

Significance
The transport of ions through the plant cell membrane establishes the key physicochemical parameters for cell function. Stress situations such as those created by soil salinity or low potassium conditions alter the ion transport across the membrane producing dramatic changes in the cell turgor, the membrane potential, and the intracellular pH and concentrations of toxic cations such as sodium and lithium. As a consequence, fundamental metabolic routes are inhibited. The CIPK family of 26 protein kinases regulates the function of several ion transporters at the cell membrane to restore ion homeostasis under stress situations. Our analyses provide an explanation on how the CIPKs are differentially activated to coordinate the adequate cell response to a particular stress.
Keywords: signaling, ion transport, abiotic stress
Abstract
Plant cells have developed specific protective molecular machinery against environmental stresses. The family of CBL-interacting protein kinases (CIPK) and their interacting activators, the calcium sensors calcineurin B-like (CBLs), work together to decode calcium signals elicited by stress situations. The molecular basis of biological activation of CIPKs relies on the calcium-dependent interaction of a self-inhibitory NAF motif with a particular CBL, the phosphorylation of the activation loop by upstream kinases, and the subsequent phosphorylation of the CBL by the CIPK. We present the crystal structures of the NAF-truncated and pseudophosphorylated kinase domains of CIPK23 and CIPK24/SOS2. In addition, we provide biochemical data showing that although CIPK23 is intrinsically inactive and requires an external stimulation, CIPK24/SOS2 displays basal activity. This data correlates well with the observed conformation of the respective activation loops: Although the loop of CIPK23 is folded into a well-ordered structure that blocks the active site access to substrates, the loop of CIPK24/SOS2 protrudes out of the active site and allows catalysis. These structures together with biochemical and biophysical data show that CIPK kinase activity necessarily requires the coordinated releases of the activation loop from the active site and of the NAF motif from the nucleotide-binding site. Taken all together, we postulate the basis for a conserved calcium-dependent NAF-mediated regulation of CIPKs and a variable regulation by upstream kinases.
Cell perception of extracellular stimuli is followed by a transient variation in cytosolic calcium concentration. Plants have evolved to produce the specific molecular machinery to interpret this primary information and to transmit this signal to the components that organize the cell response (1–4). The plant family of serine/threonine protein kinases PKS or CIPKs (hereinafter CIPKs) and their activators, the calcium-binding proteins SCaBPs or CBLs (hereinafter CBLs) (5, 6) function together in decoding calcium signals caused by different environmental stimuli. Available data suggest a mechanism in which calcium mediates the formation of stable CIPK–CBL complexes that regulate the phosphorylation state and activity of various ion transporters involved in the maintenance of cell ion homeostasis and abiotic stress responses in plants. Among them, the Arabidopsis thaliana CIPK24/SOS2-CBL4/SOS3 complex activates the Na+/H+ antiporter SOS1 to maintain intracellular levels of the toxic Na+ low under salt stress (7–9), the CIPK11–CBL2 pair regulates the plasma membrane H+-ATPase AHA2 to control the transmembrane pH gradient (10), the CIPK23–CBL1/9 (11, 12) regulates the activity of the K+ transporter AKT1 to increase the plant K+ uptake capability under limiting K+ supply conditions (12, 13), and CIPK23–CBL1 mediates nitrate sensing and uptake by phosphorylation of the nitrate transporter CHL1 (14). Together these findings show that understanding the molecular mechanisms underling CIPKs function provides opportunities to increase plant tolerance to abiotic stress and to improve plants for human benefit.
CIPKs and CBLs contain discrete structural modules that are involved in the calcium-dependent regulation of the activity of the system and ensure the colocalization of the CIPK–CBL interacting pairs with their substrates at particular sites within the cell (15–17). CIPKs include an N-terminal kinase catalytic domain followed by a characteristic self-inhibitory motif known as FISL or NAF motif (hereinafter NAF, Pfam no. PF03822) (1, 6) and a protein phosphatase 2C binding domain designated as PPI (11, 18, 19). The NAF motif directly interacts with the catalytic domain and inhibits the kinase activity. The calcium-dependent interaction of CBLs with the NAF motif relieves the self-inhibition and activates the CIPKs (5, 6, 19, 20). The calcium binding to CBLs is mediated by four EF hand-like calcium binding motives. In addition, several CBLs are myristoylated and/or palmitoylated. These modifications are essential for recruiting their interacting CIPK partner to the plasma or vacuolar membrane (17, 21–23), and they may also be involved in the interaction of the CIPK–CBL complexes with their substrates (24). In addition, the phosphorylation of a conserved serine residue at the C terminus of CBLs by its interacting CIPK is required for activation of transporter substrates. It has been proposed that this process may stabilize the CIPK–CBL complex and trigger conformational changes to the binary complex that enhance its specificity toward target proteins (13, 25).
Like many other kinases, CIPKs are also regulated by the phosphorylation of the activation loop by upstream kinases. This loop undergoes large conformational changes upon phosphorylation, allowing the entrance and the stabilization of substrates at the kinase active site (26). The activation loop of the CIPKs contains three conserved Tyr, Thr, or Ser residues. For some members of the family, the mutation of one of these residues to Asp mimics phosphorylation and produces the activation of the kinase, partly overcoming the effect of the self-inhibitory NAF motif. In fact, these phosphorylation-mimicking mutations and the deletion of the inhibitory domain produce a synergistic effect on the CIPK activity (6, 27–29). Transgenic plants expressing these CIPK24/SOS2 mutant proteins show improved salt tolerance (30).
The kinase self-phosphorylation is another regulatory mechanism used by CIPKs. CIPK24/SOS2 is able to self-phosphorylate, and the autophosphorylation is important for its activity (31). Although the default state of CIPKs is inactive, some degree of autophosphorylation activity has been observed even for dephosphorylated and CBL-unbound CIPKs, which suggests that some CIPKs display basal activity (6). Indeed, it has been shown that the general regulatory factor 14-3-3 proteins (32) interact with CIPK24/SOS2 and repress its basal kinase activity when plants are grown in the absence of salt stress (33).
The crystal structure of the binary complex of Ca2+-CBL4/SOS3 with the C-terminal regulatory moiety of CIPK24/SOS2 revealed the molecular mechanism underlying CBL-mediated activation of the CIPKs. The structure showed that the CIPK24/SOS2 self-inhibitory NAF motif is bound to CBL4/SOS3 and, consequently, it is not accessible to the kinase domain (19, 20). However, whether the CBL-unbound NAF blocks the active site or inhibits the enzyme by an allosteric mechanism is not known. To determine the molecular and structural basis for the CIPKs autoinhibition by the NAF and the activation by upstream kinases, we solved the structures of CIPK23 and CIPK24/SOS2. Our data show that inactivation of the kinases relies on the blockage of the active site by the NAF motif and the activation loop, which constitutes the basis for the conserved NAF-mediated self-inhibition of the CIPKs.
Results and Discussion
The Crystal Structures of CIPK23 and CIPK24/SOS2 Reveal a Canonical Kinase Fold.
To determine the molecular architecture of A. thaliana CIPKs, we cloned and expressed in Escherichia coli a set of constructs containing several combinations of the functional domains of CIPK23 and CIPK24/SOS2 (Fig. 1A and Materials and Methods). After extensive optimization of the purification and crystallization conditions, a CIPK23 fragment comprising residues 24–331 (CIPK23ΔC) produced good diffracting crystals to 2.3 Å resolution. This protein chain included the kinase catalytic domain plus the junction region to the regulatory domain (Fig. 1A). In addition, we also solved the structure of the Thr190Asp (CIPK23ΔC T190D) mutant protein at 1.9 Å resolution (Fig. 1B, Table S1, and Materials and Methods). This version of CIPK23 mimics the observed phosphorylation of Thr190 (34) located at the activation loop. Both structures are nearly identical [Cα backbone root mean square deviation (rmsd) of 0.3 Å] with local differences around the mutated residue. Hence fore, unless otherwise stated, all of the results presented here will refer to the CIPK23 T190D mutant as it was resolved at higher resolution. The equivalent version of pseudophosphorylated recombinant double mutant CIPK24/SOS2 Thr168Asp Ser228Asp, which also mimics a self-phosphorylated protein at Ser228, severely aggregated and did not crystallize under any tested condition. The application of the consensus-based approach to introduce stabilizing mutations (35, 36) was successful. Crystals of the CIPK24/SOS2 variant Glu107Lys/Ser109Asp/Cys127Ser/Pro81Lys/Leu266Lys (herein after CIPK24/SOS2ΔC T168D) were obtained and the structure was resolved at 3.4 Å resolution (Fig. 1B, Table S1, and Materials and Methods). These point mutations are not close to the active site and are located at the molecular surface of the protein, where they enhance the protein solubility and reduce unproductive crystal packing interactions. The overall fold of CIPK24/SOS2ΔC T168D and CIPK23ΔC T190D is very similar (Cα rmsd of 1.0 Å for 263 atoms).
Fig. 1.

The crystal structures of CIPK23 and CIPK24/SOS2. (A) Domain structure of CIPKs. (B) A ribbon representation of the crystal structures of CIPK23ΔC T190D (Left) and CIPK24/SOS2ΔC T168D (Right). The key structural features are labeled and highlighted in different colors. (C) The ribbon representation of the active site section of the CIPK23ΔC T190D, CIPK24/SOS2ΔC T168D (semitransparent overlaid on the left), and the related active kinase PKA (Right) (PDB ID code: 1ATP). The catalytically relevant residues and the ATP are displayed in a stick representation; CIPK23 and CIPK24/SOS2 residues are displayed in white and cyan, respectively.
Overall, the structure of the catalytic domains of CIPK23 and CIPK24/SOS2 display the canonical Ser/Thr protein kinase fold, similar to other Snf1 kinase domains (37) (Fig. 1B). To summarize, the protein folds into two separate lobes connected by the activation loop. The N lobe consists of a six-stranded β-sheet plus two α-helices. The C lobe is mainly helical. All of the residues involved in ATP, metal, and substrate binding are placed between these domains. The junction region docks on the kinase domain opposite to the active site (Fig. 1B). The junction spans from the C to the N lobe where it packs against αB and αC helices and the central β-sheet. A sequence analysis suggests that these features are conserved among the members of the CIPK family (Fig. S1). A molecule of CHAPS detergent was found in the ATP binding pocket of CIPK23 (Fig. 1B).
The crystal structures of CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D contain four molecules in the asymmetric unit forming two head-to-tail dimers (Fig. S2A). The CIPK23ΔC T190D dimeric structures are similar to that described for the yeast Snf1 kinase domain (38) that has been suggested to represent a physiological inactive form of the kinase as they bury kinase active sites. CIPK23ΔC T190D dimers are further stabilized by a CHAPS molecule, used in the crystallization procedure, and by a disulfide bond between two equivalent Cys192 residues. However, gel filtration chromatography shows that CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D are monomeric (Fig. S2B); hence, it seems that the crystal packing interactions between independent molecules are not physiologically relevant.
CIPK23 and CIPK24/SOS2 Adopt a Conserved Inactive Conformation Stabilized by the Activation Loop.
Protein kinases display a universal regulatory mechanism involving the optimal organization of the residues that are essential for recognition of ATP and peptide substrates, and for the subsequent catalysis (39, 40). This arrangement is normally associated with a conformational change in the relative position of the N lobe with respect to the C lobe that moves from an “open” inactive state to a “closed” active state. In CIPKs, this transition is regulated by both the phosphorylation of the activation loop and by the interaction between the self-inhibitory NAF motif and the catalytic domain (6). Such processes are common to other kinases (26, 37, 41, 42) and rely on the stabilization of the kinase conformation either in the active or inactive state according to cell necessities.
To establish the molecular and structural basis of the regulatory mechanism mediated by the activation loop phosphorylation and by the NAF interaction with the catalytic domain, we compared the CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D structures with the structure of the active catalytic subunit of cAMP-dependent protein kinase (43) and analyzed the structural determinants that yield kinase activation (39, 40) (Fig. 1C and Fig. S3). This comparison is possible because the internal architecture among all of the active eukaryotic kinase families, including the conformation of residues involved in substrate and cofactor binding, is remarkably well conserved (39). Our analysis shows that CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D display an inactive open conformation in which the N-terminus of the activation loop forms a short α-helical turn (αT1) that is totally buried between the N lobe and C lobe and pushes the helix αC harboring the conserved catalytically relevant Glu79/Glu59 (for CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D) away. The C-terminus of the activation loops of CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D display different structures. Although the CIPK24/SOS2ΔC T168D loop spans out of the active site, the CIPK23ΔC T190D loop contains another well-ordered α-helical turn (αT2) followed by a long loop that blocks the access of ATP and peptide substrate to the active site and drives the nucleotide binding loop (the G loop) apart (Fig. 1B). The observed inactive conformations of CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D are surprising because the constructions used for protein crystallization lack the NAF self-inhibitory motif and include the T190D mutation (T168D for CIPK24/SOS2), mimicking a phosphorylated state of the activation loop that leads to hyperactive forms of CIPK24/SOS2 and some other CIPK proteins (6, 27). The fact that CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D display largely identical overall conformations, although we solved them in two different space groups with different crystal packing contacts, indicates that their conformations are unlikely to be affected by crystal packing.
To determine the biological significance of our structural observations, we performed kinase activity assays comparing the CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D constructs lacking the regulatory C-terminal domain that were used for crystallography (Fig. 2). Our results showed that although the phosphomimicking mutation T168D greatly enhanced the kinase activity of CIPK24/SOS2ΔC, the corresponding T190D mutation produced little effect on CIPK23ΔC. These data correlate well with those obtained with full-length proteins expressed by using a wheat germ extract-based in vitro transcription/translation protocol (13). In these experiments, the catalytic efficiency (kcat/Km) was roughly eightfold lower for CIPK23 than for CIPK24/SOS2. To understand the basis for such different activities, we performed a structurally guided sequence analysis of the activation loop of the A. thaliana CIPKs (Fig. 3A). The remarkable conservation of αT1 structure and sequence suggests that the inactive conformation observed for CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D is a general feature of CIPKs. By contrast, CIPKs display the highest variability at αT2. In particular, the residue composition at the solvent inaccessible CIPK23ΔC Val182 and Arg183 permits us to classify most of the CIPKs in four groups: (i) those like CIPK23 containing a hydrophobic residue followed by a positively charged one, (ii) those containing two positively charged or polar residues and (iii) those containing two hydrophobic residues. In addition, CIPK24/SOS2 and CIPK8 form the fourth group that displays the Gly-Val dipeptide at these positions plus a two-residue deletion adjacent to this area.
Fig. 2.

Comparative kinase activity analyses of CIPK23 and CIPK24/SOS2 proteins. The average specific activities expressed in nmol⋅min−1⋅μg−1 of protein units determined by a kinase spectrophotometric assay of the proteins expressed in E. coli. Error bars indicate the SD calculated from four independent measurements. All of the distributions are different compared with the CIPK23ΔC with a confidence level of 99% but those corresponding to CIPK23ΔC T190D and CIPK23-FILS in which the confidence level is 95% (Student’s t test).
Fig. 3.

The activation loop. (A) Sequence alignment of the activation loop of CIPKs together with a schematic representation of CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D secondary structures. Sequences are grouped according to their sequence similarity. Residues are colored according to their conservation (77). The arrows highlight the totally conserved and potentially phosphorylable residues. The rectangular box highlights the residues at αT2 that serve to classify the CIPKs into four groups. (B) A detailed view of CIPK23ΔC T190D showing the structural environment of the conserved Ser176, Thr190, and Tyr197. The view highlights the role of Val182 and Arg183 in the stabilization of the loop at the active site. Inset shows a comparison of CIPK23ΔC (white sticks) and CIPK23ΔC T190D (cyan sticks) structures showing that the point mutation is accommodated without a significant change in the loop conformation.
In CIPK23ΔC T190D, the Val182 and Arg183 side chains stabilize the activation loop at the active site (Fig. 3B). These two residues interact with a hydrophobic pocket at the N lobe and, additionally, the charged guanidinium moiety of Arg182 is hydrogen bonded to the catalytic Asp154 and other residues in the activation loop, suggesting that these two residues stabilize the inactive conformation of the activation loop and may explain why CIPK24/SOS2ΔC T168D, which lacks αT2, is more active than CIPK23ΔC T190D. There is no available experimental structure for representative proteins of the other two αT2 groups to establish a structure function relationship. However, it is likely that the replacement of the totally buried Val182 by a charged residue will promote the release of the activation loop from the active site and enhance the kinase activity (Fig. 3B). To test this hypothesis, we generated the CIPK23ΔC V182K T190D mutant protein to convert it into a group of those CIPKs with two positively charged residues in the αT2. Our results showed that this single point mutant displays enhanced kinase activity relative to the wild type CIPK23ΔC and CIPK23ΔC T190D protein (Fig. 2). This is in agreement with the available biochemical data of a natural CIPK protein of this group which is known to be stimulated by the phosphorylation of the activation loop (44). Likewise, the replacement of CIPK23 activation loop with that of CIPK24/SOS2 enhances the kinase activity of CIPK23ΔC T190D (Fig. S4).
The available data indicate that full activation of CIPK23 may require additional and yet-undetermined phosphorylation events by upstream plant kinases. In most kinase families, the phosphorylation of the activation loop at Ser, Thr, or Tyr residues shifts the equilibrium toward an active conformation that promotes substrate binding (45). The comparison of the CIPK activation loop sequences shows that residues Ser176, Thr190, and Tyr197 of CIPK23 are conserved (Fig. 3A). Thus, several phosphorylation events at the activation loop may be required to activate CIPK23. Indeed, the analyses of the structures of CIPK23ΔC and CIPK23ΔC T190D show that this mutation is accommodated without any change in the conformation of the activation loop (Fig. 3B). By contrast, the equivalent T168D of CIPK24/SOS2ΔC is solvent accessible, indicating that this mutation promotes the release of the activation loop from the active site. Residues Ser176 and Tyr197 in CIPK23 are structurally conserved with CIPK24/SOS2 Ser156 and Tyr175, and it has been shown that the individual mutation of these two residues in CIPK24/SOS2 to Asp increases the activity of recombinant CIPK24/SOS2 (28). The analysis of these structures shows that Ser176/Ser156 are placed at αT1 and are solvent inaccessible, whereas the phenol rings of Tyr197/Tyr175 are stabilized at a hydrophobic pocket in the C lobe with the hydroxyl group pointing toward solvent (Fig. 3B). The phosphorylation of any of these hydroxyl groups could promote the separation of the activation loop from the active site. In support of this idea, kinase activity assays showed that the CIPK23ΔC S176D T190D Y197E triple mutant displayed enhanced kinase activity compared with the wild-type CIPK23ΔC and CIPK23ΔC T190D single mutant (Fig. 2).
Thus, the combined structural and biochemical data reveal an additional level of complexity in the regulation of the CIPK–CBL network wherein, in addition to the CBL-mediated activation, some kinases would require a different degree of phosphorylation by upstream kinases to achieve full activity, and this phosphorylation level depends on the natural sequence variability of the activation loop.
The Structure of CIPK23 Defines a Large Cavity That Accommodates the Self-Inhibitory NAF Motif.
A remarkable characteristic of the CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D structures is that the relative position of the N lobe with respect to the C lobe defines a wide-open cavity at the ATP binding site. In fact, there is well-defined electron density for a CHAPS molecule, used for the crystallization experiments, that spans the ATP binding pocket of CIPK23ΔC T190D (Fig. S5A). The hydrophobic face of the CHAPS cholate ring is oriented toward the nonpolar side of the N lobe β-sheet. In addition, it makes hydrophobic contacts with Leu187 and His189 side chains at the activation loop and with Phe110 at the hinge region between the N and C lobes. The polar face of CHAPS is solvent accessible and forms three hydrogen bonds to water molecules (Fig. S5B). Because many ATP competitive kinase inhibitors target this binding site, we performed an automated search on the entire Protein Data Bank (PDB) using the geometry and the chemical properties of CIPK23ΔC T190D cavity as search criteria (Relibase+; ref. 46). Our analysis reveals that, in contrast with the chemical nature of CHAPS, the inhibitory molecules bound to this site consist of planar aromatic rings that mimic the adenine ring of ATP. In addition, the study shows that the CIPK23 cavity is wider than other occupied cavities (Fig. S5C). This observation, together with the fact that a similar cavity is present in CIPK24/SOS2 (Fig. S5D), show that CHAPS binding is indicative of the hydrophobic nature and of the characteristic enlarged size of the ATP binding site of CIPKs with respect to the size and shape of other inactive kinase molecules. In addition, the fact that CIPK23ΔC T190D activation loop is mainly stabilized by contacts with the kinase domain suggests that CHAPS binding does not affect the conformation of the activation loop, although local effects in the vicinity of Leu-187 cannot be discarded (Fig. S5E).
The fact that the kinase activation via phosphorylation of the activation loop and via CBL binding are synergic processes indicates that the release of both the activation loop and the NAF motif from the catalytic domain are coupled (6, 27–29). A plausible model to account for these observations would imply that the inactive kinase bears two different but interacting sites for NAF and activation loop. Thus, we explored the possibility that the ATP binding pocket of CIPK23 could accommodate the NAF self-inhibitory domain, blocking ATP entrance and stabilizing a closed conformation of the activation loop, thereby hindering the substrate entrance. The crystal structures of the regulatory moiety of CIPK24/SOS2 and CIPK14 in complex with their interacting CBLs revealed that the NAF motif consists of two amphipathic helices connected by a variable loop (19, 47) (Fig. 4A). Interestingly, the visual and automatic analyses of the CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D molecular surfaces using CAVER (48) reveal that the ATP cavity spans linearly, wrapping the kinase domain around the hinge region between the N and C lobes (Fig. 4A). This cavity is wide enough to accommodate the two amphipathic helical segments of the NAF motif and it is connected to the activation loop, thus suggesting that it constitutes the CIPKs self-inhibitory binding pocket. Remarkably, the cavity overlaps well with the calmodulin binding helix of the regulatory domain of the calcium/calmodulin-dependent protein kinase I (49) (Fig. S6).
Fig. 4.

(A) Ribbon and schematic representations of the CIPK24/SOS2 regulatory domain structures in complex with CBL4/SOS3 (PDB ID code: 2EHB) (Left) and CIPK23ΔC T190D together with a surface representation of the cavity connecting the ATP binding site and the hinge region between N and C lobes (Right). The cavity is wide enough to accommodate the two amphipathic helices forming the NAF motif. (B) Comparison of the far-UV CD spectra of CIPK23ΔC and CIPK23-NAF; the arrows indicate the characteristic α-helical maximum at 195, and minima at 208 and 222 nm. (C) Comparison of the thermal stability profiles monitored by CD for CIPK23ΔC (Left) and CIPK23-NAF (Right) at increasing CHAPS concentration.
To corroborate this hypothesis, we first checked whether the self-inhibitory NAF extension inhibits the kinase activity. The unphosphorylated CIPK23-NAF protein expressed in E. coli (comprising residues 24–349) displayed slightly lower activity than CIPK23ΔC and CIPK23ΔC T190D. This effect is more noticeable when comparing the highly active CIPK23ΔC S176D T190D Y197E triple mutant with the corresponding CIPK23-NAF S176D T190D Y197E enlarged protein (Fig. 2). Nevertheless, it is in agreement with the biochemical data on CIPK24/SOS2 (6), CIPK20 (44), and CIPK8 (27) in which a moderated reduction of the kinase activity of the full-length active CIPK compared with a mutated version lacking the NAF was observed. Second, we checked whether the NAF motif folds into a α-helix in the absence of CBL by comparing and analyzing the circular dichroism spectra of CIPK23ΔC and CIPK23-NAF. This spectroscopic technique provides a direct relationship between the α-helical content of a protein and the spectral signal intensity at 195, 208, and 222 nm (50). Our data showed that CIPK23-NAF has a higher helical content than CIPK23ΔC (Fig. 4B). Using DichroWeb, a server to analyze circular dichroism data using several algorithms, this increase was estimated to be approximately 5–8% in accordance the expected increase if the NAF folds as a α-helix (51). In addition, we tested whether CHAPS and the NAF compete for the same site at the CIPK23 catalytic domain. Hence, we monitored the thermal denaturation of CIPK23ΔC and CIPK23-NAF at increasing concentrations of CHAPS by circular dichroism spectroscopy. Our results showed that CHAPS was able to bind CIPK23ΔC and CIPK23-NAF because it induced a change in the thermal denaturation temperature (Tm) of these proteins. However, analysis of the variation of Tm with the concentration of the ligand shows that Tm variations occur at lower CHAPS concentration for CIPK23ΔC than for CIPK23-NAF. These data show that CIPK23ΔC displays higher affinity for CHAPS than CIPK23-NAF and indicate that the NAF motif and CHAPS bind to the same site in the catalytic domain of CIPK23 (Fig. 4C).
Thus, the joined structural and biochemical data suggest a model in which the NAF segment folds as a α-helix bound to the interface between the N and C lobes, blocking ATP access to the active site and hindering the conformational change required for kinase activation. This regulatory mechanism strongly resembles the inhibition by the C-terminal pseudosubstrate segment observed in the CaMKI, another calcium calmodulin-dependent protein kinase (49). In this case, an inhibitory helical segment, equivalent to the NAF, competes with ATP for the same site and induces a pronounced distortion of the kinase structure toward an inactive conformation. Like CBLs and CIPKs, the calcium-dependent interaction of calmodulin with the CaMKI kinase releases the pseudosubstrate from the active site and activates the kinase.
The Junction Domain of CIPK23 Is Involved in the Stabilization of the Catalytically Relevant α-C helix.
The characterization of the functional domains of CIPK24/SOS2 revealed that the junction region placed between the catalytic and the self-inhibitory NAF motif is required for kinase activity (6, 30, 33). In addition, it has been shown that the phosphorylation by an uncharacterized kinase of Ser294 at this junction region enhances the interaction with the general regulatory protein 14-3-3 and produces repression of CIPK24/SOS2 basal activity (33). Despite the high sequence variability of this region among CIPKs (Fig. S7A), we investigated whether the structures of CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D could unveil the structural basis for this additional level of regulation.
The crystallographic analysis reveals that the junction domain connects the C lobe and the N lobe through a long unstructured linker followed by a α-helix turn and a β-strand parallel to the central β-sheet (Fig. 1B). Interestingly, the CIPK23ΔC T190D residues Ile308 and Phe309 (Val287 and Phe288 in CIPK24/SOS2ΔC T168D) at the end of the α-helix turn, αT5 (αK in CIPK24/SOS2), are buried in a conserved hydrophobic surface pocket placed at the N lobe between αB and αC (Fig. 5). The interaction of a regulatory segment on a structurally equivalent hydrophobic motif is a common feature of several kinases and involves the stabilization of the helix αC to facilitate its optimal alignment for catalysis (42) or to provide the minimal structural scaffold necessary for basal activity as observed for the Arabidopsis Snf1-related SnRK2.6/OST1 (52, 53). Moreover, in some cases such as the ACG kinases (54), this regulation mechanism is triggered by the phosphorylation of a conserved Ser on the regulatory segment. Accordingly, recombinant unphosphorylated CIPK24/SOS2 displays some basal constitutive activity and the C-terminal truncation of SOS2/CIPK24 just before the Val287 (equivalent to Ile308 residue in CIPK23), but not after, produces an inactive kinase and a salt-sensitive phenotype in planta (6, 30). Thus, it seems that at least for CIPK24/SOS2, the interaction of these two hydrophobic residues with N lobe is sufficient for basal activity and is necessary for activation of the kinase. Moreover, it appears clear that phosphorylation of Ser294 and 14-3-3 binding would counteract this basal level of activation. To investigate the importance of Ile308 and Phe309 in CIPK23, we mutated them to Asp and performed kinase activity assays. Our results show that the I308D F309D mutated versions of the CIPK23ΔC and of the highly active CIPK23ΔC S176D T190D Y197E triple mutant do not display kinase activity (Fig. 2), corroborating the role of these two residues in catalytic stabilization of the kinases. The sequence analysis of the CIPK family of proteins reveals that this hydrophobic motif is conserved in 10 of 26 members of the family (Fig. S7A), suggesting an important role in CIPKs activity, although its precise role remains to be investigated.
Fig. 5.
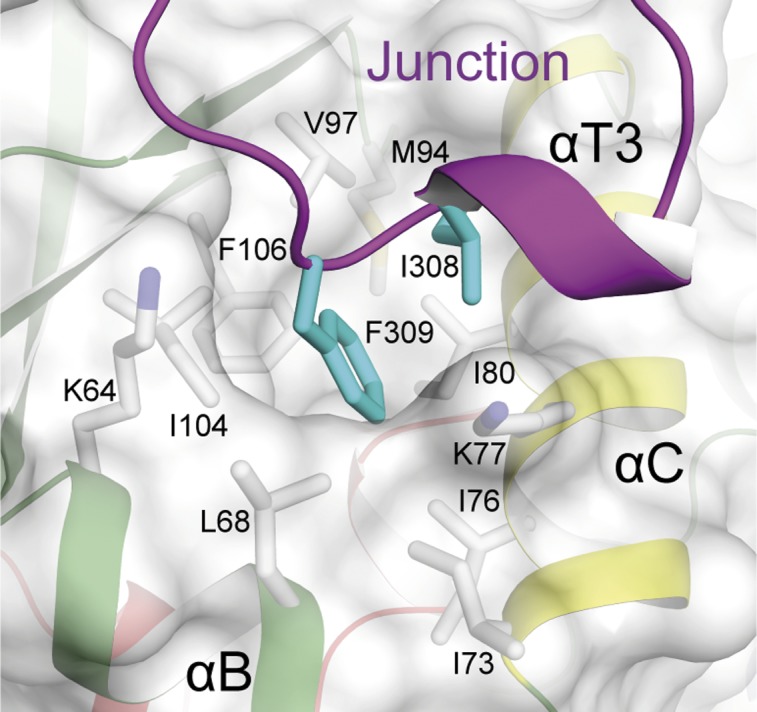
A section of the CIPK23ΔC T190D structure showing the interaction between Ile308 and Phe309 with the hydrophobic pocket at the N lobe of CIPK23. A cartoon representation of the kinase is displayed together with a semitransparent surface.
The presence of a structurally equivalent linker between the N and C lobes like the one observed in the structures of CIPK23ΔC T190D and CIPK24/SOS2ΔC T168D is another recurrent theme in kinase structures. Remarkably, Zap-70 kinase autoinhibition relies on the blockage of the transition from an “open” inactive state to a “closed” active state by an equivalent linker connecting N and C lobes (55). In CIPKs, the N to C lobe linker seems to be flexible because it forms few intramolecular contacts with the catalytic domain and has high crystallographic temperature factors (Fig. S7B). Thus, it does not seem to be hindering the kinase activation process.
The Regulatory Mode of CIPKs.
The structural data presented here show that the activation loop and the self-inhibitory pocket are spatially connected, providing a working model in which the activation loop at the active site and the NAF segment at the self-inhibitory pocket are mutually stabilized. In this scenario, the CIPK activation necessarily implies the simultaneous release of these two elements from the catalytic domain. The release can be effectively achieved by the calcium-dependent CBL interaction, or through phosphorylation of the activation loop by an upstream kinase, or both, thereby explaining the additive effects on activity achieved by mimicking phosphorylation by putative upstream kinases and deletion of the NAF domain (Fig. 6). Moreover, the reported basal kinase activity for some CIPKs indicates that these processes need not always be concurrent for activation. In accordance with the biochemical data, the activity of a particular CIPK will depend on its ability to stabilize these active molecular species and/or on the balance between them and the inactive form of the kinase. Consequently, the maximum activity will be observed for CBL-bound and phosphorylated CIPKs, because the active form will be fully stabilized. The required phosphorylation of the C-terminal region of CBLs by CIPKs to achieve fully functional activation of the CBL–CIPKs complexes may be central to understand why the moderate NAF motif-mediated stimulation of the in vitro CIPK23 (Fig. 2) is absolutely required for in vivo activation of AKT1 K+ channel (13). This data additionally supports the idea that CBL phosphorylation may affect the structure of the CBL-CIPKs complexes and consequently, their activity. Besides, an additional layer of CIPK regulation comes from the cofactor preference of Mn2+ compared with Mg2+ (13, 27, 28). The fact that one of the metal ion binding sites of CBL4/SOS3 displays higher affinity for Mn2+ than for Ca2+ suggests that CBLs could act as a carrier for this cofactor or, alternatively, could buffer the availability of free Mn2+ to prevent a constitutive activation of the CIPKs (56).
Fig. 6.

A working model for CIPKs regulation. CIPK is schematically represented with the N lobe and the C lobe depicted in green and blue, respectively, the junction as a purple line, the activation loop in red, and the NAF motif as a cyan rectangle. Activation of CIPKs implies necessarily the release of both the activation loop and the self-inhibitory NAF motif. The calcium-dependent CBL interaction and/or phosphorylation of the activation loop set the equilibrium between the inactive and the active forms. The activity of CIPKs depends on the balance between active and inactive forms. Some CIPKs (for instance CIPK24/SOS2) display basal activity. The junction region may be responsible for the stabilization of a CBL unbound and unphosphorylated CIPK form with basal activity. Once CIPK is active, the fully functional activation is achieved by the phosphorylation of the CBL.
The available structural and biochemical data support a model wherein CIPK23 is intrinsically inactive and requires the calcium dependent CBL1/9 binding and/or the phosphorylation by upstream kinases for activation, while CIPK24/SOS2 does not. This differential behavior may be related to the physiology roles of these proteins. To our knowledge, CIPK23 targets solely the K+ channel AKT1 and the nitrate transporter CHL1 (11, 14). By contrast, in addition to SOS1, CIPK24/SOS2 is involved in the regulation of a number of antiporters and H+-ATPases (57–59) and it has been reported to participate in signaling pathways regulating flowering time and redox metabolism (60, 61). Consequently, compared with CIPK23, CIPK24/SOS2 will require a wider range of activation states and a more specialized regulation. In contrast to CIPK23, which is intrinsically inactive and shows an absolute requirement for external stimulation, CIPK24/SOS2 displays basal activity that is finely regulated by phosphorylation of Ser294 and 14-3-3 binding, thus adding a further layer of biochemical regulation (33).
In conclusion, the analysis of the CIPK structure reported here provides the basis for comprehending the regulatory mechanism of a major signaling network in plant response to environmental cues. Understanding this mechanism is central to enhance crop species production by augmenting the limited capacities of plants to cope with environmental stresses.
Materials and Methods
Gene Cloning and Site-Directed Mutagenesis.
The sequences of the primers used in the cloning and the site-directed mutagenesis of all constructs are listed in Table S2. The kinase domain of CIPK23 (CIPK23ΔC; residues 24–331) was cloned between NdeI and NotI restriction sites of the pGEX4T2 expression vector (GE Healthcare) that had been previously modified to contain an NdeI restriction site between EcoRI and SmaI (forward primer 23N and reverse primer 23C). CIPK23ΔC T190D mutant was generated by PCR using pGEX4T2-CIPK23 as a template (forward primer 23T190D5 and reverse primer 23T190D3) and CIPK23ΔC V182K T190D mutant using CIPK23ΔC T190D as a template (forward primer 23V182K5 and reverse primer 23V182K3). CIPK23 S176D T190D Y197E was generated successively by PCR using CIPK23ΔC T190D as a template, and the resulted constructs were used as a template for the following ones (forward primers 23S176D5 and 23Y197E5 and reverse primers 23S176D3 and 23Y197E3). CIPK23ΔC T190D I308D F309D and CIPK23ΔC S176D T190D Y197E I308D F309D were prepared by using pGEX4T2-CIPK23ΔC T190D and pGEX4T2-CIPK23ΔC S176D T190D Y197E as a template, respectively, (forward primer 23I308DF309D5 and reverse primer 23I308DF309D3) and CIPK23-SOS2-like mutant was generated by using pGEX4T2-CIPK23ΔC as a template (forward primer 23SOS2like5 and reverse primer 23SOS2like3).
The kinase domain of CIPK23 (residues 1–331) was amplified by PCR using the primers 23N2 and 23C and cloned as SmaI/NotI fragment in the yeast expression vector pEG(KT) (62). To subclone the mutant allele CIP23-SOS2-like in pEG(KT), a 0.69-kb BglII/NotI fragment encoding amino acids Lys103 to Val331 of CIPK23 was obtained from the corresponding pGEX4-T2 construct and ligated in the pEG(KT)-CIPK23 digested with the same restriction enzymes.
The kinase domain of CIPK24/SOS2 (CIPK24/SOS2ΔC residues 7–308) was cloned between BamHI and EcoRI restriction sites of the pGEX4T2 expression vector (GE Healthcare) (forward primer 24N and the reverse primer 24C). CIPK24/SOS2 mutants were generated successively by PCR using this construct as the first template, and the resulted constructs were used as a template for the following ones. CIPK24/SOS2ΔC T168D (forward primer 24T168D5 and reverse primer 24T168D3), S228D (forward primer 24S228D5 and reverse primer 24S228D3), E107K and S109D (forward primer 24E107KS109D5 and reverse primer 24E107KS109D3), C127S (forward primer 24C127S5 and reverse primer 24C127S3), P81K (forward primer 24P81K5 and reverse primer 24P81K3), and L266K (forward primer 24L266K5 and reverse primer 24L266K3). All sequences were confirmed by plasmid DNA sequencing.
CIPK23-NAF (CIPK23; residues 24–352) was cloned between BamHI and HindIII restriction sites of the multiple cloning sites I of pETDuet-1 expression vector (Novagen) (forward primer 23FN and reverse primer 23FC), previously modified to contain CBL1 (residues 1–213) between NdeI and KpnI restriction sites of multiple cloning site II (forward primer CBL1N and reverse primer CBL1C). CIPK23-NAF S176D T190D Y197E was generated successively by PCR using CIPK23-NAF as a template, and the resulted constructs were used as a template for the following ones (forward primers 23S176D5, 23T190D5, and 23Y197E5 and reverse primers 23S176D3, 23T190D3, and 23Y197E3).
Protein Preparation, Crystallization, and Data Collection.
Recombinant proteins were expressed in Rosetta 2 (DE3) pLysS cells (Stratagene) by induction at OD600 = 0.7 with 0.3 mM isopropyl-β-d-thiogalactoside for 16 h at 16 °C. Following isolation by centrifugation, the CIPK23ΔC and CIPK23ΔC T190D bacterial pellets were then resuspended in a 150 mM NaCl, 20 mM Hepes pH 7.0, and 0.5 mM Tris(2-carboxyethyl)phosphine (TCEP) buffer, whereas the CIPK24/SOS2ΔC T168D bacterial pellet was resuspended in a 20 mM NaCl, 20 mM Tris⋅HCl pH 8.5, and 5 mM DTT buffer. Cells were lysed by sonication and purified by using glutathione Sepharose 4B (GE Healthcare) following overnight thrombin cleavage for CIPK23 and an overnight preScission cleavage for CIPK24/SOS2ΔC T168D. The protein was dialyzed overnight in 20 mM NaCl, 20 mM Tris⋅HCl pH 9.0, and 5 mM DTT. Proteins were further purified by gel filtration chromatography on a Hi Load 16/60 Superdex 200 prep grade column (GE Healthcare). Chromatography yields single peaks, which correspond to the monomeric proteins. CIPK23ΔC, CIPK23ΔC T190D, and CIPK24/SOS2ΔC T168D constructs were concentrated to 14.0 mg⋅ml−1, 14.0 mg⋅ml−1, and 5.0 mg⋅ml−1, respectively, using an Amicon Ultra 10 K centrifugal filter device (Millipore). The sample purity was determined by SDS/PAGE.
Initial crystallization screens were set up as sitting-drop vapor-diffusion experiments. We used an Innovadine crystallization robot and crystallization kits from Qiagen, Hampton Research, and Jena Bioscience. Initial screenings of CIPK23ΔC lead to spherulites in many conditions. We selected condition number 4 (0.1 M Tris⋅HCl pH 8.0 and 2.0 M ammonium sulfate) from Crystal Screen HT (Hampton Research) as a starting point for crystal optimization. CIPK23ΔC spherulites were screened against a set of detergents (Detergent Screen HT; Hampton Research). Needle-shaped crystals grew after 3 d using the under oil microbatch method. The final crystallization condition was obtained by mixing 1 μL of 3.5 M ammonium sulfate, 0.1 M Hepes pH 7.5, 16 mM CHAPS, and 1 μL of protein preparation. These crystals were cryoprotected by adding a saturated solution of ammonium sulfate to the crystallization drop until saturation. Identical crystallization protocol was followed for CIPK23ΔC T190D. Initial screenings of CIPK24/SOS2ΔC T168D lead to thin plates in condition number 47 (30% wt/vol PEG 4000, 0.1 M Tris⋅HCl pH 8.5, and 0.2 M magnesium chloride) from JBScreen Classic: 1, 2, 3, and 4 (Jena Bioscience). CIPK24/SOS2ΔC T168D crystallyzation was further optimized by screening against a matrix of pH (7.0, 8.2, 8.5, 8.8, and 9.0) versus PEG 4K concentration (12–30%) using the under oil microbatch method. Thicker plates appeared between pH 5.8 and 9.0 and PEG 4K concentration between 20 and 24%. In-well dehydrated crystals were used for crystal structure determination.
Crystals from CIPK23ΔC, CIPK23ΔC T190D, and CIPK24/SOS2ΔC T168D were mounted in fiber loops and then flash cooled in liquid nitrogen. X-ray diffraction data from CIPK23ΔC and T190D mutant were collected at the European Syncrotron Radiation Facility beamlines ID14-4 and ID23-2, respectively. CIPK24/SOS2ΔC T168D diffraction data were collected at the Petra-III synchrotron, Hamburg, beamline P13.
CIPK23ΔC, CIPK23ΔC T190D, and CIPK24/SOS2ΔC T168D diffraction datasets were processed by using MOSFLM (63) and XDS (64) respectively, and scaled using SCALA (65) from the CCP4 package (Collaborative Computational Project, Number 4, 1994). A summary of the data collection statistics is given in Table S1.
Structure Determination.
The X-ray structures of CIPK23ΔC and CIPK23ΔC T190D were solved by molecular replacement with the program MolRep (66) using the coordinates of a kinase domain (PDB ID code: 3H4J) (37). The electron density map calculated by using these phases was good enough to manually build and refine the structure of CIPK23ΔC T190D. Several cycles of restrained refinement with PHENIX (67) and REFMAC5 (68) and iterative model building with COOT (69) were carried out. The X-ray structure of CIPK24/SOS2ΔC T168D was solved by molecular replacement with the program MolRep (66) using the coordinates of a kinase domain of CIPK23. Several cycles of Jelly-body and restrained refinement with REFMAC5 (68) and iterative model building with COOT (69) were carried out. Calculations were performed by using CCP4 programs (70). The stereochemistry of the models was verified with MolProbity (71). Ribbon figures were produced by using PyMOL (72). The refinement statistics are summarized in Table S1.
In Vitro Kinase Assays.
Kinetic studies of kinases were conducted by coupling reaction method as described (73, 74). In brief, 3 μM kinase was added to a reaction mixture containing 1 mM MgCl2, 100 mM phosphoenolpyruvate (Sigma), 0.28 mM NADH (Calbiochem), 10–20 units of pyruvate kinase and 20–30 units of lactate dehydrogenase (Sigma), 1 mg/mL histone (Sigma), and 0.5 mM ATP (Sigma). The reaction was performed in a 96-well flat-bottom plate (Sarstedt). The NADH depletion, which is coupled to the rate of the kinase reaction, was followed by measuring the decrease in absorbance at 340 nm for 40 min at 22 °C in a Synergy HT BioTek microplate spectrophotometer. Blank reactions without enzyme were carried out for each enzyme. Four replicas were collected for each reaction mixture, and the average activities were quantified.
[γ-32P] ATP Kinase Assays.
For radioactive in vitro phosphorylation assays, wild-type and mutant CIPK23ΔC and GST-CIPK23-SOS2–like proteins were purified from yeast cells as described (62). Cells were collected by centrifugation and lysed with glass beads in PBS buffer (10 mM Na2HPO4, 2 mM KH2PO4, 2.7 mM KCl, and 137 mM NaCl, pH 7.4). Recombinant proteins were affinity-purified on glutathione-Sepharose (GE Healthcare). Myelin basic protein (Sigma) (∼1 µg) was subjected to phosphorylation by recombinant CIPK23 protein kinases (∼100 ng) in 30 μL of buffer (66.7 mM Tris·HCl, pH 8, 100 mM NaCl, 5 mM MnSO4, 0.5 mM CaCl2, and 2 mM DTT). Reactions were started by adding ATP (10 μM with 20 μCi of [γ-32P]ATP), which was incubated at 30 °C for 45 min, and stopped with 10 μL of 4× SDS/PAGE sample buffer. Proteins were separated by SDS/PAGE by using a 12% (wt/vol) acrylamide gel. Radioactivity was detected on the dry gels by using Biomax Light-1 (Kodak) films.
Circular Dichroism Spectra.
Protein samples were prepared in a buffer containing 20 mM sodium phosphate at pH 8.2 and 0.5 mM TCEP to optimize signal-to-noise ratio. Special attention was paid to the instrument calibration (50). Circular dichroism spectra were recorded in a JASCO J-810 spectropolarimeter. Far-UV spectra were recorded in a 0.02 cm-pathlength quartz cell at a protein concentration of 0.7 mg⋅ml−1 (20 μM). The spectra were analyzed by using CONTIN (75) included in the DICROWEB web page (76). Thermal denaturation experiments were carried out in a 0.1 cm-pathlength quartz cell at a protein concentration of 0.2 mg⋅ml−1 (5.6 μM) and monitored at wavelength of 220 nm by increasing the temperature from 30 °C to 70 °C at 50 °C⋅h−1.
Supplementary Material
Acknowledgments
A.A. thanks Dr. Douglas Vinson Laurents for critical reading of the manuscript, and the European Syncrotron Radiation Facility (beamlines ID14-4 and ID23-2) and PETRAIII (beamline P13, BIOSTRUCTX_3100.5) for the access to the synchrotron radiation source. This work was funded by Ministerio de Economía y Competitividad Grants BFU2011-25384 and CSD2006-00015 (to A.A.), BIO2011-28184-C02-02 (to M.J.S.-B.), and BIO2012-36533 (to F.J.Q.), which was cofinanced by the European Regional Development Fund, and Comunidad de Madrid Grant S2010/BMD-2457 (to A.A.). A.C.-S. is supported by a Formación de Personal Investigador Predoctoral Fellowship, and M.J.S.-B. is supported by Ramón y Cajal Contract RYC-2008-03449 from MINECO.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank (PDB), www.pdb.org (PDB ID codes 4CZT, 4CZU, and 4D28).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1407610111/-/DCSupplemental.
References
- 1.Albrecht V, Ritz O, Linder S, Harter K, Kudla J. The NAF domain defines a novel protein-protein interaction module conserved in Ca2+-regulated kinases. EMBO J. 2001;20(5):1051–1063. doi: 10.1093/emboj/20.5.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi J, et al. Novel protein kinases associated with calcineurin B-like calcium sensors in Arabidopsis. Plant Cell. 1999;11(12):2393–2405. doi: 10.1105/tpc.11.12.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batistič O, Kudla J. Analysis of calcium signaling pathways in plants. Biochim Biophys Acta. 2012;1820(8):1283–1293. doi: 10.1016/j.bbagen.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Steinhorst L, Kudla J. Calcium and reactive oxygen species rule the waves of signaling. Plant Physiol. 2013;163(2):471–485. doi: 10.1104/pp.113.222950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luan S, Kudla J, Rodriguez-Concepcion M, Yalovsky S, Gruissem W. Calmodulins and calcineurin B-like proteins: Calcium sensors for specific signal response coupling in plants. Plant Cell. 2002;14(Suppl):S389–S400. doi: 10.1105/tpc.001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo Y, Halfter U, Ishitani M, Zhu JK. Molecular characterization of functional domains in the protein kinase SOS2 that is required for plant salt tolerance. Plant Cell. 2001;13(6):1383–1400. doi: 10.1105/tpc.13.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu QS, Guo Y, Dietrich MA, Schumaker KS, Zhu JK. Regulation of SOS1, a plasma membrane Na+/H+ exchanger in Arabidopsis thaliana, by SOS2 and SOS3. Proc Natl Acad Sci USA. 2002;99(12):8436–8441. doi: 10.1073/pnas.122224699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintero FJ, et al. Activation of the plasma membrane Na/H antiporter Salt-Overly-Sensitive 1 (SOS1) by phosphorylation of an auto-inhibitory C-terminal domain. Proc Natl Acad Sci USA. 2011;108(6):2611–2616. doi: 10.1073/pnas.1018921108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Núñez-Ramírez R, et al. Structural insights on the plant salt-overly-sensitive 1 (SOS1) Na(+)/H(+) antiporter. J Mol Biol. 2012;424(5):283–294. doi: 10.1016/j.jmb.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Fuglsang AT, et al. Arabidopsis protein kinase PKS5 inhibits the plasma membrane H+ -ATPase by preventing interaction with 14-3-3 protein. Plant Cell. 2007;19(5):1617–1634. doi: 10.1105/tpc.105.035626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SC, et al. A protein phosphorylation/dephosphorylation network regulates a plant potassium channel. Proc Natl Acad Sci USA. 2007;104(40):15959–15964. doi: 10.1073/pnas.0707912104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, et al. A protein kinase, interacting with two calcineurin B-like proteins, regulates K+ transporter AKT1 in Arabidopsis. Cell. 2006;125(7):1347–1360. doi: 10.1016/j.cell.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto K, et al. Phosphorylation of calcineurin B-like (CBL) calcium sensor proteins by their CBL-interacting protein kinases (CIPKs) is required for full activity of CBL-CIPK complexes toward their target proteins. J Biol Chem. 2012;287(11):7956–7968. doi: 10.1074/jbc.M111.279331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho CH, Lin SH, Hu HC, Tsay YF. CHL1 functions as a nitrate sensor in plants. Cell. 2009;138(6):1184–1194. doi: 10.1016/j.cell.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Kim BG, et al. The calcium sensor CBL10 mediates salt tolerance by regulating ion homeostasis in Arabidopsis. Plant J. 2007;52(3):473–484. doi: 10.1111/j.1365-313X.2007.03249.x. [DOI] [PubMed] [Google Scholar]
- 16.Quan R, et al. SCABP8/CBL10, a putative calcium sensor, interacts with the protein kinase SOS2 to protect Arabidopsis shoots from salt stress. Plant Cell. 2007;19(4):1415–1431. doi: 10.1105/tpc.106.042291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishitani M, et al. SOS3 function in plant salt tolerance requires N-myristoylation and calcium binding. Plant Cell. 2000;12(9):1667–1678. doi: 10.1105/tpc.12.9.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohta M, Guo Y, Halfter U, Zhu JK. A novel domain in the protein kinase SOS2 mediates interaction with the protein phosphatase 2C ABI2. Proc Natl Acad Sci USA. 2003;100(20):11771–11776. doi: 10.1073/pnas.2034853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sánchez-Barrena MJ, et al. The structure of the C-terminal domain of the protein kinase AtSOS2 bound to the calcium sensor AtSOS3. Mol Cell. 2007;26(3):427–435. doi: 10.1016/j.molcel.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sánchez-Barrena MJ, Martínez-Ripoll M, Albert A. Structural biology of a major signaling network that regulates plant abiotic stress: The CBL-CIPK mediated pathway. Int J Mol Sci. 2013;14(3):5734–5749. doi: 10.3390/ijms14035734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batistič O, et al. S-acylation-dependent association of the calcium sensor CBL2 with the vacuolar membrane is essential for proper abscisic acid responses. Cell Res. 2012;22(7):1155–1168. doi: 10.1038/cr.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintero FJ, Ohta M, Shi H, Zhu JK, Pardo JM. Reconstitution in yeast of the Arabidopsis SOS signaling pathway for Na+ homeostasis. Proc Natl Acad Sci USA. 2002;99(13):9061–9066. doi: 10.1073/pnas.132092099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckert C, et al. The vacuolar calcium sensors CBL2 and CBL3 affect seed size and embryonic development in Arabidopsis thaliana. Plant J. 2014;78(1):146–156. doi: 10.1111/tpj.12456. [DOI] [PubMed] [Google Scholar]
- 24.Held K, et al. Calcium-dependent modulation and plasma membrane targeting of the AKT2 potassium channel by the CBL4/CIPK6 calcium sensor/protein kinase complex. Cell Res. 2011;21(7):1116–1130. doi: 10.1038/cr.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin H, et al. Phosphorylation of SOS3-LIKE CALCIUM BINDING PROTEIN8 by SOS2 protein kinase stabilizes their protein complex and regulates salt tolerance in Arabidopsis. Plant Cell. 2009;21(5):1607–1619. doi: 10.1105/tpc.109.066217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15(5):661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 27.Gong D, Gong Z, Guo Y, Chen X, Zhu JK. Biochemical and functional characterization of PKS11, a novel Arabidopsis protein kinase. J Biol Chem. 2002;277(31):28340–28350. doi: 10.1074/jbc.M107719200. [DOI] [PubMed] [Google Scholar]
- 28.Gong D, Guo Y, Jagendorf AT, Zhu JK. Biochemical characterization of the Arabidopsis protein kinase SOS2 that functions in salt tolerance. Plant Physiol. 2002;130(1):256–264. doi: 10.1104/pp.004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao P, Kolenovsky A, Cui Y, Cutler AJ, Tsang EW. Expression, purification and analysis of an Arabidopsis recombinant CBL-interacting protein kinase3 (CIPK3) and its constitutively active form. Protein Expr Purif. 2012;86(1):45–52. doi: 10.1016/j.pep.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Gong D, Guo Y, Schumaker KS, Zhu JK. The SOS3 family of calcium sensors and SOS2 family of protein kinases in Arabidopsis. Plant Physiol. 2004;134(3):919–926. doi: 10.1104/pp.103.037440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujii H, Zhu JK. An autophosphorylation site of the protein kinase SOS2 is important for salt tolerance in Arabidopsis. Mol Plant. 2009;2(1):183–190. doi: 10.1093/mp/ssn087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oecking C, Jaspert N. Plant 14-3-3 proteins catch up with their mammalian orthologs. Curr Opin Plant Biol. 2009;12(6):760–765. doi: 10.1016/j.pbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 33.Zhou H, et al. Inhibition of the Arabidopsis salt overly sensitive pathway by 14-3-3 proteins. Plant Cell. 2014;26(3):1166–1182. doi: 10.1105/tpc.113.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, et al. Quantitative phosphoproteomics after auxin-stimulated lateral root induction identifies an SNX1 protein phosphorylation site required for growth. Mol Cell Proteomics. 2013;12(5):1158–1169. doi: 10.1074/mcp.M112.021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steipe B. Consensus-based engineering of protein stability: From intrabodies to thermostable enzymes. Methods Enzymol. 2004;388:176–186. doi: 10.1016/S0076-6879(04)88016-9. [DOI] [PubMed] [Google Scholar]
- 36.Mesa-Torres N, et al. The consensus-based approach for gene/enzyme replacement therapies and crystallization strategies: The case of human alanine-glyoxylate aminotransferase. Biochem J. 2014;462(3):453–463. doi: 10.1042/BJ20140250. [DOI] [PubMed] [Google Scholar]
- 37.Chen L, et al. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature. 2009;459(7250):1146–1149. doi: 10.1038/nature08075. [DOI] [PubMed] [Google Scholar]
- 38.Nayak V, et al. Structure and dimerization of the kinase domain from yeast Snf1, a member of the Snf1/AMPK protein family. Structure. 2006;14(3):477–485. doi: 10.1016/j.str.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Taylor SS, Kornev AP. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36(2):65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meharena HS, et al. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 2013;11(10):e1001680. doi: 10.1371/journal.pbio.1001680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams JA. Activation loop phosphorylation and catalysis in protein kinases: Is there functional evidence for the autoinhibitor model? Biochemistry. 2003;42(3):601–607. doi: 10.1021/bi020617o. [DOI] [PubMed] [Google Scholar]
- 42.Jura N, et al. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42(1):9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng J, et al. 2.2 A refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr D Biol Crystallogr. 1993;49(Pt 3):362–365. doi: 10.1107/S0907444993000423. [DOI] [PubMed] [Google Scholar]
- 44.Gong D, Zhang C, Chen X, Gong Z, Zhu JK. Constitutive activation and transgenic evaluation of the function of an arabidopsis PKS protein kinase. J Biol Chem. 2002;277(44):42088–42096. doi: 10.1074/jbc.M205504200. [DOI] [PubMed] [Google Scholar]
- 45.Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: Structural basis for regulation. Cell. 1996;85(2):149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 46.Hendlich M, Bergner A, Günther J, Klebe G. Relibase: Design and development of a database for comprehensive analysis of protein-ligand interactions. J Mol Biol. 2003;326(2):607–620. doi: 10.1016/s0022-2836(02)01408-0. [DOI] [PubMed] [Google Scholar]
- 47.Akaboshi M, et al. The crystal structure of plant-specific calcium-binding protein AtCBL2 in complex with the regulatory domain of AtCIPK14. J Mol Biol. 2008;377(1):246–257. doi: 10.1016/j.jmb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Chovancova E, et al. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLOS Comput Biol. 2012;8(10):e1002708. doi: 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldberg J, Nairn AC, Kuriyan J. Structural basis for the autoinhibition of calcium/calmodulin-dependent protein kinase I. Cell. 1996;84(6):875–887. doi: 10.1016/s0092-8674(00)81066-1. [DOI] [PubMed] [Google Scholar]
- 50.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751(2):119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers. 2008;89(5):392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 52.Yunta C, Martínez-Ripoll M, Zhu JK, Albert A. The structure of Arabidopsis thaliana OST1 provides insights into the kinase regulation mechanism in response to osmotic stress. J Mol Biol. 2011;414(1):135–144. doi: 10.1016/j.jmb.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ng LM, et al. Structural basis for basal activity and autoactivation of abscisic acid (ABA) signaling SnRK2 kinases. Proc Natl Acad Sci USA. 2011;108(52):21259–21264. doi: 10.1073/pnas.1118651109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gold MG, Barford D, Komander D. Lining the pockets of kinases and phosphatases. Curr Opin Struct Biol. 2006;16(6):693–701. doi: 10.1016/j.sbi.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 55.Deindl S, et al. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell. 2007;129(4):735–746. doi: 10.1016/j.cell.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 56.Sánchez-Barrena MJ, Martínez-Ripoll M, Zhu JK, Albert A. The structure of the Arabidopsis thaliana SOS3: Molecular mechanism of sensing calcium for salt stress response. J Mol Biol. 2005;345(5):1253–1264. doi: 10.1016/j.jmb.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 57.Batelli G, et al. SOS2 promotes salt tolerance in part by interacting with the vacuolar H+-ATPase and upregulating its transport activity. Mol Cell Biol. 2007;27(22):7781–7790. doi: 10.1128/MCB.00430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng NH, Pittman JK, Zhu JK, Hirschi KD. The protein kinase SOS2 activates the Arabidopsis H(+)/Ca(2+) antiporter CAX1 to integrate calcium transport and salt tolerance. J Biol Chem. 2004;279(4):2922–2926. doi: 10.1074/jbc.M309084200. [DOI] [PubMed] [Google Scholar]
- 59.Qiu QS, et al. Regulation of vacuolar Na+/H+ exchange in Arabidopsis thaliana by the salt-overly-sensitive (SOS) pathway. J Biol Chem. 2004;279(1):207–215. doi: 10.1074/jbc.M307982200. [DOI] [PubMed] [Google Scholar]
- 60.Kim WY, et al. Release of SOS2 kinase from sequestration with GIGANTEA determines salt tolerance in Arabidopsis. Nat Commun. 2013;4:1352. doi: 10.1038/ncomms2357. [DOI] [PubMed] [Google Scholar]
- 61.Verslues PE, et al. Interaction of SOS2 with nucleoside diphosphate kinase 2 and catalases reveals a point of connection between salt stress and H2O2 signaling in Arabidopsis thaliana. Mol Cell Biol. 2007;27(22):7771–7780. doi: 10.1128/MCB.00429-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mitchell DA, Marshall TK, Deschenes RJ. Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast. 1993;9(7):715–722. doi: 10.1002/yea.320090705. [DOI] [PubMed] [Google Scholar]
- 63.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 66.Vagin AA, Isupov MN. Spherically averaged phased translation function and its application to the search for molecules and fragments in electron-density maps. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 10):1451–1456. doi: 10.1107/s0907444901012409. [DOI] [PubMed] [Google Scholar]
- 67.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 70.Anonymous; Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 71.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schrödinger LLC (2013) The PyMOL molecular graphics system (Schrödinger, LLC, New York), Version 1.3.
- 73.Lietha D, et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129(6):1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.LaFevre-Bernt M, et al. Intramolecular regulatory interactions in the Src family kinase Hck probed by mutagenesis of a conserved tryptophan residue. J Biol Chem. 1998;273(48):32129–32134. doi: 10.1074/jbc.273.48.32129. [DOI] [PubMed] [Google Scholar]
- 75.Lees JG, Miles AJ, Wien F, Wallace BA. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics. 2006;22(16):1955–1962. doi: 10.1093/bioinformatics/btl327. [DOI] [PubMed] [Google Scholar]
- 76.Lobley A, Whitmore L, Wallace BA. DICHROWEB: An interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics. 2002;18(1):211–212. doi: 10.1093/bioinformatics/18.1.211. [DOI] [PubMed] [Google Scholar]
- 77.Gouet P, Courcelle E, Stuart DI, Métoz F. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15(4):305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.