

Abstract
Endothelium dysfunction has been understood primarily in terms of abnormal vasomotor function, which plays an important role in the pathogenesis of diabetes and chronic diabetic complications. However, it has not been fully studied that the endothelium may regulate metabolism itself. The response gene to complement 32 (RGC-32) has be considered as an angiogenic inhibitor in the context of endothelial cells. We found that RGC-32 was induced by high fat diet in vivo and by glucose or insulin in endothelial cells, and then we set out to investigate the role of endothelial RGC-32 in metabolism. DNA array analysis and qPCR results showed that glutamine-fructose-6-phosphate aminotransferase [isomerizing] 1 (GFPT1), solute carrier family 2 (facilitated glucose transporter), member 12 (SLC2A12, GLUT12) and glucagon-like peptide 2 receptor (GLP2R) may be among possible glucose metabolism related downstream genes of RGC-32. Additionally, in the mice with endothelial specific over-expressed RGC-32, the disposal of carbohydrate was improved without changing insulin sensitivity when mice were faced with high fat diet challenges. Taken together, our findings suggest that RGC-32 in the endothelial cells regulates glucose metabolism related genes and subsequent helps to maintain the homeostasis of blood glucose.
Keywords: The response gene to complement 32, endothelial cells, glucose homeostasis
Introduction
The endothelium is now recognized as a complex organ with a multitude of properties involved in both physiologic and pathologic processes. Endothelial cells (ECs) facilitate metabolic exchange between the circulation and tissues [1]. Endothelial dysfunction is a common feature in diabetes, insulin resistance and obesity [2,3]. Previously, endothelium dysfunction has been understood primarily in terms of abnormal vasomotor function, which plays an important role in the pathogenesis of diabetes and chronic diabetic complications, such as microangiopathy and macroangiopathy [4]. However, specific mechanisms through which the endothelium itself may directly modulate glucose metabolism or insulin sensitivity have not been fully studied.
The response gene to complement 32 (RGC-32) protein is localized in the cytoplasm and has multiple functions in different cells. RGC-32 plays a dual role in cancers through activating Akt and the CDC2 kinase [5], mediating TGF-β-induced epithelial-mesenchymal transition [6] or regulating chromatin assembly [7]. RGC-32 plays as a promoter of TGF-beta-mediated profibrotic effects in multiple sclerosis [8] and renal fibrosis [9]. RGC-32 is essential for both the proliferation and migration of vascular smooth muscle cells in vascular injury [10]. In the context of endothelial cells, RGC-32 is a novel hypoxia-regulated angiogenic inhibitor [11] and causes foetal growth restriction through interrupting the placental angiogenesis [12]. However, the role of RGC-32 in glucose metabolism has not been investigated yet.
In this report, we examined RGC-32 involvement in the endothelium-related effect of glucose regulation by using cultured endothelial cells and high fat diet treated transgenic mice. We demonstrate that RGC-32 is induced by high fat diet in vivo and by glucose or insulin in vitro. Accordingly, glutamine-fructose-6-phosphate aminotransferase [isomerizing] 1 (GFPT1), solute carrier family 2 (facilitated glucose transporter), member 12 (SLC2A12, GLUT12) and glucagon-like peptide 2 receptor (GLP2R) may be among possible glucose metabolism related downstream genes of RGC-32. Additionally, in the mice with over-expressed endothelial RGC-32, the disposal of carbohydrate was improved without changing insulin sensitivity when mice were faced with high fat diet (HFD) challenges. Our findings suggest that RGC-32 in the ECs regulates glucose metabolism related genes and subsequent helps to maintain the homeostasis of blood glucose.
Material and methods
The investigation related with animals conforms to the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, 1996) and was approved by the Institutional Animal Care and Use Committee at Beth Israel Deaconess Medical Center.
Cell culture
Human microvascular endothelial cells (HMEC-1, Centers for Disease Control and Prevention, Atlanta, GA, USA) were cultured in MCDB-131( Lonza, Walkersville, MD, USA), and human embryonic kidney cells 293 (HEK 293, American Type Culture Collection, Manassas, VA, USA) were cultured in DMEM (Invitrogen, Carlsbad, CA, USA), both supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 100 μg/ml streptomycin and 100 units/ml penicillin (Life technology, Lonza, Walkersville, MD, USA), at 37°C in 95% air and 5% CO2 atmosphere.
Retroviral construction
The RGC-32 (NM_014059.1) coding sequence was cloned into a pBMN-green fluorescence protein (GFP) vector (Obigen, San Diego, CA, USA) for retrovirus packaging. pBMN-GFP or pBMN-GFP-RGC-32 were transfected to 293T using Polyethylenimine (PEI, Polysciences, Inc. Warrington, PA, USA) with pVSV-G, pJK3, and pCMVtat (Obigen, San Diego, CA, USA). The medium with retrovirus/RGC-32 was collected and filtered before being used to infect HMEC-1 cells.
siRNA transfection
siRNAs targeting human RGC-32 were synthesized by GenePharma Co, Ltd. (ZhangJiang, Shanghai, China). Knockdown efficiency of the duplexes of siRNA of RGC-32 (siRNA: 5’-GAUUCACUUUAUAGGAACATT-3’, 5’-UGUUCCUAUAAAGUGAAUCTG-3’) or a nontarget control (5’-UUC UCC GAA CGU GUC ACG UTT-3’, 5’-ACG UGA CAC GUU CGG AGA ATT-3’) was determined by transfection into HMEC-1 cells at a final concentration of 50 nM according to the manufacturer’s protocol. Briefly, a master mix of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was diluted with 1 ml of OPTI-MEM (Invitrogen, Carlsbad, CA, USA) and incubated for 5 min. Lipofectamine 2000 dilution was added to the siRNA dilution, incubated for 20 min, and added dropwise to the cells. Five hours after transfection, media was changed. 72 hours later, cells were collected for the check of knockdown efficiency.
Hyperglycemia and hyperinsulinmia
For hyperglycemic conditions, endothelial cells were cultured with 25 mM additional D-glucose (Sigma, St. Louis, MO, USA) adding to the 5 mM in baseline of MCDB. For hyperinsulinmia conditions, endothelial cells were cultured with 100 nM insulin (Sigma, St. Louis, MO, USA) in MCDB.
DNA array analysis
siRNAs targeting human RGC-32 or nontarget control was transfected to HMEC-1 cells. 72 hours later, total RNA was isolated from the HEMC cells using TRIzol (Invitrogen), and 15 μg of each RNA sample was used for microarray analysis on the Human Whole Genome One Array (HOA 4.3) by Phalanx Biotech Group, Inc.
Signal pathway inhibitor treatment
After cells had grown to confluence, they were placed in a starvation medium (0.5% FBS) for 16 hours. Starved cells were pretreated for 1 hour with vehicle or selective PI3-K inhibitor LY294002 (Sigma; St. Louis, MO, USA), or IKK α/β inhibitor Wedelolactone (Calbiochem; Billerica, MA, USA).
Generation of RGC-32 transgenic mice
RGC-32 transgenic mice were generated on the FVB background at the Beth Israel Deaconess Medical Center (BIDMC) Transgenic Core Facility using the vascular endothelial (VE)-cadherin promoter to drive endothelial-specific expression of human RGC-32.
High fat diet treatment
Five-week-old endothelial-specific RGC-32 o/e mice and their littermate control mice were housed in a temperature controlled facility (25°C) with a 12 h light-dark cycle. Mice were fed with a HFD (Research Diets, 45% kcal from fat) (Harlan Laboratories, Inc. Madison, WI, USA) for 6 weeks.
Metabolic studies
Blood was obtained from feed-deprived (overnight), restrained unanesthetized mice via submandibular bleed for fasting glucose and insulin test. After submandibular bleeds, feed was returned to the cages and mice were allowed to recover. Blood glucose was quantified using a CVS TRUEtrack glucose monitor (Home Diagnostics, Ft. Lauderdale, FL, USA). Plasma insulin concentration was measured by enzyme-linked immunosorbent assay following the instructions of the manufacturer (Crystal Chem Inc., Downers Grove, IL, USA).
Glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed in conscious transgenic and littermate control mice (n = 3-5 for each genotype) at the end point of high fat diet treatment. For GTT, mice were fasted overnight, yet given free access to water, and glucose (1 g/kg body weight) was injected intraperitoneally. Blood glucose was measured by tail bleeding using CVS TRUEtrack glucometer at 0, 30, 60, and 120 minutes after glucose injection. For ITT, mice were injected with insulin (0.75 U/kg body weight; Eli Lilly and Co.) intraperitoneally, and blood glucose was measured at the same time points for GTT.
RNA isolation, cDNA synthesis and quantitative real-time PCR analysis
Total RNA was extracted from the stable RGC-32 o/e and control HMEC-1 cell lines using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. A total of 2.0 μg of RNA were reverse-transcribed using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA, USA) with random primers according to the manufacturer’s protocol. Quantitative real-time PCR (QPCR) amplification was done using SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s protocol with the following primers (Table 1).
Table 1.
Sequences of qPCR primers
| Gene | Forward primer | Reverse Primer |
|---|---|---|
| GLP2R | 5’-GGAAGTGGGCTCAGTACAAAC-3’ | 5’-GTCCCGTTACAAAATATGCCAGA-3’ |
| GLUT12 | 5’-GAGGCTGCGGCATGTTTAC-3’ | 5’-CCAAGTTCATAACCCACCAGG-3’ |
| GFPT1 | 5’-AACTACCATGTTCCTCGAACGA-3’ | 5’-CTCCATCAAATCCCACACCAG-3’ |
| RGC-32 | 5’-TCCAACCAACTCCTCCTCTCCAG | 5’-GTCACCTAATTTGGCTTTCCGA-3’ |
| GAPDH | 5’-TGGTGAAGCAGGCATCTGAG-3’ | 5’-CTCCTGCGACTTCAACAGCA-3’ |
Real time quantitative PCR was performed in the SDS 7000 System (Applied Biosystems, Carlsbad, CA, USA). For all individual cDNAs, amplification of each specific mRNA sequence was performed in at least 3 independently performed PCR experiments. For each reaction, expression was calculated as 2-DCt, where DCt is the difference between the Ct for the gene of interest and the Ct for the housekeeping gene, GAPDH.
Immunoblot analysis
RGC-32 and GFP-stably transfected HMEC-1 cells were washed twice with cold phosphate-buffered saline (PBS) and lysed in cold RIPA buffer (Boston Bio-Products, Inc., Ashland, MA, USA) containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Roche, Basel, Switzerland). Protein concentrations were determined with the DC Protein Standard Assay (Bio-Rad, Munich, Germany). Samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes (Whatman, Springfield Mill, UK) and subsequently blocked in TBS-Tween 20 containing 5% non-fat milk for 1 h. The membranes were incubated with the indicated primary antibodies: polyclonal anti-RGC-32 (a kind gift from Dr. Rus, University of Maryland and custom synthesized by Genemed, CA, USA), polyclonal anti-AKT (Cell Signaling Technology, Inc., Danvers, MA, USA), polyclonal anti-p-AKT (Cell Signaling Technology, Inc., Danvers, MA, USA), monoclonal anti-vinculin(Sigma, Germany); followed by incubation with horseradish peroxidase-conjugated secondary antibodies anti-rabbit IgG with 1:2000 dilution (Calbiochem, La Jolla, CA, USA) or anti-mouse IgG with 1:2000 dilution (Vector Labs, Burlingame, CA, USA). Blots were developed using the chemiluminescence detection system according to the instructions of the manufacturer (Thermo Fisher, Pittsburgh, PA, USA). Densitometric analysis was done using the NIH software program, Image J 4.5.
Statistical analysis
Data was obtained from at least three independent cell cultures or animals, as denoted in the figure legends. For statistical analysis, if differences were established, the values were compared using a Student’s t-test. The values were expressed as mean ± SEM. The results were considered significant if P < 0.05.
Results
RGC-32 expression is increased in high fat diet treated mice
To investigate the role of RGC-32 in metabolism, we initially examined RGC-32 expression in wild type mice fed with high fat diet comparing to normal chow diet. HFD treated mice demonstrated an increase in RGC-32 levels in adipose tissue and liver comparing to normal chow diet treated mice (Figure 1A, 1B). These results demonstrate that RGC-32 is induced by high fat diet and may be important to the homeostasis of metabolism to some extent.
Figure 1.
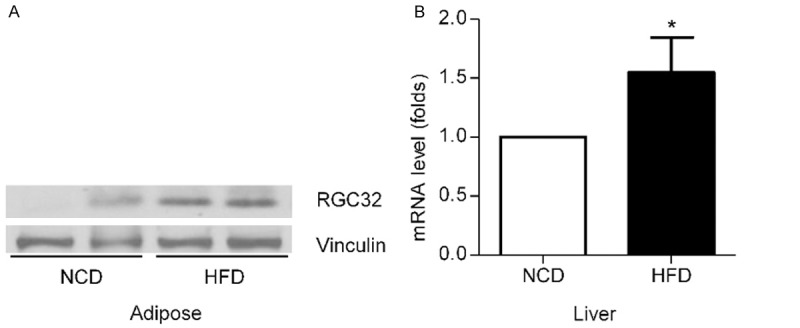
Effect of high fat diet on RGC-32 expression in wild type mice. A. RGC-32 expression in adipose tissue was assessed after either high fat diet or normal chow diet treated mice by western blot. B. RGC-32 expression in liver was assessed after either high fat diet or normal chow diet treated mice by qPCR. Results are mean ± SEM, *P<0.05.
RGC-32 is induced by glucose and insulin in HMEC-1 through PI3K pathway
To determine whether RGC-32 in endothelial cells can be induced by hyperglycemia or hyperinsulinmia, HMEC-1 cells were treated by additional 25 mM D-glucose or 100 nM insulin for 1, 3 or 5 days respectively. When HMEC-1 cells were treated with 25 mM D-glucose, RGC-32 protein and mRNA levels were increased comparing to control cells (Figure 2A, 2B). When HMEC-1 cells were exposed to 100 nM insulin for 1 to 5 days, RGC-32 was increased as well (Figure 2A). Phosphate-AKT was also increased during the period of high glucose or insulin treatment (Figure 2A). To verify that RGC-32 was induced through PI3K-AKT pathway, HMEC-1 cells were pretreated with vehicle or selective PI3-K inhibitor LY294002 or IKK α/β inhibitor Wedelolactone. Not NF-κB inhibitor but PI3K inhibitor dramatically attenuated the RGC-32 expression (Figure 2C). These results suggest that RGC-32 is induced by hyperglycemia or hyperinsulinmia in endothelial cells through PI3K- AKT pathway.
Figure 2.
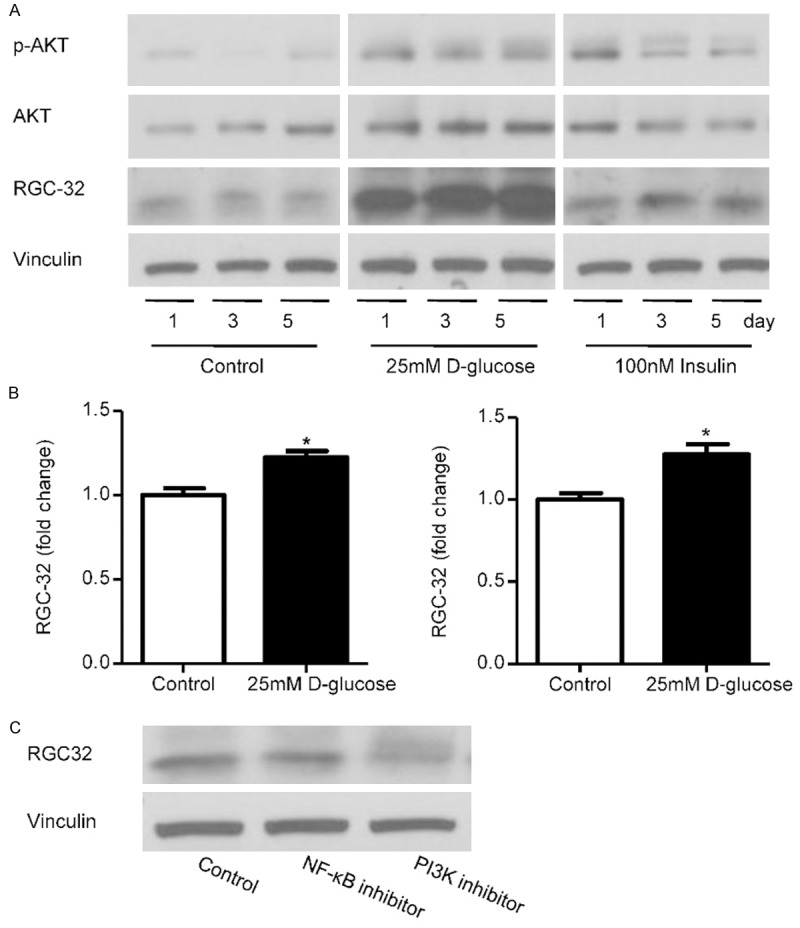
Effect of glucose and insulin in RGC-32 expression in HMEC-1. A. Immunoblot analysis of RGC-32, phosphate-AKT and AKT in HMEC-1 cells after 25 mM D-glucose or 100 nM insulin treated for 1, 3 or 5 days. Upper panel showed represent blot and the lower panel showed the result analyzed by Image J. B. Quantitative real-time polymerase chain reaction (QPCR) analysis of RGC-32 mRNA levels in human microvascular endothelial cells (HMEC-1) after 1 or 3 days treatment by 25 mM D-glucose. C. Immunoblot analysis of RGC-32 in HMEC-1 cells treated with selective PI3-K inhibitor LY294002 or IKK α/β inhibitor Wedelolactone. *P<0.05.
RGC-32 regulates glucose metabolism related genes
To test whether RGC-32 in HEMC regulates glucose metabolism related genes, RGC-32 was either knocked down by transfection of siRNAs or overexpressed by retroviral infection in HMEC-1. Originally, DNA microarray data from RGC-32 siRNA treated HMEC-1 illustrated that a handful of metabolism related genes had significant changes, including GFPT1, GLUT12 and GLP2R (Figure 3A). GFPT1 and GLUT12 mRNA expression were decreased markedly in RGC-32 o/e HMEC cells comparing with control cells, while GLP2R mRNA expression was increased dramatically (Figure 3B). Thus, results from both RGC-32 knockdown and overexpression HMEC cells confirmed that RGC-32 regulates GFPT1, GLUT12 and GLP2R.
Figure 3.
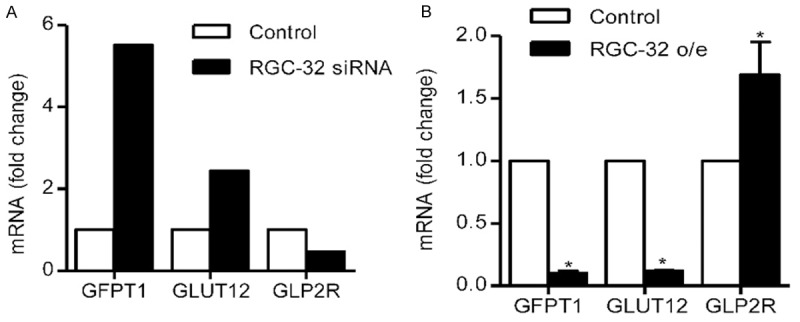
Effect of RGC-32 on glucose metabolism related genes. A. DNA array analysis of GFPT1, GLUT12 and GLP2R mRNA levels in RGC-32 siRNA infected HMEC-1 cells. B. qPCR analysis of GFPT1, GLUT12 and GLP2R mRNA levels in RGC-32 o/e HEMC cells. Results are mean ± SEM based on 3 experiments. *P<0.05.
Effect of endothelial specific RGC-32 overexpression in high fat diet treated mice
We next examined the metabolic effect of endothelial specific RGC-32 overexpression on mice fed a high fat diet. Five-week-old endothelial-specific RGC-32 o/e mice and their littermate control mice were fed HFD (42% kcal from fat) for 6 weeks. RGC-32 did not significantly alter body weight (Figure 4A). Overnight fasting blood glucose and 6-hour fasting plasma insulin concentrations were not significantly altered in RGC-32 o/e mice (Figure 4B). We evaluated insulin sensitivity and glucose homeostasis in RGC-32 o/e and littermate controls fed HFD. As shown in Figure 4C, the ability of glucose disposal in response to an intraperitoneal glucose tolerance test (1 g/kg body weight) was significantly increased in RGC-32 o/e mice compared with their littermate controls. Additionally, the area under the curve (AUC) was calculated and was significantly smaller in the transgenic RGC-32 o/e mice. In contrast, insulin induced hypoglycemia (after intraperitoneal administration of insulin (0.75 U/kg)), was not significantly different in RGC-32 o/e mice versus littermate control (Figure 4D), indicating similar insulin sensitivity between two groups. Thus, the mice with RGC-32 overexpression in endothelial cells appear to have increased ability of blood glucose disposal on condition of high fat diet.
Figure 4.
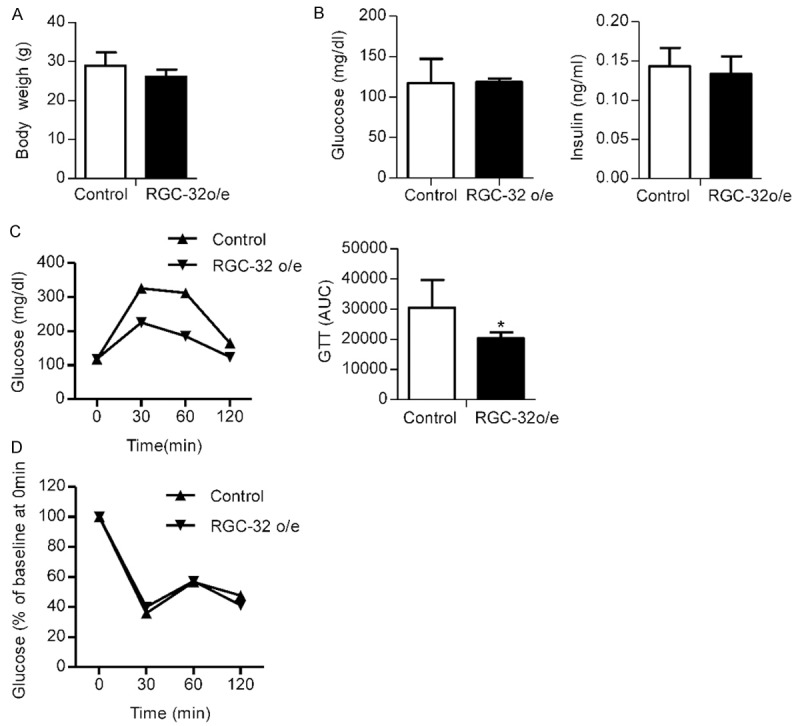
Effect of RGC-32 on glucose homeostasis and insulin sensitivity. A. Body weight was obtained on endothelial-specific RGC-32 o/e and their littermate control mice after being fed with a high-fat diet for 6 weeks. B. Fasting blood glucose (left panel) and insulin (right panel) concentration in endothelial-specific RGC-32 o/e and their littermate control mice. C. Glucose tolerance test in endothelial-specific RGC-32 o/e and their littermate control mice. Glucose was measured before and 30, 60, and 120 minutes after intraperitoneal glucose injection at 11 weeks of age (left panel). The AUC of glucose levels during glucose tolerance tests is shown (right panel). D. Insulin induced hypoglycemia in endothelial-specific RGC-32 o/e and their littermate control mice was analyzed; glucose was measured before and 30, 60, and 120 minutes after intraperitoneal insulin injection, and then glucose disposal rate comparing to baseline at different time points was shown.
Discussion
The role of the endothelial organ in governing physiologic homeostasis and fostering disease pathogenesis has been recognized [1]. Endothelial dysfunction is a common and early finding in diabetes, which is promoted by a bunch of metabolic factors such as dyslipidemia, elevated free fatty acids, hypertension, and obesity [13]. To date, endothelial dysfunction has been understood primarily in the context of abnormal vasomotor function [14]. Our results showed that the over expression of RGC-32 in endothelial will increase the ability of blood glucose disposal, although the insulin sensitivity was not changed. The mechanisms linking endothelium RGC-32 with glucose homeostasis have not been investigated yet.
We previously reported that related transcriptional enhancer factor-1 (RTEF-1) in endothelial cells not only plays an important role via regulation of angiogenesis [15] but also decrease blood glucose via stimulating IGFBP-1 expression [16]. It was reported that PPARγ in the endothelium integrates metabolic and vascular responses [17]. These findings provide the basis for novel insights into the role of vascular endothelial cells in metabolism.
The present studies revealed an important role of RGC-32 in the regulation of glucose homeostasis. First, we demonstrated that RGC-32 is induced by high fat diet in mice adipose and liver. Second, we identified RGC-32 is induced by hyperglycemia and hyperinsulinemia in endothelial cells through PI3K-AKT pathway. Third, we found that carbohydrate metabolic genes, such as GFPT1, GLUT12, GLP2R, were regulated by RGC-32. Finally, endothelial-specific overexpression of RGC-32 results in imp-roved disposal of carbohydrate.
RGC-32 was induced in high fat diet treated mice and glucose or insulin treated HMEC-1 cells, which helped to link RGC-32 particularly in endothelial cells to glucose metabolism. Furthermore, when we used DNA array to screen potential RGC-32 regulated genes, a few carbohydrate metabolisms related genes came into our sight which was verified by qPCR. Among these genes, the expression of GFPT1 and GLUT12 were decreased and GLP2R increased.
Control of glucose homeostasis is a tightly regulated process involving the interplay of gut and pancreatic hormones, gastric motility, insulin sensitivity, neural signals, and regulation of hepatic glucose production. An endothelial cell- specific overexpressing (VE-Cad) transgenic model was used to clarify the effects of endothelial RGC-32 on glucose homeostasis. RGC-32 has no significant impact on fasting glucose. RGC-32 ameliorates the glucose excursion after carbohydrate stress without changing the circulation insulin level or insulin sensitivity. Some downstream genes of RGC-32 may be involved in the maintaining of glucose homeostasis. GLPT1, GLUT12 and GLP2R were included but not limited to.
GFPT1, the rate-limiting enzyme of the first step, controls the flux of glucose into the hexosamine pathway. Glucose shunted into the hexosamine pathway is not only regarded as a cellular nutrient sensor but also responsible for glucotoxicity [18]. Decreased expression of human GFPT would result in higher insulin sensitivity in animal models [19]. Thus, human GFPT is recognized as an interesting potential therapeutic target for type II diabetes [20]. Attenuating GFPT1 may be partial mechanism by which RGC-32 improves disposal of blood glucose.
Glucose uptake from the bloodstream is a rate-limiting step in glucose utilization, and is regulated by a family of membrane proteins called glucose transporters (GLUTs). Although GLUT4 is the predominant isoform in insulin-sensitive tissues, previous evidence presented that GLUT12 could be a novel insulin-sensitive GLUT [21]. Other evidences showed that GLUT12 worked as a basal and insulin-independent glucose transporter in heart and active cell surface. The expression of GLUT12 was increased in the diabetic myocardium [22]. Although the detailed function of GLUT12 was disputable, RGC-32 down-regulate GLUT12 may play a role in the regulation of glucose homeostasis.
Glucagon-like peptide-2 (GLP-2) is a 33-amino acid proglucagon-derived peptide, which stimulates glucagon secretion through GLP2R (known as the receptor of GLP-2) present on the alpha cell in rats with no effect on insulin secretion [23]. GLP-2 can also increase circulating levels of glucagon in human volunteers [24]. Knockdown of GLP2R in diabetic mice exhibited elevated levels of glucagon and blood glucose, impaired glucose utilization after intraperitoneal glucose injection and abnormal allocation of β- and α-cell lineages [25]. Therefore, we deduced that increased expression of GLP2R by over-expressed endothelial RGC-32 may help to increase the utility of carbohydrates after carbohydrate tolerance test.
Taken together, we demonstrated that RGC-32 in the endothelium plays an important role in regulating glucose homeostasis and over expression of RGC-32 may increase the utility of blood glucose after carbohydrate stress. The underlying mechanisms may be involved in down-regulated GFPT1 and GLUT12, and up-regulated GLP2R. The interaction and balance between GFPT1, GLUT12 and GLP2R remain to be further examined.
Acknowledgements
This work was supported in part by a National Institutes of Health grant HLR01082837 (Li), an AHA grant-in-aid 0265494T (Li), China Program for New Century Excellent Talents in University NCET-13-0692 (Guo) and the China Scholarship Council (Guo). The authors would like to thank Dr. Jian Li for all guidance and support in the design of manuscript and interpretation of data during Dr. Shuzhen Guo worked in Dr. Li’s laboratory at the CardioVascular Institute, Beth Israel Deaconess Medical Center, Harvard Medical School.
Disclosure of conflict of interest
This work was performed while Dr. Jian Li was employed at the CardioVascular Institute, Beth Israel Deaconess Medical Center, Harvard Medical School and Dr. Shuzhen Guo worked in Dr. Jian Li’s laboratory. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States Government. The authors declare that they have no competing interests.
Abbreviations
- ECs
endothelial cells
- GFPT1
glutamine-fructose-6-phosphate aminotransferase [isomerizing] 1
- GLUT12
solute carrier family 2 (facilitated glucose transporter), member 12 (SLC2A12)
- GLP-2
glucagon-like peptide 2
- GLP2R
glucagon-like peptide 2 receptor
- HFD
high fat diet
- GFP
green fluorescence protein
- GTT
glucose tolerance test
- ITT
insulin tolerance test
References
- 1.Cleaver O, Melton DA. Endothelial signaling during development. Nat Med. 2003;9:661–668. doi: 10.1038/nm0603-661. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97:2601–2610. doi: 10.1172/JCI118709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meigs JB, Hu FB, Rifai N, Manson JE. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA. 2004;291:1978–1986. doi: 10.1001/jama.291.16.1978. [DOI] [PubMed] [Google Scholar]
- 4.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 5.Vlaicu SI, Tegla CA, Cudrici CD, Danoff J, Madani H, Sugarman A, Niculescu F, Mircea PA, Rus V, Rus H. Role of C5b-9 complement complex and response gene to complement-32 (RGC-32) in cancer. Immunol Res. 2013;56:109–121. doi: 10.1007/s12026-012-8381-8. [DOI] [PubMed] [Google Scholar]
- 6.Zhu L, Qin H, Li PY, Xu SN, Pang HF, Zhao HZ, Li DM, Zhao Q. Response gene to complement-32 enhances metastatic phenotype by mediating transforming growth factor beta-induced epithelial-mesenchymal transition in human pancreatic cancer cell line BxPC-3. J Exp Clin Cancer Res. 2012;31:29. doi: 10.1186/1756-9966-31-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vlaicu SI, Tegla CA, Cudrici CD, Fosbrink M, Nguyen V, Azimzadeh P, Rus V, Chen H, Mircea PA, Shamsuddin A, Rus H. Epigenetic modifications induced by RGC-32 in colon cancer. Exp Mol Pathol. 2010;88:67–76. doi: 10.1016/j.yexmp.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tegla CA, Cudrici CD, Azimzadeh P, Singh AK, Trippe R 3rd, Khan A, Chen H, Andrian-Albescu M, Royal W 3rd, Bever C, Rus V, Rus H. Dual role of Response gene to complement-32 in multiple sclerosis. Exp Mol Pathol. 2013;94:17–28. doi: 10.1016/j.yexmp.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Li Z, Xie WB, Escano CS, Asico LD, Xie Q, Jose PA, Chen SY. Response gene to complement 32 is essential for fibroblast activation in renal fibrosis. J Biol Chem. 2011;286:41323–41330. doi: 10.1074/jbc.M111.259184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang JN, Shi N, Xie WB, Guo X, Chen SY. Response gene to complement 32 promotes vascular lesion formation through stimulation of smooth muscle cell proliferation and migration. Arterioscler Thromb Vasc Biol. 2011;31:e19–26. doi: 10.1161/ATVBAHA.111.230706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An X, Jin Y, Guo H, Foo SY, Cully BL, Wu J, Zeng H, Rosenzweig A, Li J. Response gene to complement 32, a novel hypoxia-regulated angiogenic inhibitor. Circulation. 2009;120:617–627. doi: 10.1161/CIRCULATIONAHA.108.841502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui XB, Guo X, Chen SY. Response gene to complement 32 deficiency causes impaired placental angiogenesis in mice. Cardiovasc Res. 2013;99:632–639. doi: 10.1093/cvr/cvt121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 14.Sena CM, Pereira AM, Seica R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta. 2013;1832:2216–2231. doi: 10.1016/j.bbadis.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Jin Y, Wu J, Song X, Song Q, Cully BL, Messmer-Blust A, Xu M, Foo SY, Rosenzweig A, Li J. RTEF-1, an upstream gene of hypoxia-inducible factor-1alpha, accelerates recovery from ischemia. J Biol Chem. 2011;286:22699–22705. doi: 10.1074/jbc.M111.237024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messmer-Blust AF, Philbrick MJ, Guo S, Wu J, He P, Li J. RTEF-1 attenuates blood glucose levels by regulating insulin-like growth factor binding protein-1 in the endothelium. Circ Res. 2012;111:991–1001. doi: 10.1161/CIRCRESAHA.112.268110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanda T, Brown JD, Orasanu G, Vogel S, Gonzalez FJ, Sartoretto J, Michel T, Plutzky J. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J Clin Invest. 2009;119:110–124. doi: 10.1172/JCI36233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McClain DA. Hexosamines as mediators of nutrient sensing and regulation in diabetes. J Diabetes Complications. 2002;16:72–80. doi: 10.1016/s1056-8727(01)00188-x. [DOI] [PubMed] [Google Scholar]
- 19.Hebert LF Jr, Daniels MC, Zhou J, Crook ED, Turner RL, Simmons ST, Neidigh JL, Zhu JS, Baron AD, McClain DA. Overexpression of glutamine: fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance. J Clin Invest. 1996;98:930–936. doi: 10.1172/JCI118876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakaishi Y, Bando M, Shimizu H, Watanabe K, Goto F, Tsuge H, Kondo K, Komatsu M. Structural analysis of human glutamine: fructose-6-phosphate amidotransferase, a key regulator in type 2 diabetes. FEBS Lett. 2009;583:163–167. doi: 10.1016/j.febslet.2008.11.041. [DOI] [PubMed] [Google Scholar]
- 21.Purcell SH, Aerni-Flessner LB, Willcockson AR, Diggs-Andrews KA, Fisher SJ, Moley KH. Improved insulin sensitivity by GLUT12 overexpression in mice. Diabetes. 2011;60:1478–1482. doi: 10.2337/db11-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waller AP, George M, Kalyanasundaram A, Kang C, Periasamy M, Hu K, Lacombe VA. GLUT12 functions as a basal and insulin-independent glucose transporter in the heart. Biochim Biophys Acta. 2013;1832:121–127. doi: 10.1016/j.bbadis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 23.de Heer J, Pedersen J, Orskov C, Holst JJ. The alpha cell expresses glucagon-like peptide-2 receptors and glucagon-like peptide-2 stimulates glucagon secretion from the rat pancreas. Diabetologia. 2007;50:2135–2142. doi: 10.1007/s00125-007-0761-6. [DOI] [PubMed] [Google Scholar]
- 24.Meier JJ, Nauck MA, Pott A, Heinze K, Goetze O, Bulut K, Schmidt WE, Gallwitz B, Holst JJ. Glucagon-like peptide 2 stimulates glucagon secretion, enhances lipid absorption, and inhibits gastric acid secretion in humans. Gastroenterology. 2006;130:44–54. doi: 10.1053/j.gastro.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Bahrami J, Longuet C, Baggio LL, Li K, Drucker DJ. Glucagon-like peptide-2 receptor modulates islet adaptation to metabolic stress in the ob/ob mouse. Gastroenterology. 2010;139:857–868. doi: 10.1053/j.gastro.2010.05.006. [DOI] [PubMed] [Google Scholar]