

Abstract
Alpha 1-adrenergic receptors (α1-ARs) control the activity of dorsal raphe nucleus (DRn) serotonin (5-HT) neurons and play crucial role in the regulation of arousal and stress homoeostasis. However, the precise role of these receptors in regulating glutamate synapses of rat DRn 5-HT neurons and whether chronic stress exposure alters such regulation remain unknown. In the present study, we examined the impact of chronic restraint stress on α1-AR-mediated regulation of glutamate synapses onto DRn 5-HT neurons. We found that, in the control condition, activation of α1-ARs induced an inward current and long-term depression (LTD) of glutamate synapses of DRn 5-HT neurons. The α1-AR LTD was initiated by postsynaptic α1-ARs but mediated by a decrease in glutamate release. The presynaptic expression of the α1-AR LTD was signaled by retrograde endocannabinoids (eCBs). Importantly, we found that chronic exposure to restraint stress profoundly reduced the magnitude of α1-AR LTD but had no effect on the amplitude of α1-AR-induced inward current. Chronic restraint stress also reduced the CB1 receptor-mediated inhibition of EPSC and the eCB-mediated depolarization-induced suppression of excitation. Collectively, these results indicate that chronic restraint stress impairs the α1-AR LTD by reducing the function of presynaptic CB1 receptors and reveal a novel mechanism by which noradrenaline controls synaptic strength and plasticity in the DRn. They also provide evidence that chronic stress impairs eCB signaling in the DRn, which may contribute, at least in part, to the dysregulation of the stress homeostasis.
Keywords: dorsal raphe, endocannabinoid, glutamate, LTD, serotonin, stress
Introduction
Serotonin (5-hydroxytryptamine [5-HT]) neurons in the dorsal raphe nucleus (DRn) (Dahlström and Fuxe, 1964) provide major serotonergic projections to brain areas controlling the behavioral and neuroendocrine responses to stress (Petrov et al., 1994). By modulating the stress-associated neuronal circuits, DRn 5-HT neurons control stress homeostasis and mood (Joëls and Baram, 2009). Indeed, animal studies have shown that the behavioral responses to various stressors are mediated, at least in part, by the activation of 5-HT system. For instance, exposure to uncontrollable stressors (e.g., tail shock) activates DRn 5-HT neurons and enhances 5-HT transmission (Amat et al., 1998; Maswood et al., 1998; Grahn et al., 1999). Activation of DRn 5-HT neurons also regulates uncontrollable stress-induced learned helplessness (Grahn et al., 1999), characterized by a set of behaviors, including reduced escape to aversive stimuli, increased fear conditioning, and anxiety (Maier et al., 1994, 1995). Conversely, inhibition of DRn 5-HT neurons reduces the behavioral responses to uncontrollable stressors (Maier et al., 1994, 1995), indicating that DRn 5-HT neurons play a key role in modulating the behavioral responses to uncontrollable stress (Maier and Watkins, 2005). Furthermore, results from clinical studies have established that stress-induced dysregulation of the 5-HT system is a major contributing factor for the development of mood disorders, such as depression and anxiety (Southwick et al., 2005; Lupien et al., 2009).
The DRn receives a major noradrenergic input from the locus ceruleus (Baraban and Aghajanian, 1981), which activates DRn 5-HT neurons (Baraban and Aghajanian, 1981) and regulates arousal and stress homeostasis (Morilak et al., 2005; Stone et al., 2007). Previous studies have shown that exposure to various stressors increases noradrenaline release in the DRn (Tanaka et al., 1983; Shimizu et al., 1994) and induces anxiety-like behaviors (Chiba et al., 2012; Kim et al., 2012), at least in part, via the activation of α1-ARs located on DRn 5-HT neurons (Stone et al., 2007). α1-AR signaling in the DRn also regulates fear conditioning, as blockade of these receptors prevents conditioned fear and impairs escape performance (Grahn et al., 2002). Furthermore, disruption of DRn α1-AR signaling alters the behavioral effects of antidepressant and antianxiety drugs (O'Leary et al., 2007; Doze et al., 2009). Collectively, these studies indicate that α1-AR-mediated control of DRn 5-HT neurons plays an important role in the regulation of stress homoeostasis and that the alteration of α1-AR signaling in the DRn might contribute to stress-related mood disorders.
Remarkably, despite the crucial role of α1-AR signaling in the DRn in controlling the behavioral responses to stress, the effects of chronic stress on α1-AR-mediated control of the excitability of 5-HT neurons and synaptic transmission in the DRn remain unknown. In this study, we show that exposure to chronic restraint stress (CRS) impairs α1-AR LTD of glutamate synapses in the DRn but has no effects on α1-AR-induced membrane depolarization/inward current in DRn 5-HT neurons. The CRS-induced impairment of α1-AR LTD is mediated by a downregulation of eCB signaling. Such results unravel a novel cellular mechanism by which chronic stress could induce long-lasting changes in the function of the 5-HT system.
Materials and Methods
Brain slice preparation.
All the experimental procedures used in this study were approved by the University at Buffalo Animal Care and Use Committee and follow the guidelines in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Brain slices containing the DRn were prepared from 6- to 8-week-old male Sprague Dawley rats (Harlan Laboratories) using standard procedures (Haj-Dahmane, 2001). In brief, rats were anesthetized with isoflurane and killed by decapitation. The brainstem area was isolated, and coronal sections (300 μm) containing the DRn were cut in ice-cold modified ACSF of the following composition (in mm): 110 choline chloride, 2.5 KCl, 0.5 CaCl2, 7 MgSO4, 1.25 NaH2PO4, 26.2 NaHCO3, 11.6 sodium l-ascorbate, 3.1 sodium pyruvate, 25 glucose, and equilibrated with 95% O2/5% CO2 using a vibrating-blade microtome (Lancer series 1000; Leica Biosystem). Slices were incubated for 45 min at 35°C and then at room temperature for at least 1 h in a holding chamber filled with regular ACSF (in mm): 119 NaCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, 11 glucose, and continuously bubbled with 95% O2/5% CO2. After recovery, slices were transferred to a recording chamber (Warner Instruments) mounted on a fixed upright microscope. In the chamber, the slice is continuously perfused (2–3 ml/min) with ACSF saturated with 95% O2/5% CO2 and heated to 30 ± 1°C using a solution heater (Warner Instruments).
Electrophysiological recordings.
DRn neurons were visualized using a BX 51 Olympus microscope (Olympus) equipped differential interference contrast and infrared optical filter. Somatic whole-cell recordings were obtained from putative DRn 5-HT neurons with patch electrodes (3–5 mΩ) filled with a solution containing the following (in mm): 120 potassium gluconate, 10 KCl, 10 Na2-phosphocreatine, 10 HEPES, 1 MgCl2, 1 EGTA, 2 Na2-ATP, 0.25 Na-GTP, pH 7.3, osmolarity 280–290 mOsmol. To examine the role of postsynaptic G-proteins in mediating the effects of α1-ARs, GTP was replaced with GDPβS. To examine the depolarization-induced suppression of excitation (DSE) at glutamate synapses of the DRn, whole-cell recordings were performed with patch electrodes filled with cesium methanesulfonate-based solution of the following composition (in mm): 120 cesium methanesulfonate, 10 CsCl, 10 Na2-phosphocreatine, 10 HEPES, 1 MgCl2, 1 EGTA, 2 Na2-ATP, 0.25 Na-GTP, pH 7.3, osmolarity 280–290 mOsmol. The use of this internal solution facilitates the induction of the DSE in the DRn (Haj-Dahmane and Shen, 2009). DRn 5-HT neurons were identified using previously well established electrophysiological criteria (Haj-Dahmane, 2001).
All recordings were performed from neurons located in the dorsomedial and ventromedial subdivisions of the DRn. EPSCs were evoked with single square-pulses (100–200 μs duration) delivered at 0.1 Hz with patch pipettes (3–5 mΩ) filled with ACSF and placed 50–100 μm dorsolateral to the recorded neuron. In some experiments, to assess the change in paired-pulse ratio (PPR), pairs of EPSCs were evoked with an interstimulus interval of 20–30 ms. The intensity of the stimulus was adjusted to evoke 75% of the maximal amplitude of EPSCs. α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-mediated EPSCs were recorded in neurons voltage-clamped at −70 mV and in the continuous presence of GABAA and glycine receptor antagonists picrotoxin (100 μm) and strychnine (20 μm), respectively. To determine the AMPAR/NMDAR ratio, neurons were recorded with cesium methylsulfonate-based internal solution in the presence of GABAA and glycine receptor antagonists and voltage-clamped at 50 mV. EPSCs were recorded in the absence (mixed EPSCs) and presence of APV (50 μm: AMPAR-EPSCs). NMDAR-EPSCs were determined by digital subtraction of AMPAR-EPSCs from mixed EPSCs. The AMPAR/NMDAR ratio was determined by dividing the average (30 traces) amplitude of AMPAR-EPSC by the average (30 traces) amplitude of NMDAR-EPSCs. Membrane currents were amplified with an Axoclamp 2B or Multiclamp 700B amplifier (Molecular Devices). The membrane currents were filtered at 3 kHz, digitized at 20 kHz with Digidata 1440, and acquired using the pClamp 10.0 software (Molecular Devices). Access resistance (10–20 mΩ) was monitored online using 5 mV hyperpolarizing voltage steps (200 ms duration). Recordings were discarded when the access resistance increased by >10% to 20%.
Stress procedures.
The stress paradigm used in this study is the chronic inescapable restraint stress (CRS). Male rats were physically restrained using rodent restrainers (IITC Life Science) for a 45 min session, three times per day for 7 consecutive days. After each session, rats were returned to their home cage. Control animals were handled briefly (5–10 min) for 7 consecutive days. Electrophysiological studies were performed 24 h after the last restraint session.
Data analysis.
AMPAR-EPSCs were analyzed using the Clampfit 10.2 software (Molecular Devices). The amplitude of AMPAR-EPSCs was determined by measuring the average current during a 2 ms time window at the peak of each EPSC and subtracted from the baseline current determined during a 5 ms time window before the stimulus artifact. All AMPAR-EPSC amplitudes were normalized to the mean baseline amplitude recorded for at least 10 min before drug application. The PPR (EPSC2/EPSC1) was averaged for at least 60 trials in the absence and presence of phenylephrine (PHE). To determine the coefficient of variation (CV) and the SD, the mean amplitude of AMPAR-EPSCs was calculated for at least 60 consecutive trials in control condition and during the LTD. The CV was then given by the following ratio: (SD)/(EPSC mean amplitude). Statistical analysis was performed using the Origin 8.0 software (Microcal Software). The results in the text and figures are expressed as mean ± SEM. Statistical comparisons were conducted using the Student's paired t test for within-group comparisons and the independent t test for comparisons between groups. Statistical significance was set at p < 0.05.
Chemicals and drugs.
Most chemicals were obtained from Fisher Scientific. PHE, prazosin, picrotoxin, strychnine, N-(piperidin-1-yl)-5-(4-iodophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM 251), (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate (WIN 55212-2), and 1-[6-[[(17β)-3-methoxyestra1,2,5(10)-trien-17-yl]amino[hexyl]-1H-pyrole-2,5-dione (U73122) were purchased from Tocris Cookson. GDPβS and N-formyl-l-leucine(1S)-1-[[(2S,3S)-3=hexyl-4-oxo-2-oxetanyl]methyl]dodecyl ester (tetralhydrolipstatin; THL) were obtained from Sigma-Aldrich.
Results
α1-ARs induce LTD of glutamate synapses in the DRn
To assess the impact of α1-ARs on glutamatergic synaptic transmission onto putative DRn 5-HT neurons, we examined the effect the α1-AR agonist PHE on the amplitude of AMPAR-EPSCs. As reported previously (Pan et al., 1994), bath administration of PHE (100 μm) induced a significant inward shift of the holding current in all putative DRn 5-HT neurons voltage clamped at their resting membrane potential (i.e., −70 mV; Fig. 1A). On average, the amplitude of the PHE-induced inward current (IPHE) was 53.5 ± 5.9 pA (n = 15). In addition to this postsynaptic effect, PHE (100 μm) profoundly reduced the amplitude of evoked AMPAR-EPSCs. A representative experiment depicting the effect of PHE on the amplitude of AMPAR-EPSCs is represented in Figure 1B. On average, PHE (100 μm) reduced the amplitude of AMPAR-EPSCs to 36.5 ± 5.2% at the peak of PHE response (Fig. 1D, n = 15). Washout of PHE resulted in a partial recovery of the amplitude of AMPAR-EPSCs. On average, AMPAR-EPSCs recovered to 58.4 ± 2.9% of baseline after extensive (>40 min) washout of PHE (Fig. 1C,D; n = 15). Treatment of slices with prazosin (10 μm), a selective α1-AR antagonist, abolished IPHE (n = 8, data not shown) and blocked the acute and the long-lasting depression of AMPA-EPSC amplitude (98.1 ± 8.2% of baseline, n = 8, p < 0.05; Fig. 2A), indicating that both the IPHE and the depression of AMPAR-EPSCs are signaled by α1-ARs.
Figure 1.

PHE induces an inward current and a long-lasting depression of AMPAR-EPSCs in DRn 5-HT neurons. A, Illustration of a typical inward current induced by bath application of PHE (100 μm) in putative DRn 5-HT neurons. B, Depiction of the effect of bath application of PHE (100 μm) on the amplitude of AMPAR-EPSCs. C, Left, Summary graph of the effect of PHE (100 μm) on the amplitude of AMPAR-EPSCs (n = 15). Right, Superimposed EPSC traces taken at time points indicated by numbers in C. D, Illustration of the average amplitude of EPSCs measured during bath application of PHE (PHE acute) and after washout of PHE (PHE-LTD). PHE induces an LTD of AMPAR-EPSCs. Calibration: 50 pA, 10 ms.
Figure 2.

α1-Adrenergic receptors mediate the PHE-LTD. A, Blockade of α1-ARs with prazosin (10 μm) abolishes the PHE-LTD. Left, Summary graph of the PHE-LTD in control condition (white circles, n = 8) and in the presence of prazosin (10 μm, filled red circles, n = 8). Right, Superimposed EPSC traces taken at the time points indicated by numbers in the left panel. B, The PHE-LTD is not caused by persisting stimulation of α1-ARs. Left, Summary graph of the PHE-LTD obtained during washout of PHE with prazosin. Right, Superimposed EPSC traces collected during the time point indicated by numbers in the left panel. Calibration: 50 pA, 10 ms.
Unlike the IPHE, which was reversible after washout of PHE, the depression of AMPAR-EPSC amplitude was long-lasting, suggesting that the persistent depression of AMPA-EPSCs could not be attributed to an incomplete washout of PHE or to a persistent activation of α1-ARs. To further test this notion, we examined whether prazosin (10 μm) could reverse the α1-AR-induced persistent depression of AMPAR-EPSCs. As illustrated in Figure 2B, washout of PHE with prazosin failed to reverse the PHE-induced persistent depression of AMPAR-EPSCs (57.8 ± 7.6% of baseline, n = 8, p < 0.05; Fig. 2B). Collectively, these results indicate that the activation of α1-ARs induces an LTD of glutamate synapses onto DRn 5-HT neurons.
Previous studies in other brain areas have reported that α1-ARs induce LTD of glutamate synapses by postsynaptic mechanisms involving an alteration of the function and/or the subunit composition of AMPARs (Kirkwood et al., 1999; McElligott and Winder, 2008; McElligott et al., 2010). To determine the mechanism underlying the α1-AR LTD of glutamate synapses in the DRn, we monitored the PPRs of AMPAR-EPSCs, a synaptic parameter inversely correlated with the release probability of neurotransmitters, before and during the PHE-LTD. We found that the α1-AR LTD was accompanied by a persistent increase in the PPR (Fig. 3B; n = 7). The average PPR increased from 1.10 ± 0.07 in control condition to 1.40 ± 0.06 during the α1-AR LTD (Fig. 3C; p < 0.05, n = 7). In addition, the α1-AR LTD was also associated with a significant increase in the CV of AMPAR-EPSC amplitude, another synaptic parameter that reflects changes in the probability of neurotransmitter release. The increase in the CV resulted in a decrease in 1/CV2 (Fig. 3D; n = 7), which was correlated with the magnitude of the α1-AR LTD. Together, these results indicate that the α1-AR LTD of glutamate synapses in the DRn is caused by a decrease in the probability of synaptic glutamate release.
Figure 3.

The α1-AR LTD is mediated by a decrease in glutamate release. A, Summary graph of the α1-AR LTD recorded using a pair of stimuli with an interstimulus interval of 30 ms (n = 7). Left, Superimposed EPSC traces (average of 60 trials) collected before (1) and (2) during the α1-ARs LTD. B, α1-AR LTD is accompanied by a persistent and significant increase in PPR (n = 7). C, Summary histogram of average PPR obtained in baseline condition and during LTD (n = 7). *p < 0.05. D, Change in 1/CV2 as a function of EPSC amplitude. α1-AR LTD is associated with a significant increase in the CV (n = 7). *p < 0.05. Calibration: 50 pA, 20 ms.
G-protein-driven endocannabinoid production mediates the α1-AR LTD in the DRn
In principle, α1-AR LTD could be mediated by activation of presynaptic α1-ARs, which could induce a persistent decrease in glutamate release. Alternatively, it is possible that the LTD could be initiated by the activation of postsynaptic α1-ARs but mediated presynaptically via retrograde messengers. To determine whether presynaptic or postsynaptic α1-ARs mediate the LTD, we examined the effect of inhibiting postsynaptic α1-AR signaling with intracellular application of the membrane impermeable G-proteins inhibitor GDPβS on the α1-AR LTD. To ensure that this manipulation was effective in blocking postsynaptic α1-ARs, we also monitored the amplitude of IPHE, a response signaled by postsynaptic α1-ARs. As expected for a postsynaptic response, intracellular application of GDPβS (250 μm) totally blocked IPHE (Fig. 4A; IPHE control = 53.5 ± 5.9 pA; IPHE GDPβS = 4.65 ± 5.6 pA, p < 0.05 vs control). More importantly, GDPβS also blocked the α1-AR LTD (LTD control = 42.24 ± 2.32% of baseline, LTD GDPβS = 95.46 ± 4.37% of baseline, p < 0.05 vs control, n = 8; Fig. 4B), indicating that the activation of postsynaptic α1-ARs initiates the LTD induction. Such results, along with the observation that α1-AR LTD is caused by a persistent decrease in glutamate release, also imply that the presynaptic expression of α1-AR LTD is mediated by retrograde messengers.
Figure 4.
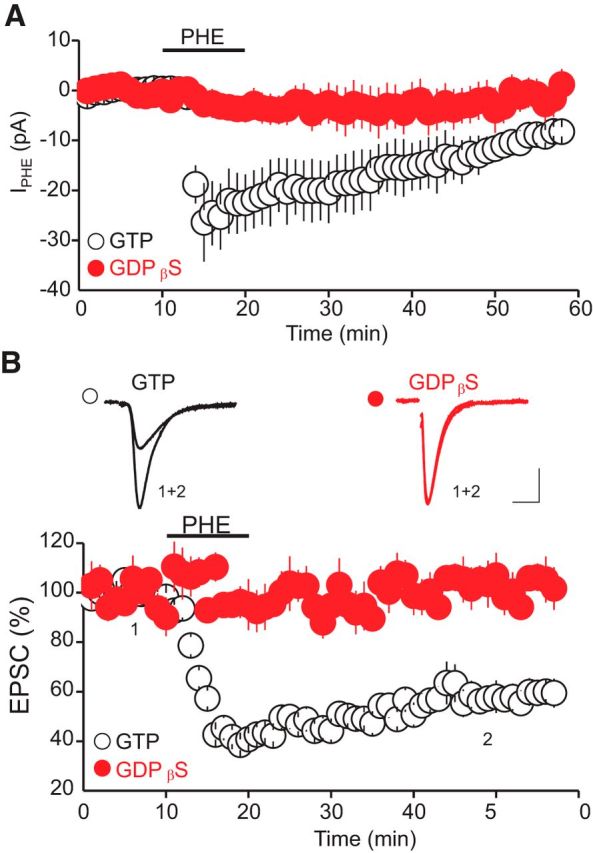
The α1-AR LTD is initiated by the activation of postsynaptic α1-ARs. A, Summary graph of the amplitude of IPHE induced by PHE (100 μm) and recorded with an internal solution containing GTP (red circles, n = 8) or GDPβS (white circles, n = 8). Postsynaptic intracellular application of the G-protein inhibitors GDPβS abolishes the IPHE. B, Blockade of postsynaptic G-protein signaling prevents the induction of the α1-AR LTD. Bottom, Summary graph of the PHE-LTD recorded with an internal solution containing GTP (0.25 mm, white circles, n = 10) or GDPβS (0.25 mm, red circles, n = 8). Top, Superimposed AMPAR-EPSCs (average 30 trials) recorded with GTP, or GDPβS containing internal solution and collected before (1) and during (2) PHE-LTD. Calibration: 50 pA, 10 ms.
Classically, α1-ARs are coupled to the canonical Gq-11/PLCβ signaling cascade. Activation of this signaling cascade elicits the hydrolysis of phosphatidylinositol (4,5) biphosphate (PIP2) and the formation of 1,2 diacyl-glycerol (1,2-DAG) (Cotecchia et al., 1992; Wu et al., 1992, 1995), a major precursor of the eCB 2-arachidonyl-glycerol (2-AG) (Stella et al., 1997). Because stimulation of Gq-11 coupled receptors enhances the synthesis/release of 2-AG (Maejima et al., 2001; Kim et al., 2002; Ohno-Shosaku et al., 2003; Haj-Dahmane and Shen, 2005), which mediates retrograde modulation of synaptic transmission and plasticity via presynaptic CB1 receptors (Katona and Freund, 2012), we hypothesized that the α1-AR LTD in the DRn is most likely mediated by retrograde eCB signaling. To test this hypothesis, we examined the effect of CB1 receptor antagonist AM 251 on the magnitude of α1-AR LTD. We found that treatment of brain slices with AM 251 (3 μm), a manipulation that has been shown to block CB1 receptor-mediated inhibition of glutamate release in the DRn (Haj-Dahmane and Shen, 2005, 2009), strongly reduced the acute depression of AMPAR-EPSCs induced by PHE (100 μm) (control = 41.7 ± 2.9% of baseline; AM 251 = 82.3 ± 7.3% of baseline; p < 0.05, n = 8) and the magnitude of α1-AR LTD (control = 57.9 ± 6.5% of baseline; AM 251 = 89.9 ± 8.9% of baseline; p < 0.05, n = 8; Fig. 5B). In contrast, blockade of CB1 receptors had no significant effect on the amplitude of IPHE (IPHE control = 56.5 ± 8.9 pA; IPHE AM 251 = 53.89 ± 6.8 pA; n = 8, p > 0.05; data not shown), indicating that the inhibition of α1-AR-mediated acute depression of AMPAR-EPSCs and LTD induced by AM 251 could not be attributed to a blockade of α1-AR signaling, but to a blockade of CB1 receptors. These results also suggest that the α1-AR LTD in the DRn is mediated by retrograde eCBs acting through presynaptic CB1 receptors.
Figure 5.
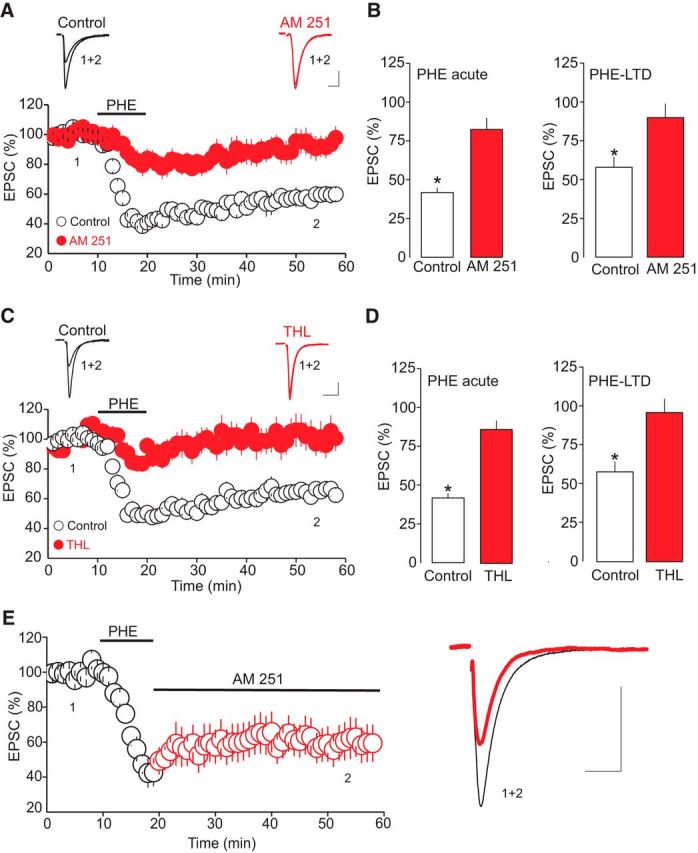
The presynaptic expression of the α1-AR LTD is mediated by 2-AG. A, Blockade of the CB1 receptor with AM 251 markedly reduces both the acute inhibition of AMPAR-EPSCs and the magnitude of the α1-AR LTD. Bottom, Summary graph of the effect of PHE (100 μm) on the amplitude of AMPAR-EPSCs obtained in the control condition (white circles, n = 12) and in the presence of AM 251 (3 μm, filled red circles, n = 8). Top, Superimposed AMPAR-EPSC traces (30 trials) collected before (1), during α1-AR LTD (2), and recorded in the absence (white circles) and presence of AM 251 (filled red circles). B, Summary graphs of the α1-AR-induced acute depression of AMPAR-EPSCs (left) and LTD (right) obtained in the control condition (open bars) and in the presence of AM 251 (filled bars). *p < 0.05. C, Blockade of 2-AG synthesis inhibits both the acute depression of AMPAR-EPSCs and LTD induction by α1-ARs. Bottom, Summary of the effect of PHE (100 μm) on the amplitude of AMPAR-EPSCs obtained in the control condition (white circles, n = 12) and in slices treated with THL (10 μm, filled red circles, n = 8). Top, AMPAR-EPSC traces recorded before (1) and during (2) α1-AR LTD. D, Summary histograms of the α1-AR-induced acute depression of AMPAR-EPSCs (left) and LTD (right) obtained in the control condition (open bars) and in the presence of THL (filled bars). E, The α1-AR LTD is not mediated by a sustained activation of CB1 receptors. Left, The effect of AM 251 (3 μm) applied after PΗΕ administration on the magnitude and time course of the α1-AR LTD. Blockade of CB1 receptors after the initiation of α1-AR LTD did not reverse the LTD (n = 8). Right, Superimposed AMPAR-EPSC traces at time points indicated by number in left graph. Calibration: 50 pA, 10 ms.
To determine whether 2-AG was the eCB messenger that mediated the acute inhibition of AMPAR-EPSCs and the LTD induced by α1-ARs, we explored the effects of inhibiting sn-1-diacylglycerol lipase (DGL-α/β), an enzyme that converts 1,2-DAG into 2-AG, on the α1-AR-induced acute inhibition of AMPAR-EPSCs and LTD. We found that treatment of brain slices with THL (10 μm), a DGL-α/β inhibitor, profoundly reduced the acute depression of AMPAR-EPSCs induced by PHE (100 μm, THL = 86.3 ± 5.5% of baseline, n = 8, p < 0.05). Blockade of a DGL-α/β also abolished the α1-AR-LTD induced by PHE (control = 64.11 ± 3.4% of baseline; THL = 96.8 ± 8.6% of baseline; Fig. 5B; n = 8, p < 0.05). These results suggest that DGL-α/β is required for both the acute depression of AMPAR-EPSCs and the LTD induced by activation of α1-ARs. Importantly, such results also indicate that 2-AG is the most likely retrograde messenger that mediates α1-AR LTD in DRn 5-HT neurons.
We next tested whether a sustained increase in 2-AG release and activation of CB1 receptors are necessary for the maintenance of the α1-AR LTD. To that end, we examined the effect of AM 251 on the duration and magnitude of the α1-AR LTD after it was initiated. As illustrated in Figure 5E, administration of AM 251 (3 μm) after initiation of α1-AR LTD induced by bath application of PHE (100 μm) failed to reverse and reduce the magnitude of the LTD (Fig. 5E; 58.3 ± 8.4% of baseline, p > 0.05, n = 6). Similarly, administration of the DGL-α/β inhibitor THL (10 μm) after initiation of the α1-AR LTD did not alter the time course or the magnitude of the LTD (n = 3; data not shown). Collectively, these results indicate that the maintenance of the α1-AR LTD is not mediated by a sustained increase in tonic 2-AG release and persistent activation of CB1 receptors. Instead, these findings indicate that a transient activation of CB1 receptors triggers a persistent decrease in the probability of glutamate release, which mediates the LTD.
Chronic restraint stress impairs the α1-AR LTD in DRn 5-HT neurons
Exposure to CRS is known to increase noradrenaline release in the DRn, which regulates the behavioral responses to stress, at least in part, via α1-AR signaling (Tanaka et al., 1983; Shimizu et al., 1994). More importantly, behavioral studies have suggested that alterations of α1-AR signaling in the DRn contribute to chronic stress-induced anxiety and depression-like behaviors (Christiansen et al., 2011; Chiba et al., 2012; Kim et al., 2012). However, the impact of chronic stress, including restraint stress on the function of α1-ARs in the DRn, remains unknown. Therefore, we explored the effect of exposure to CRS on the α1-AR-mediated control of the strength and plasticity of glutamate synapses onto DRn 5-HT neurons. To that end, we first examined the impact of CRS on the baseline glutamate release and the overall strength of glutamate synapses. As illustrated in Figure 6A, we found that exposure to CRS did not significantly affect the PPR (PPR control = 1.13 ± 0.03, n = 12; PPR stress = 1.11 ± 0.06; n = 12), indicating that exposure to CRS had no significant effect on the probability of glutamate release in the DRn. Similarly, CRS had no significant effect on the AMPA/NMDA ratio (AMPA/NMDA control = 0.842 ± 0.12, n = 11; AMPA/NMDA stress = 0.796 ± 0.13; Fig. 6B; n = 11), a measure of the strength of glutamate synapses, indicating that CRS did not alter the overall strength of glutamate synapses impinging onto DRn 5-HT neurons. We next examined the impact of CRS on the α1-AR-induced acute depression of AMPR-EPSCs and LTD. Compared with the control condition, we found that CRS exposure significantly reduced the acute depression of AMPAR-EPSCs induced by PHE (100 μm). On average, the amplitude of AMPAR-EPSCs was reduced to 50.4 ± 6.8% and 76.9 ± 4.6% of baseline in control and CRS-exposed rats, respectively (Fig. 6C,D; n = 13, p < 0.05). CRS exposure also significantly reduced the magnitude of α1-AR LTD (Fig. 6C,D; 89.3 ± 9.7% of baseline, p < 0.05, n = 13). Increasing the concentration of PHE to 300 μm still failed to increase the magnitude of the α1-AR LTD (data not shown; n = 4). In contrast to CRS, acute exposure to restraint stress did not significantly affect the magnitude α1-AR LTD (control = 62.5 ± 7.5% of baseline; acute restraint = 65.3 ± 8.5 of baseline p > 0.05 vs control; data not shown). Collectively, these results indicate that exposure to CRS impairs the ability of the α1-ARs to control the strength and plasticity of glutamate synapses onto DRn 5-HT neurons.
Figure 6.

Exposure to CRS has no effect on baseline glutamatergic transmission but profoundly reduces both the α1-AR-induced acute inhibition of AMPAR-EPSCs and the magnitude of α1-AR LTD. A, Exposure to CRS fails to alter the probability of glutamate release. Left, Sample pairs of AMPAR-EPSCs recorded in control and CRS-exposed rats. Right, Summary histogram of the average PPR obtained from control (open bar, n = 12) and CRS-exposed rats (filled bar, n = 12). Calibration: 50 pA, 20 ms. B, Exposure to CRS has no significant effect on the AMPA/NMDA ratio. Left, Superimposed AMPAR-EPSCs and NMDA-EPSCs recorded from putative DRn 5-HT neurons voltage clamped at 50 mV from control (top) and CRS-exposed rats (bottom). Right, Summary graph of the average AMPA/NMDA ratio obtained in control (open bar, n = 11) and CRS-exposed rats (filled bar, n = 11). Calibration: 20 pA, 50 ms. C, Exposure to CRS profoundly reduces the acute inhibition of AMPAR-EPSCs and the α1-AR LTD. Top, Superimposed AMPAR-EPSC traces obtained before (1) and during (2) the LTD in control slices (left) and in slices from CRS-exposed rats (right). Calibration: 50 pA, 10 ms. Bottom, Summary of the time course and magnitude of the inhibition of AMPAR-EPSC amplitude induced by PHE (100 μm) in control (white circles, n = 14) and CRS-exposed rats (red circles, n = 13). D, Summary histograms of the acute effect and LTD induced by PHE (100 μm) obtained in control (open bars) and in CRS-exposed rats (filled bars). CRS significantly reduces the acute depression of AMPAR-EPSC and the LTD. *p < 0.05. E, Summary graph of the average amplitude of IPHE induced by PHE (100 μm) in control (white circles, n = 12) and CRS-exposed rats (red circles, n = 13). Exposure to CRS has no significant effect on the amplitude of IPHE (p > 0.05).
Results from previous studies have reported that chronic exposure to stress or to corticosterone downregulates α-adrenoceptors in the brain areas controlling stress homeostasis (Joëls and de Kloet, 1989; Miyahara et al., 1999). Therefore, it is possible that the CRS-induced impairment of α1-AR LTD could be attributed to an alteration of α1-AR function in the DRn. To test this possibility, we monitored the effect of CRS on the amplitude of the IPHE, a response signaled by the activation of α1-ARs. Surprisingly, we found that exposure to CRS, which strongly inhibited the magnitude of the α1-AR LTD (Fig. 6C; n = 13), failed to alter the amplitude of IPHE (IPHE control = 32.17 ± 3.9 pA; IPHE CRS = 29.22 ± 5.2 pA; Fig. 6D; n = 13). Such results suggest that the CRS-induced inhibition of the α1-AR LTD is unlikely to be attributed to downregulation/desensitization of DRn α1-ARs. However, it remains possible that alterations of downstream signaling cascade from α1-ARs could mediate the CRS-induced inhibition of the α1-AR LTD.
Because the α1-AR LTD is signaled by an increase in eCB synthesis/release and the subsequent activation of presynaptic CB1 receptors, CRS could inhibit the magnitude of the LTD by reducing the α1-AR-driven eCB synthesis or by downregulating presynaptic CB1 receptors. If the CRS-induced inhibition of α1-AR LTD were to be mediated by a reduction in eCB synthesis/release, CRS should have no significant effect on the depression of AMPAR-EPSCs induced by exogenous CB1 receptor agonists. We tested this notion by examining the effect of CRS on the inhibition of AMPA-EPSCs induced by WIN 55212-2, a CB1 receptor agonist (Haj-Dahmane and Shen, 2009). We found that, compared with control, CRS exposure markedly reduced the ability of WIN 55212-2 (10 μm) to inhibit AMPAR-EPSCs. On average, bath application of WIN 55212-2 reduced the amplitude of AMPAR-EPSCs to 46.8 ± 4.5% of baseline (Fig. 7A; n = 13) and 88.89 ± 9.8% of baseline in control and CRS-exposed rats (Fig. 7A; n = 12, p < 0.01, unpaired t test), respectively. Detailed dose–response curves of the inhibition of AMPAR-EPSCs revealed that the efficacy of high (3, 10, and 30 μm), but not low concentrations (0.1 and 1 μm) of WIN 55212-2 was significantly reduced in CRS-exposed rats compared with control (Fig. 7B; n = 8). These results support the idea that the impairment of the α1-AR LTD after CRS exposure can be attributed to functional downregulation of presynaptic CB1 receptors.
Figure 7.
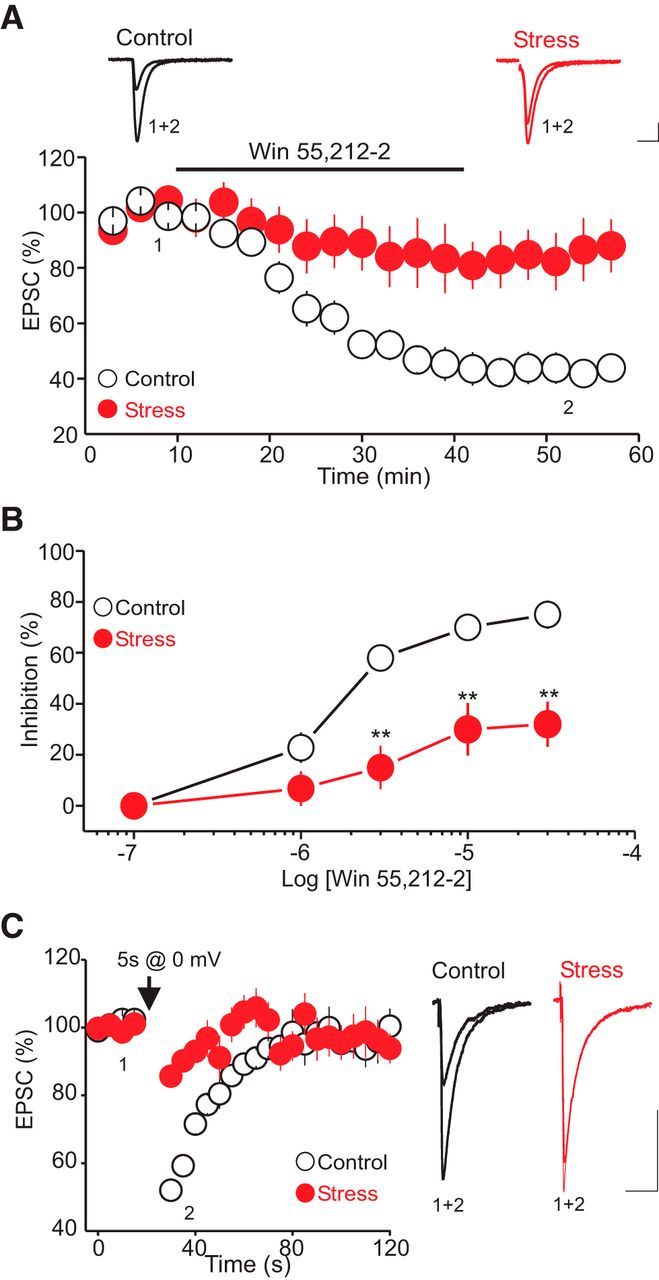
Exposure to CRS impairs the function of presynaptic CB1 receptors in the DRn. A, CRS reduces the ability of presynaptic CB1 receptors to inhibit glutamate release. Top, Superimposed AMPA-EPSCs taken before and during bath application of the CB1 receptor agonist WIN 55212-2 (10 μm) in control (left) and CRS-exposed rats (right). Calibration: 50 pA, 10 ms. Bottom, Summary graph of the inhibition of AMPA-EPSC amplitude induced by WIN 55212-2 (10 μm) in control (white circles, n = 13) and CRS-exposed rats (red circles, n = 10). CRS exposure profoundly reduces the efficacy of WIN 55212-2 in inhibiting the amplitude of AMPAR-EPSCs. B, Dose–response curves of the effect of WIN 55212-2 on AMPAR-EPSCs obtained in control (white circles) and CRS-exposed rats (red circles). **p < 0.01. C, Exposure to CRS profoundly reduces the magnitude of the eCB-mediated DSE. Left, DSE induced in control (white circles, n = 14) and CRS-exposed rats (red circles, n = 12). Right, Superimposed AMPAR-EPSCs traces taken before and during the DSE in putative DRn 5-HT neurons from control and CRS-exposed rats. Calibration: 50 pA, 10 ms.
To further test that a downregulation of presynaptic CB1 receptors mediates the CRS-induced impairment of α1-AR LTD, we also examined the impact of CRS exposure on the magnitude of the eCB-mediated DSE, a CB1 receptor-mediated short-term synaptic plasticity (Haj-Dahmane and Shen, 2009). Consistent with previous reports (Haj-Dahmane and Shen, 2009), in slices from control rats, a brief (5 s) depolarization (from −70 to 0 mV) of DRn 5-HT neurons elicited a robust DSE (DSE control = 53.5 ± 4.7% of baseline, n = 12; Fig. 7C). Remarkably, exposures to CRS profoundly reduced the magnitude of the DSE (DSE stress = 89.9 ± 3.5% of baseline, p < 0.05, n = 12; Fig. 7C). These results, along with the observation that CRS reduces the efficacy of CB1 agonist to inhibit the amplitude of AMPAR-EPSCs, strongly indicate that CRS impairs α1-AR LTD by reducing the function of presynaptic CB1 receptors.
Discussion
The present study shows that activation of α1-ARs elicits LTD of glutamate synapses onto DRn 5-HT neurons. This form of synaptic plasticity is initiated by the activation of postsynaptic α1-ARs but expressed presynaptically by a decrease in glutamate release. The α1-AR LTD is signaled by retrograde eCB messengers acting on presynaptic CB1 receptors. More importantly, we report that exposure to CRS profoundly reduces of the magnitude of α1-AR LTD. The CRS-induced impairment of α1-AR LTD is essentially mediated by a downregulation of presynaptic CB1 receptors. As such, our study reveals a novel cellular mechanism by which noradrenaline controls the function of DRn 5-HT. It also provides the first direct evidence that chronic stress reduces eCB signaling at glutamate synapses of DRn 5-HT neurons, which could have important functional implications for stress-induced maladaptation of the 5-HT system.
Previous studies have examined the regulation of glutamate synapses by α1-ARs in various brain areas (Scanziani et al., 1993; Scheiderer et al., 2004; Choi et al., 2005; McElligott and Winder, 2008; Marzo et al., 2010; McElligot et al., 2010). Generally, these studies have reported that activation of α1-ARs elicits a transient inhibition of glutamatergic transmission. However, in some brain areas, such as the cerebral cortex (e.g., visual cortex and prefrontal cortex), hippocampus, and the bed nucleus of striata terminalis, activation of α1-ARs induces LTD of glutamate synapses (Scheiderer et al., 2004; Choi et al., 2005; McElligott and Winder, 2008; Marzo et al., 2010; McElligot et al., 2010). Depending on the brain area studied, the α1-AR LTD seems to be mediated by different cellular mechanisms. In the hippocampus and cerebral cortex, the α1-AR LTD is mediated by a postsynaptic mechanism that involves α1-AR-induced activation of the extracellular signal regulating kinase (ERK1/2) pathways (Scheiderer et al., 2008; Marzo et al., 2010). Activation of ERK 1/2 induces LTD by reducing the function and/or number of AMPARs. At glutamate synapses of the bed nucleus of striata terminalis, the α1-AR LTD is mediated by a switch of the subunit composition of AMPARs from GluA2-lacking, which exhibit higher unitary conductance and calcium permeability (Kamboj et al., 1995; Dingledine et al., 1999), to GluA2 containing AMPARs (McElligott et al., 2010). In the DRn, the present study shows that the α1-AR LTD is initiated by the activation of postsynaptic α1-ARs but mediated by a decrease in glutamate release induced by retrograde eCB messengers. This cellular mechanism is supported by multiple lines of evidence. First, inhibition of postsynaptic α1-AR signaling with G-protein inhibitors abolishes the LTD. Second, α1-AR LTD is associated with a persistent decrease in glutamate release as indicated by the increase in PPR and CV. Finally, blockade of presynaptic CB1 receptors or inhibition of 2-AG synthesis abolishes the α1-AR LTD. The conclusion that the α1-AR LTD of glutamate synapses onto DRn 5-HT neurons is mediated by 2-AG acting at presynaptic CB1 receptors is consistent with numerous studies showing that activation of Gq/11-coupled receptors, such as Group I metabotropic glutamate receptors (mGluR1/5), M1/M5 muscarinic receptors, and orexin receptors, increase the synthesis/release of 2-AG in various brain areas (Maejima et al., 2001; Kim et al., 2002; Ohno-Shosaku et al., 2003), including the DRn (Haj-Dahmane and Shen, 2005). Generally, Gq/11 coupled receptor-driven 2-AG synthesis and release mediate transient inhibition of excitatory and inhibitory synaptic transmission (Maejima et al., 2001; Kim et al., 2002; Haj-Dahmane and Shen, 2005). However, growing evidence indicates that this mode of 2-AG synthesis/release also mediates the presynaptic form of LTD at glutamate and GABA synapses and, hence, plays an ubiquitous role in regulating synaptic plasticity in the brain (Castillo et al., 2012).
Results from previous studies have reported that chronic exposure to various stressors increases the expression in the DRn of various synaptic proteins, such as synaptosomal-associated protein 25 and synaptic vesicle glycoprotein 2B (Abumaria et al., 2006, 2007), suggesting that chronic stress can induce a long-lasting alteration of synaptic function and plasticity in the DRn. Consistent with this idea, we report that CRS impairs the α1-AR LTD of glutamate synapses onto DRn 5-HT neurons. The alteration of α1-AR-mediated synaptic plasticity in the DRn may represent an important cellular mechanism by which chronic stress can induce a long-lasting alteration of the 5-HT system. The effects of restraint stress on the α1-AR-mediated control of synaptic transmission have also been examined in several other brain areas. In the amygdala, exposure to acute restraint stress combined with tail shock blocks the α1-AR-mediated facilitation of GABA-ergic transmission (Braga et al., 2004). The mechanisms underlying this effect remain unknown. Here, we find that acute exposure to restraint stress has no effect on the ability of α1-ARs to control the function of glutamate synapses. In contrast, exposure to CRS profoundly impairs the α1-AR LTD of glutamate synapses in the DRn by blocking the induction and maintenance of the LTD. Such finding is in agreement with a previous study showing that exposure to CRS, but not to acute restraint stress, also impairs the LTD of glutamate synapses induced by α1-ARs (McElligott et al., 2010) in the basal nucleus of striata terminalis. Collectively, these studies suggest that the impairment of the α1-AR-mediated control of the strength and plasticity of glutamate synapses may represent a common response to chronic stress exposure. Importantly, such alterations of synaptic plasticity may mediate the maladaptive behavioral responses to chronic stress, including depression and anxiety.
An interesting finding of the present study is that exposure to CRS has no significant effect on the amplitude of the α1-AR-induced inward current but reduces the effect of presynaptic CB1 receptors on glutamate release. These results strongly indicate that the CRS-induced impairment of the α1-AR LTD is not mediated by a downregulation of α1-ARs but by a profound reduction of presynaptic CB1 receptor function. However, it remains possible that CRS could also reduce eCB synthesis/release, which may contribute to the impairment of the α1-AR LTD. The conclusion that CRS reduces the function of presynaptic CB1 receptors is consistent with previous studies showing that chronic exposure to various stressors, including CRS, downregulates CB1 receptors and impairs eCB-mediated control of glutamatergic (Rossi et al., 2008; Wang et al., 2010; Reich et al., 2013) and GABAergic synaptic transmission in other brain areas (Wamsteeker et al., 2010; Hu et al., 2011). Importantly, in the nucleus accumbens, exposure to chronic stress has also been shown to block the eCB-mediated LTD induced by mGluR1 at glutamate synapses onto medium spiny neurons (Wang et al., 2010). As in the DRn, the blockade of the mGluR1 LTD is mainly attributed to a reduction of presynaptic CB1 receptor function (Wang et al., 2010). Together, the results of these studies indicate that reduced retrograde eCB signaling (e.g., downregulation of CB1 receptors) may represent a common mechanism by which chronic stress impairs Gq/11-coupled receptor-mediated control of synaptic function and plasticity in the brain.
Although exposure to chronic stress has been shown to impair the function of presynaptic CB1 receptors in various brain areas (Hill et al., 2005; Wang et al., 2010), the precise molecular mechanisms underlying this effect remain unknown. It is well established that exposure to chronic stress increases the circulating levels of corticosterone and noradrenaline (Krugers et al., 2012). Because both of these stress mediators stimulate eCB synthesis and release in the DRn (Wang et al., 2012; present study) and other brain areas (Di et al., 2003), it is tempting to speculate that the high circulating levels of noradrenaline and corticosterone during daily stress lead to chronic increase in eCB release and activation of CB1 receptors, which could induce downregulation of these receptors. Consistent with this idea, results from previous studies have shown that chronic exposure to stress enhances the release of 2-AG in various brain regions (Patel and Hillard, 2008; Patel et al., 2009). More importantly, chronic activation of CB1 receptors with eCBs or exogenous cannabinoids has been shown to reduce the function of presynaptic CB1 receptors (Sim et al., 1996; Breivogel et al., 1999). Thus, it is possible that the CRS-induced functional downregulation of presynaptic CB1 receptors reported in this study could be attributed to an agonist-induced downregulation. However, future studies are required to further test this notion and determine the precise cellular mechanisms underlying the downregulation of presynaptic CB1 receptors.
Extensive work has established that noradrenergic inputs from the locus ceruleus provide a major excitatory drive to the DRn, which is mediated by α1-ARs. Activation of these receptors increases the excitability of DRn 5-HT neurons by inducing membrane depolarization (Aghajanian, 1985; Pan et al., 1994) and reducing the amplitude of after hyperpolarizing potential (Pan et al., 1994). In addition to these excitatory effects, the present study shows that activation of postsynaptic α1-ARs enhances 2-AG release, which in turn reduces the strength of glutamate synapses onto DRn 5-HT neurons (Fig. 8). Combined, these studies indicate that the noradrenergic modulation of 5-HT neurons is more complex than initially thought and that α1-AR signaling in the DRn exerts a bidirectional control on the excitability of 5-HT neurons. The bidirectional control exerted by α1-AR could play an important role in maintaining the activity of 5-HT neurons within desirable range and prevent excessive excitation of DRn 5-HT neurons, especially during heightened arousal (e.g., stress), which is characterized by increased noradrenergic tone (Krugers et al., 2012). As such, the reduction of eCB signaling and the impairment of α1-AR LTD induced by chronic stress may lead to an abnormal increase in the excitability of DRn 5-HT neurons and persistent alteration of central 5-HT transmission. Furthermore, the impairment of eCB signaling in the DRn could mediate, at least in part, some of the behavioral consequences of chronic stress exposure, such as depression-like behaviors. It is noteworthy that pharmacological manipulation that increases eCB signaling has been shown to block chronic stress-induced depression-like behaviors (Zhong et al., 2014).
Figure 8.

A model of α1-ARs mediated regulation of glutamate synapses onto DRn 5-HT neurons. Activation of α1-ARs elicits an increase in the synthesis/release of the eCB messenger 2-AG. The release of 2-AG reduces the strength of glutamate synapses by the activation of presynaptic CB1 receptors. Exposure to CRS impairs α1-AR-mediated depression of glutamate synapses by reducing the function of presynaptic CB1 receptors.
Footnotes
This work was supported by National Institutes of Health Grant MH 078009. We thank Dr. James Rice for reading the manuscript.
The authors declare no competing financial interests.
References
- Abumaria N, Rygula R, Havemann-Reinecke U, Rüther E, Bodemer W, Roos C, Flügge G. Identification of genes regulated by chronic social stress in the rat dorsal raphe nucleus. Cell Mol Neurobiol. 2006;26:145–162. doi: 10.1007/s10571-006-9024-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abumaria N, Rygula R, Hiemke C, Fuchs E, Havemann-Reinecke U, Rüther E, Flügge G. Effect of chronic citalopram on serotonin-related and stress-regulated genes in the dorsal raphe nucleus of the rat. Eur Neuropsychopharmacol. 2007;17:417–429. doi: 10.1016/j.euroneuro.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK. Modulation of a transient outward current in serotoninergic neurons by alpha1-adrenoreceptors. Nature. 1985;315:501–503. doi: 10.1038/315501a0. [DOI] [PubMed] [Google Scholar]
- Amat J, Matus-Amat P, Watkins LR, Maier SF. Escapable and inescapable stress differentially and selectively alter extracellular levels of 5-HT in the ventral hippocampus and dorsal periaqueductal gray of the rat. Brain Res. 1998;797:12–22. doi: 10.1016/S0006-8993(98)00368-0. [DOI] [PubMed] [Google Scholar]
- Baraban JM, Aghajanian GK. Noradrenergic innervation of serotonergic neurons in the dorsal raphe: demonstration by electron microscopic autoradiography. Brain Res. 1981;204:1–11. doi: 10.1016/0006-8993(81)90646-6. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Manion ST, Hough CJ, Li H. Stress impairs α1A adrenoceptor-mediated noradrenergic facilitation of GABAergic transmission in the basolateral amygdala. Neuropsychopharmacology. 2004;29:45–58. doi: 10.1038/sj.npp.1300297. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ, Sim-Selley LJ. Chronic δ-9-tetrahydrocanabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J Neurochem. 1999;73:2447–2459. doi: 10.1046/j.1471-4159.1999.0732447.x. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S, Numakawa T, Ninomiya M, Richards MC, Wakabayashi C, Kunugi H. Chronic restrain stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39:112–119. doi: 10.1016/j.pnpbp.2012.05.018. [DOI] [PubMed] [Google Scholar]
- Choi SY, Chang J, Jiang B, Seol GH, Min SS, Han JS, Shin HS, Gallagher M, Kirkwood A. Multiple receptors coupled to phospholipase C gate long-term depression in visual cortex. J Neurosci. 2005;25:11433–11443. doi: 10.1523/JNEUROSCI.4084-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen SH, Olesen MV, Wörtwein G, Woldbye DP. Fluoxetine reverts chronic restraint stress-induced depression like behaviour and increases neuropeptides Y and galanin expression in mice. Behav Brain Res. 2011;216:585–591. doi: 10.1016/j.bbr.2010.08.044. [DOI] [PubMed] [Google Scholar]
- Cotecchia S, Ostrowski J, Kjelsberg MA, Caron MG, Lefkowitz RJ. Discrete amino acid sequences of the α1 -adrenergic receptor determine the selectivity of coupling to phosphatidylinositol hydrolysis. J Biol Chem. 1992;267:1633–1639. [PubMed] [Google Scholar]
- Dahlström A, Fuxe K. Evidence for the existence of monoamine containing neurons in the central nervous system: I. Demonstration of monoamines in cell bodies of brain stem neurons. Acta Physiol Scand. 1964;62(Suppl 232):1–55. [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Doze VA, Handel EM, Jensen KA, Darsie B, Luger EJ, Haselton JR, Talbot JN, Rorabaugh BR. α1A- and α1B-adrenergic receptors differentially modulate antidepressant-like behavior in the mouse. Brain Res. 2009;285:148–157. doi: 10.1016/j.brainres.2009.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahn RE, Will MJ, Hammack SE, Maswood S, McQueen MB, Watkins LR, Maier SF. Activation of serotonin-immunoreactive cells in the dorsal raphe nucleus in rats exposed to an uncontrollable stressor. Brain Res. 1999;826:35–43. doi: 10.1016/S0006-8993(99)01208-1. [DOI] [PubMed] [Google Scholar]
- Grahn RE, Hammack SE, Will MJ, O'Connor KA, Deak T, Sparks PD, Watkins LR, Maier SF. Blockade of alpha1 adrenoreceptors in the dorsal raphe nucleus prevents enhanced conditioned fear and impaired escape performance following uncontrollable stressor exposure in rats. Behav Brain Res. 2002;134:387–392. doi: 10.1016/S0166-4328(02)00061-X. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S. D2-like dopamine receptor activation excites rat dorsal raphe 5-HT neurons in vitro. Eur J Neurosci. 2001;14:125–134. doi: 10.1046/j.0953-816x.2001.01616.x. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Shen RY. The wake-promoting peptide orexin-B inhibits glutamatergic transmission to dorsal raphe nucleus serotonin neurons through retrograde endocannabinoid signaling. J Neurosci. 2005;25:896–905. doi: 10.1523/JNEUROSCI.3258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Shen RY. Endocannabinoids suppress excitatory synaptic transmission to dorsal raphe serotonin neurons through the activation of presynaptic CB1 receptors. J Pharmacol Exp Ther. 2009;331:186–196. doi: 10.1124/jpet.109.153858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ, Gorzalka BB. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 2005;30:508–515. doi: 10.1038/sj.npp.1300601. [DOI] [PubMed] [Google Scholar]
- Hu W, Zhang M, Czéh B, Zhang W, Flügge G. Chronic restraint stress impairs endocannabinoid mediated suppression of GABAergic signaling in the hippocampus of adult rats. Brain Res Bull. 2011;85:374–379. doi: 10.1016/j.brainresbull.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, de Kloet ER. Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science. 1989;245:1502–1505. doi: 10.1126/science.2781292. [DOI] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainite receptors. J Physiol. 1995;15:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Kwon HJ, Baek IS, Han PL. Repeated short-term (2hX14d) emotional stress induces lasting depression-like behavior in mice. Exp Neurobiol. 2012;21:16–22. doi: 10.5607/en.2012.21.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugers HJ, Karst H, Joels M. Interactions between noradrenaline and corticosteroids in the brain: from electrical activity to cognitive performance. Front Cell Neurosci. 2012;6:15. doi: 10.3389/fncel.2012.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behavior and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/S0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Maier SF, Watkins LR. Stressor controllability and learned helplessness: the role of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neurosci Biobehav Rev. 2005;29:829–841. doi: 10.1016/j.neubiorev.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Maier SF, Kalman BA, Grahn RE. Chlordiazepoxide microinjected into the region of the dorsal raphe nucleus eliminates the interference with escape responding produced by inescapable shock or escape testing. Behav Neurosci. 1994;108:121–130. doi: 10.1037/0735-7044.108.1.121. [DOI] [PubMed] [Google Scholar]
- Maier SF, Grahn RE, Watkins LR. 8-OH-DPAT microinjection in the region of the dorsal raphe nucleus blocks and reverses the enhancement of fear conditioning and interference with escape produced by exposure to inescapable shock. Behav Neurosci. 1995;109:404–412. doi: 10.1037/0735-7044.109.3.404. [DOI] [PubMed] [Google Scholar]
- Marzo A, Bai J, Caboche J, Vanhoutte P, Otani S. Cellular mechanisms of long-term depression induced by noradrenaline in rat prefrontal neurons. Neuroscience. 2010;169:74–86. doi: 10.1016/j.neuroscience.2010.04.046. [DOI] [PubMed] [Google Scholar]
- Maswood S, Barter JE, Watkins LR, Maier SF. Exposure to inescapable but not escapable shock increases extracellular levels of 5-HT in the dorsal raphe nucleus of the rat. Brain Res. 1998;786:115–120. doi: 10.1016/S0006-8993(97)01313-9. [DOI] [PubMed] [Google Scholar]
- McElligott ZA, Winder DG. α1-Adrenergic receptor-induced heterosynaptic long-term depression in the bed nucleus of the striata terminalis is disrupted in mouse models of affective disorders. Neuropsychopharmacology. 2008;33:2313–2323. doi: 10.1038/sj.npp.1301635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElligott ZA, Klug JR, Nobis WP, Patel S, Grueter BA, Kash TL, Winder DG. Distinct forms of Gq-receptor-dependent plasticity of excitatory transmission in the BNST are differentially affected by stress. Proc Natl Acad Sci U S A. 2010;107:2271–2276. doi: 10.1073/pnas.0905568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara S, Komori T, Fujiwara R, Shizuya K, Yamamoto M, Ohmori M, Okazaki Y. Effects of single and repeated stress on the expression of mRNA for alpha 1-adrenoceptors in the rat hypothalamus and midbrain. Eur J Pharmacol. 1999;379:111–114. doi: 10.1016/S0014-2999(99)00498-7. [DOI] [PubMed] [Google Scholar]
- Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S, Petre CO. Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1214–1224. doi: 10.1016/j.pnpbp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Matsui M, Fukudome Y, Shosaku J, Tsubokawa H, Taketo MM, Manabe T, Kano M. Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoids signaling in the hippocampus. Eur J Neurosci. 2003;18:109–116. doi: 10.1046/j.1460-9568.2003.02732.x. [DOI] [PubMed] [Google Scholar]
- O'Leary OF, Bechtholt AJ, Crowley JJ, Valentino RJ, Lucki I. The role of noradrenergic tone in the dorsal raphe nucleus of the mouse in the acute behavioral effects of antidepressant drugs. Eur Neuropsychopharmacol. 2007;17:215–226. doi: 10.1016/j.euroneuro.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Grudt TJ, Williams JT. Alpha1-adrenoceptors in rat dorsal raphe neurons: regulation of two potassium conductances. J Physiol. 1994;478:437–447. doi: 10.1113/jphysiol.1994.sp020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Hillard CJ. Adaptations in endocannabinoid signaling in response to repeated homotypic stress: a novel mechanism for stress habituation. Eur J Neurosci. 2008;27:2821–2829. doi: 10.1111/j.1460-9568.2008.06266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG. Repeated homotypic stress elevated 2-arachidonylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34:2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov T, Krukoff TL, Jhamandas JH. Chemically defined collateral projections from the pons to the central nucleus of the amygdale and hypothalamic paraventricular nucleus in the rat. Cell Tissue Res. 1994;277:289–295. doi: 10.1007/BF00327776. [DOI] [PubMed] [Google Scholar]
- Reich CG, Mihalik GR, Iskander AN, Seckler JC, Weiss MS. Adolescent chronic mild stress alters hippocampal CB1 receptor-mediated excitatory neurotransmission and plasticity. Neuroscience. 2013;253:444–454. doi: 10.1016/j.neuroscience.2013.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S, De Chiara V, Musella A, Kusayanagi H, Mataluni G, Bernardi G, Usiello A, Centonze D. Chronic psychoemotional stress impairs cannabinoid-receptor-mediated control GABA transmission in the striatum. J Neurosci. 2008;28:7284–7292. doi: 10.1523/JNEUROSCI.5346-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gähwiler BH, Thompson SM. Presynaptic inhibition of excitatory synaptic transmission mediated by alpha adrenergic receptors in area CA3 of the rat hippocampus in vitro. J Neurosci. 1993;13:5393–5401. doi: 10.1523/JNEUROSCI.13-12-05393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiderer CL, Dobrunz LE, McMahon LL. Novel form of long-term synaptic depression in rat hippocampus induced by activation of alpha 1 adrenergic receptors. J Neurophysiol. 2004;91:1071–1077. doi: 10.1152/jn.00420.2003. [DOI] [PubMed] [Google Scholar]
- Scheiderer CL, Smith CC, McCutchen E, McCoy PA, Thacker EE, Kolasa K, Dobrunz LE, McMahon LL. Coactivation of M(1)muscarinic and alpha1 adrenergic receptors stimulates extracellular signal-regulated protein kinase and induces long-term depression at Ca3-Ca1 synapses in rat hippocampus. J Neurosci. 2008;28:5350–5358. doi: 10.1523/JNEUROSCI.5058-06.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Tanaka M, Yokoo H, Gondoh Y, Mizoguchi K, Matsuguchi N, Tsuda A. Differential changes in rat brain noradrenaline turnover produced by continuous and intermittent restraint stress. Pharmacol Biochem Behav. 1994;49:905–909. doi: 10.1016/0091-3057(94)90242-9. [DOI] [PubMed] [Google Scholar]
- Sim LJ, Hampson RE, Deadwyler SA, Childers SR. Effects of chronic treatment with δ-9-tetrahydrocannabinol on cannabinoid-stimulated [35S] GTPγS autoradiography in rat brain. J Neurosci. 1996;16:8057–8066. doi: 10.1523/JNEUROSCI.16-24-08057.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwick SM, Vythilingam M, Charney DS. The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu Rev Clin Psychol. 2005;1:255–291. doi: 10.1146/annurev.clinpsy.1.102803.143948. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Stone EA, Quartermain D, Lin Y, Lehmann ML. Central α1-adrenergic system in behavioral activity and depression. Biochem Pharmacol. 2007;73:1063–1075. doi: 10.1016/j.bcp.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kohno Y, Nakagawa R, Ida Y, Takeda S, Nagasaki N, Noda Y. Regional characteristics of stress-induced increases in brain noradrenaline release in rats. Pharmacol Biochem Behav. 1983;19:543–547. doi: 10.1016/0091-3057(83)90132-6. [DOI] [PubMed] [Google Scholar]
- Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30:11188–11196. doi: 10.1523/JNEUROSCI.1046-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shen RY, Haj-Dahmane S. Endocannabinoids mediate the glucocorticoids-induced inhibition of excitatory synaptic transmission to dorsal raphe serotonin neurons. J Physiol. 2012;590:5795–5808. doi: 10.1113/jphysiol.2012.238659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Sun D, Pan B, Roberts CJ, Sun X, Hillard CJ, Liu QS. Deficiency in endocannabinoid signaling in the nucleus accumbens induced by chronic unpredictable stress. Neuropsychopharmacology. 2010;35:2249–2261. doi: 10.1038/npp.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Katz A, Lee CH, Simon MI. Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem. 1992;267:25798–25802. [PubMed] [Google Scholar]
- Wu D, Jiang H, Simon MI. Different alpha 1-adrenergic receptor sequences required for activating different G alpha subunits of Gq class of G proteins. J Biol Chem. 1995;270:9828–9832. doi: 10.1074/jbc.270.17.9828. [DOI] [PubMed] [Google Scholar]
- Zhong P, Wang W, Pan B, Liu X, Zhang Z, Long JZ, Zhang HT, Cravatt BF, Liu QS. Monoacylglycol lipase inhibition blocks chronic stress-induced depression-like behaviors via activation of mTOR signaling. Neuropsychopharmacology. 2014;39:1763–1776. doi: 10.1038/npp.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]