with the cardiac-specific N-terminal extension of cTnI fixes the position of the cNTnC regulatory domain relative to the rest of the troponin complex, whereas phosphorylation of cTnI Ser22/Ser23 abolishes this.
The relaxation data for cTnI[19–37] are quite informative. Upon addition of cTnC, the most striking change is the plateau of increased R2 values in this region (Fig. 4). This indicates that although cTnI[19–37] is intrinsically disordered with no secondary structure preference, there is a substantial restriction of mobility. This would be expected if Arg19–21, Arg26, and Lys35–37 are electrostatically tethered, but there is likely transient structuring occurring in the intervening segments as well. Faint intermediate-range (i,i+3 and i,i+4) intramolecular 1H-1H NOEs are scattered throughout residues 18–32, indicating a transient helix- or turn-like structure, although chemical shift analysis indicates no net helical preference. Interestingly, the MICS Protein Structural Motif Prediction program indicates some potential turn structure at residues 22–25 and 31–34. The increased R2 values in cTnI[19–37] could result from fast conformational exchange (microsecond to millisecond timescale) and/or conformational restriction on the nanosecond timescale. Support for the latter comes from accompanying changes in R1 and heteronuclear NOE values. Note that the R1 values for cTnI[19–37] are the highest in all of cTnI, and these could not arise from an equilibrium between a strongly bound structured state (which would have a correlation time of 15 ns and R1 < 1 s−1) and a free state (with R1 < 1.5 s−1 from Fig. 4). If this were the case, the R1 values would be an average of these two states and significantly lower (so long as the exchange time constants were significantly longer than the nanosecond timescale correlation times). Instead, the high R1 values (up to 2 s−1) suggest a single partially structured state (or ensemble of very rapidly interconverting partially structured states).
Residues 2–18 of cTnI appear to be very mobile with considerable subnanosecond timescale motions and increasing mobility toward the N terminus. Upon addition of cTnC, there is no change in the rapid motions (followed by R1 and NOEs) in this region, suggesting that it is not tethered in the same way as cTnI[19–37].
Calcium Titration of cTnI[1–73]–cTnC.
The calcium binding affinity of the 2-(4′- (iodoacetamido)anilino)naphthalene-6-sulfonic acid (IAANS)–Cys84 conjugate of C35S–cTnC was studied using fluorescence, in complex with either cTnI[1–73] or cTnI[34–71] (Fig. S3). cTnI[1–73]–cTnC had a calcium pCa50 of 6.04 ± 0.03, and cTnI[34–71]–cTnC, 6.04 ± 0.04. Thus, there was no measurable difference between the calcium affinities of the complexes. Thus, the electrostatic interaction between cTnI[19–37] and cNTnC does not exert a direct effect on the calcium binding affinity of cNTnC.
Discussion
The unphosphorylated cardiac-specific N-terminal extension of cTnI interacts with cNTnC while remaining in a mobile, disordered state. IDRs are believed to make up a large proportion of protein sequences in eukaryotic proteomes. Due to their high solvent accessibility, they are readily available for posttranslational modifications like proteolysis and phosphorylation (36). IDRs can be regulated by multiple weak electrostatic interactions, and multiple phosphorylations are an effective means to add to or overcome these. A good example of the former is the increasing binding affinity between Sic-1 and Cdc4 as Sic-1 is increasingly phosphorylated (26, 37). In contrast, phosphorylation of both Ser22 and Ser23 in cTnI has been shown to abolish the interaction with cNTnC (23, 35, 38), with the two phosphate groups neutralizing the +4 positive charge locally contributed by Arg19, Arg20, Arg21, and Arg26. The physiologic effect of arginine mutation or PKA phosphorylation is to reduce the calcium sensitivity of troponin (24, 38). On the other hand, the R21C (R20C by our numbering) mutation disrupts the PKA consensus phosphorylation sequence, RRXS, causing increased calcium sensitivity and hypertrophic cardiomyopathy in humans (39) and animal models (40, 41).
The direct consequence of the interaction between cTnC and unphosphorylated cTnI[1–37] is that the orientation of cNTnC becomes fixed relative to the rest of the troponin complex. Any posttranslational modification or mutation within the troponin complex that disrupts the delicate positioning of the cNTnC domain would then be expected to have an effect similar to cTnI Ser22/23 phosphorylation, releasing cNTnC and allowing it to gyrate independently of the troponin complex. This may explain why many of these modifications abolish the effect of Ser22/23 phosphorylation or have a greater influence on the unphosphorylated state (see Introduction).
The positioning of cNTnC is critically important because it binds to the switch region, cTnI[148–158], to initiate cardiac contraction. The switch region is flanked by the inhibitory and regulatory regions, cTnI[136–147] and cTnI[161–209], that are anchored to actin during diastole (42). During calcium-activated systole, hydrophobic binding of “opened” cNTnC induces helix formation in cTnI[148–158] and sequesters the inhibitory and regulatory segments from their respective binding sites on actin. The positioning of cTnI[148–158] in the context of the thin filament is currently unknown, but it is quite possible that when cTnI is unphosphorylated, cNTnC is well positioned to bind cTnI[148–158] as soon as calcium becomes available during systole. This would increase the effective concentration of the cTnI[148–158] switch peptide, driving the cNTnC equilibrium toward the calcium-stabilized open state and thereby enhancing the calcium sensitivity of the cardiac thin filament.
Optimal positioning of the cNTnC domain may also explain the steeper length dependence curve of the Frank–Starling law of the heart when cTnI is phosphorylated (9, 10). The Frank–Starling law is caused by an unknown structural change that brings myosin heads into closer proximity of actin (43, 44), increasing duty ratio, the proportion of myosin heads strongly bound to actin. This change displaces tropomyosin and the inhibitory segments of cTnI from their diastolic actin binding sites, making the switch region, cTnI[148–158], more readily available for cNTnC binding. The effective concentration of free cTnI[148–158] may be a more important factor when cNTnC is unrestrained and must randomly “search” for its binding partner. Alternatively, downstream structural changes resulting from the actin–myosin interaction may lead to a positioning of cNTnC (through an as-of-yet uncharacterized interaction) that is redundant to that set up by unphosphorylated cTnI[1–37].
One limitation of the current study is that not all of the troponin complex components are present. It is quite possible that the large volume of space accessible to the mobile cTnI[1–37] segment intersects with that of cTnI[136–147], cTnI[161–209], and cTnT[272–288], highly charged IDRs invisible in the crystal structure but known to interact with actin. Indeed, a major consequence of fixing the orientation of cNTnC via the cTnI[1–37] IDR could be that potentially nonproductive interactions with other IDRs are prevented. The extent to which all of these IDRs interact with each other and with charged surfaces on troponin, tropomyosin, or actin remains to be clarified by future NMR studies.
In conclusion, our findings contradict an earlier model that proposed a rigid structure for cTnI[1–37] that stabilizes the open state of cNTnC to directly increase its calcium affinity (16). We did not observe evidence for a rigid structure in any way or a direct increase in calcium affinity. In our proposed model, the cardiac-specific N-terminal extension of cTnI remains largely disordered while it interacts electrostatically with cNTnC. This interaction does not affect the closed–open equilibrium of cNTnC but indirectly increases calcium affinity by optimally positioning cNTnC to bind the cTnI switch peptide. Phosphorylation at cTnI Ser22/23 disrupts this delicate arrangement, which we predict will be impacted by other regulatory mechanisms as well.
Materials and Methods
Protein Production and Purification.
Wild type– or C35S,C84S–cTnC was expressed in Escherichia coli using a pET3a-derived expression vector (45) and purified according to previously published protocols (46). In brief, purification involved three chromatographic steps: anion exchange, hydrophobic, and gel filtration chromatography. cTnI[1–73] with a C-terminal His-tag was expressed in a modified pET31b vector altered to fuse cTnI[1–73] to the C terminus of the β-barrel membrane protein, PagP, as previously described (47). cTnI[1–73] was separated from PagP via cyanogen bromide cleavage in 0.1 M HCl and 6 M Gdn⋅HCl and purified using nickel affinity chromatography under denaturing conditions. cTnI[144–163] and cTnI[34–71] peptides were synthesized and purified by GL Biochem.
The cTnI[1–73] protein purified from the PagP fusion construct was stable over the course of NMR experiments. However, after addition of cTnC, it was not uncommon for cTnI[1–73] to be slowly degraded, with proteolytic cleavage occurring N-terminal to Tyr25 or Tyr28, similar to what was observed from cTnI from human heart tissue (5). The change could be monitored by following the disappearance of NMR peaks corresponding to Tyr25–Ala29. Alternatively, it could be detected in cTnC by measuring 15N R2 relaxation rates, which would show that the two domains of cTnC were no longer tethered together by cTnI[1–73].
NMR Spectroscopy Sample Preparation.
For spectral simplicity, only one component of the protein complexes was isotopically labeled at a time. Enough labeled (15N- or 2H,15N- or 13C,15N-enriched) lyophilized protein to make a 0.5–1.0 mM sample was dissolved in 450 μL of buffer, consisting of 100 mM KCl, 10 mM imidazole, 10 mM CaCl2, 0.5 mM DSS, CalBiochem Protease Inhibitor Mixture Set I, and 5% (vol/vol) D2O. When wild type–cTnC was used, the buffer was further supplemented with 20 mM DTT. The pH was corrected to about 6.2 and monitored using 1D-1H NMR by measuring the pH-sensitive downfield imidazole H2 peak (48). A 1H,15N-heteronuclear single quantum coherence (HSQC) spectrum of the initial sample was obtained at 40 °C, and a 5-mM solution of the unlabeled binding partner was titrated in 10–15-μL aliquots. The titration was followed by serial 1H,15N-HSQC spectra. The binding equilibrium was in the slow exchange regime, so the titration was continued until the peaks corresponding to the unbound form disappeared completely. A ternary complex was also made by adding an excess (2 mg) of unlabeled cTnI[144–163] switch peptide.
NMR Spectroscopy and Data Analysis.
The 3D backbone assignment spectra were recorded on a Varian Inova 600 spectrometer. All experiments were from Agilent BioPack (VnmrJ 3.2D) unless otherwise specified. The experiments used for backbone assignment were HNCA, HN(CO)CA, HN(CA)CO, and HNCO. The molecular weight of the cTnC–cTnI[1–73] complex was 28 kDa, and sensitivity was further limited by exchange broadening. Additional experiments were used to assign the flexible regions of cTnI[1–73]: HNCACB, H(CCO)NH-TOCSY, and (H)C(CO)NH-TOCSY. To obtain the complete assignments of cTnC, HN(CA)HA and HA(CACO)NH (49) experiments were recorded, in-house modified from the BioPack HNCACB and CBCA(CO)NH experiments, respectively. 15N-edited nuclear Overhauser enhancement spectroscopy (NOESY)–HSQC spectra were also used for backbone assignment. Enhanced sensitivity and gradient selection was used in the backbone assignment triple resonance experiments, but TROSY was not used for the 15N and 1H dimensions.
The 3D NOESY spectra were acquired on a Varian Inova 800 spectrometer equipped with cryoprobe. 13C-edited HMQC–NOESY spectra were run on 13C,15N-labeled samples in H2O, although the majority of intramolecular NOE data were obtained from 13C-edited NOESY–HSQC spectra acquired in D2O, and intermolecular NOEs were obtained from 12C-filtered, 13C-edited NOESY–HSQC spectra in D2O. NMR data were processed using NMRPipe (50) software and visualized and analyzed with NMRViewJ (51).
15N T1, T2, and 1H-15N NOE experiments were conducted using 2H,15N-labeled cTnI[1,73] with unlabeled aCys–cTnC. TROSY was used in the 1H and 15N dimensions. A 6-s saturation time or recycle delay was used in the NOE experiments. For T1 and T2, curves were fit using a mono exponential decay function using the simplex minimization algorithm in MATLAB. The variance for each time point was estimated from the sum of the squares of the residuals divided by (N – 2), where N is the number of data points for each curve and 2 is the number of fitting parameters. A Monte Carlo method was then applied to obtain error estimates for R1 and R2. For 1H-15N NOE, the error estimate was based on the ratio of spectral noise to signal intensity in the reference spectrum, multiplied by a factor of √2 to reflect the fact that the 1H-15N NOE is a ratio. 15N R1, R2, and 1H-15N NOE data were further analyzed using a model-free analysis of internal motions (52).
Fluorescence Spectroscopy.
IAANS reacted with C35S cTnC Cys84 in labeling buffer (in 50 mM Tris, 150 mM KCl, 1 mM EGTA, and 6 M urea, pH 7.0) for 4 h at 4 °C (32). The labeling reaction was stopped by addition of 2 mM DTT, and the labeled protein was exhaustively dialyzed against refolding buffer (600 mM Mops, 150 mM KCl, and 2 mM EGTA, pH 7.0) to remove unreacted label.
All steady-state fluorescence measurements were performed at 22 °C. IAANS fluorescence was excited at 325 nm and monitored at 450 nm. Microliter amounts of CaCl2 were titrated into a 2-mL solution containing cTnC–cTnI[1–73] or cTnC–cTnI[34–71] (0.2 μM), 200 mM Mops, pH 7.0, 150 mM KCl, 2 mM EGTA, 1 mM DTT, and 3 mM MgCl2. An excess of cTnI peptide was used for the complexes ([cTnI] / [cTnC] = 1.3), with protein concentrations quantitated by amino acid analysis following acid hydrolysis. Free [Ca2+] was calculated with the program Maxchelator developed by Chris Patton and available at http://maxchelator.stanford.edu/CaMgATPEGTA-NIST.htm. Fluorescence experiments were performed in triplicate to determine error ranges for individual measurements as well as pCa.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the work of Eric Tai Kong Chan and Cathy Qin in developing the fluorescence assay. The work was supported by Canadian Institutes of Health Research (CIHR) Grant 37769. P.M.H. is supported by a CIHR Phase I Clinician Scientist Award and an Alberta Innovates–Health Solutions (AIHS) Incentive Award. S.E.P.-S. is supported by an AIHS graduate student award.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The NMR chemical shifts have been deposited in the BioMagResBank, www.bmrb.wisc.edu (accession nos. 25118–25120).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1410775111/-/DCSupplemental.
References
- 1.Sharma K, Kass DA. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ Res. 2014;115(1):79–96. doi: 10.1161/CIRCRESAHA.115.302922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262(5569):615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 3.van der Velden J. Diastolic myofilament dysfunction in the failing human heart. Pflugers Arch. 2011;462(1):155–163. doi: 10.1007/s00424-011-0960-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solaro RJ, van der Velden J. Why does troponin I have so many phosphorylation sites? Fact and fancy. J Mol Cell Cardiol. 2010;48(5):810–816. doi: 10.1016/j.yjmcc.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res. 2011;10(9):4054–4065. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res. 2013;112(2):355–366. doi: 10.1161/CIRCRESAHA.112.268672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wattanapermpool J, Guo X, Solaro RJ. The unique amino-terminal peptide of cardiac troponin I regulates myofibrillar activity only when it is phosphorylated. J Mol Cell Cardiol. 1995;27(7):1383–1391. doi: 10.1006/jmcc.1995.0131. [DOI] [PubMed] [Google Scholar]
- 8.Hamdani N, et al. Distinct myocardial effects of beta-blocker therapy in heart failure with normal and reduced left ventricular ejection fraction. Eur Heart J. 2009;30(15):1863–1872. doi: 10.1093/eurheartj/ehp189. [DOI] [PubMed] [Google Scholar]
- 9.Hanft LM, Biesiadecki BJ, McDonald KS. Length dependence of striated muscle force generation is controlled by phosphorylation of cTnI at serines 23/24. J Physiol. 2013;591(Pt 18):4535–4547. doi: 10.1113/jphysiol.2013.258400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wijnker PJ, et al. Length-dependent activation is modulated by cardiac troponin I bisphosphorylation at Ser23 and Ser24 but not by Thr143 phosphorylation. Am J Physiol Heart Circ Physiol. 2014;306(8):H1171–H1181. doi: 10.1152/ajpheart.00580.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henze M, et al. New insights into the functional significance of the acidic region of the unique N-terminal extension of cardiac troponin I. Biochim Biophys Acta. 2013;1833(4):823–832. doi: 10.1016/j.bbamcr.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinken AC, et al. Protein kinase C depresses cardiac myocyte power output and attenuates myofilament responses induced by protein kinase A. J Muscle Res Cell Motil. 2012;33(6):439–448. doi: 10.1007/s10974-012-9294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Velden J, et al. Functional effects of protein kinase C-mediated myofilament phosphorylation in human myocardium. Cardiovasc Res. 2006;69(4):876–887. doi: 10.1016/j.cardiores.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 14.Memo M, et al. Familial dilated cardiomyopathy mutations uncouple troponin I phosphorylation from changes in myofibrillar Ca²⁺ sensitivity. Cardiovasc Res. 2013;99(1):65–73. doi: 10.1093/cvr/cvt071. [DOI] [PubMed] [Google Scholar]
- 15.Takeda S, Yamashita A, Maeda K, Maéda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424(6944):35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 16.Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J Mol Biol. 2007;373(3):706–722. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 17.Ward DG, et al. Characterization of the interaction between the N-terminal extension of human cardiac troponin I and troponin C. Biochemistry. 2004;43(13):4020–4027. doi: 10.1021/bi036128l. [DOI] [PubMed] [Google Scholar]
- 18.Ferrières G, et al. Systematic mapping of regions of human cardiac troponin I involved in binding to cardiac troponin C: N- and C-terminal low affinity contributing regions. FEBS Lett. 2000;479(3):99–105. doi: 10.1016/s0014-5793(00)01881-0. [DOI] [PubMed] [Google Scholar]
- 19.Biesiadecki BJ, et al. Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice. J Biol Chem. 2010;285(25):19688–19698. doi: 10.1074/jbc.M109.086892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camilloni C, De Simone A, Vranken WF, Vendruscolo M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry. 2012;51(11):2224–2231. doi: 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- 21.Cheung MS, Maguire ML, Stevens TJ, Broadhurst RW. DANGLE: A Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J Magn Reson. 2010;202(2):223–233. doi: 10.1016/j.jmr.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Shen Y, Bax A. Identification of helix capping and b-turn motifs from NMR chemical shifts. J Biomol NMR. 2012;52(3):211–232. doi: 10.1007/s10858-012-9602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baryshnikova OK, Li MX, Sykes BD. Modulation of cardiac troponin C function by the cardiac-specific N-terminus of troponin I: Influence of PKA phosphorylation and involvement in cardiomyopathies. J Mol Biol. 2008;375(3):735–751. doi: 10.1016/j.jmb.2007.10.062. [DOI] [PubMed] [Google Scholar]
- 24.Ward DG, Cornes MP, Trayer IP. Structural consequences of cardiac troponin I phosphorylation. J Biol Chem. 2002;277(44):41795–41801. doi: 10.1074/jbc.M206744200. [DOI] [PubMed] [Google Scholar]
- 25.Fuxreiter M. Fuzziness: Linking regulation to protein dynamics. Mol Biosyst. 2012;8(1):168–177. doi: 10.1039/c1mb05234a. [DOI] [PubMed] [Google Scholar]
- 26.Mittag T, et al. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc Natl Acad Sci USA. 2008;105(46):17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bozoky Z, et al. Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactions. Proc Natl Acad Sci USA. 2013;110(47):E4427–E4436. doi: 10.1073/pnas.1315104110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finley N, et al. NMR analysis of cardiac troponin C-troponin I complexes: Effects of phosphorylation. FEBS Lett. 1999;453(1-2):107–112. doi: 10.1016/s0014-5793(99)00693-6. [DOI] [PubMed] [Google Scholar]
- 29.Sia SK, et al. Structure of cardiac muscle troponin C unexpectedly reveals a closed regulatory domain. J Biol Chem. 1997;272(29):18216–18221. doi: 10.1074/jbc.272.29.18216. [DOI] [PubMed] [Google Scholar]
- 30.Eichmüller C, Skrynnikov NR. A new amide proton R1rho experiment permits accurate characterization of microsecond time-scale conformational exchange. J Biomol NMR. 2005;32(4):281–293. doi: 10.1007/s10858-005-0658-y. [DOI] [PubMed] [Google Scholar]
- 31.Li MX, Spyracopoulos L, Sykes BD. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38(26):8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 32.Davis JP, et al. Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys J. 2007;92(9):3195–3206. doi: 10.1529/biophysj.106.095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wijnker PJ, et al. Phosphorylation of protein kinase C sites Ser42/44 decreases Ca(2+)-sensitivity and blunts enhanced length-dependent activation in response to protein kinase A in human cardiomyocytes. Arch Biochem Biophys. 2014;554:11–21. doi: 10.1016/j.abb.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carballo S, et al. Identification and functional characterization of cardiac troponin I as a novel disease gene in autosomal dominant dilated cardiomyopathy. Circ Res. 2009;105(4):375–382. doi: 10.1161/CIRCRESAHA.109.196055. [DOI] [PubMed] [Google Scholar]
- 35.Gaponenko V, et al. Effects of troponin I phosphorylation on conformational exchange in the regulatory domain of cardiac troponin C. J Biol Chem. 1999;274(24):16681–16684. doi: 10.1074/jbc.274.24.16681. [DOI] [PubMed] [Google Scholar]
- 36.Iakoucheva LM, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32(3):1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang X, et al. Composite low affinity interactions dictate recognition of the cyclin-dependent kinase inhibitor Sic1 by the SCFCdc4 ubiquitin ligase. Proc Natl Acad Sci USA. 2012;109(9):3287–3292. doi: 10.1073/pnas.1116455109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward DG, et al. NMR and mutagenesis studies on the phosphorylation region of human cardiac troponin I. Biochemistry. 2004;43(19):5772–5781. doi: 10.1021/bi036310m. [DOI] [PubMed] [Google Scholar]
- 39.Arad M, et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation. 2005;112(18):2805–2811. doi: 10.1161/CIRCULATIONAHA.105.547448. [DOI] [PubMed] [Google Scholar]
- 40.Gomes AV, Harada K, Potter JD. A mutation in the N-terminus of troponin I that is associated with hypertrophic cardiomyopathy affects the Ca(2+)-sensitivity, phosphorylation kinetics and proteolytic susceptibility of troponin. J Mol Cell Cardiol. 2005;39(5):754–765. doi: 10.1016/j.yjmcc.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, et al. Generation and functional characterization of knock-in mice harboring the cardiac troponin I-R21C mutation associated with hypertrophic cardiomyopathy. J Biol Chem. 2012;287(3):2156–2167. doi: 10.1074/jbc.M111.294306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tripet B, Van Eyk JE, Hodges RS. Mapping of a second actin-tropomyosin and a second troponin C binding site within the C terminus of troponin I, and their importance in the Ca2+-dependent regulation of muscle contraction. J Mol Biol. 1997;271(5):728–750. doi: 10.1006/jmbi.1997.1200. [DOI] [PubMed] [Google Scholar]
- 43.Farman GP, et al. Myosin head orientation: A structural determinant for the Frank-Starling relationship. Am J Physiol Heart Circ Physiol. 2011;300(6):H2155–H2160. doi: 10.1152/ajpheart.01221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mateja RD, de Tombe PP. Myofilament length-dependent activation develops within 5 ms in guinea-pig myocardium. Biophys J. 2012;103(1):L13–L15. doi: 10.1016/j.bpj.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li MX, et al. Kinetic studies of calcium and cardiac troponin I peptide binding to human cardiac troponin C using NMR spectroscopy. Eur Biophys J. 2002;31(4):245–256. doi: 10.1007/s00249-002-0227-1. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Mercier P, Letourneau PJ, Sykes BD. Effects of Phe-to-Trp mutation and fluorotryptophan incorporation on the solution structure of cardiac troponin C, and analysis of its suitability as a potential probe for in situ NMR studies. Protein Sci. 2005;14(9):2447–2460. doi: 10.1110/ps.051595805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang PM, Pan JS, Sykes BD. A PagP fusion protein system for the expression of intrinsically disordered proteins in Escherichia coli. Protein Expr Purif. 2012;85(1):148–151. doi: 10.1016/j.pep.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Baryshnikova OK, Williams TC, Sykes BD. Internal pH indicators for biomolecular NMR. J Biomol NMR. 2008;41(1):5–7. doi: 10.1007/s10858-008-9234-6. [DOI] [PubMed] [Google Scholar]
- 49.Boucher W, Laue ED, Campbell-Burk S, Domaille PJ. Four-dimensional heteronuclear triple resonance NMR methods for the assignment of backbone nuclei in proteins. J Am Chem Soc. 1992;114(6):2262–2264. [Google Scholar]
- 50.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 51.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4(5):603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 52.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear macnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104(17):4546–4559. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.