

Abstract
Pathogen-host protein-protein interaction (PPI) plays an important role in revealing the underlying pathogenesis of viruses and bacteria. The need of rapidly mapping proteome-wide pathogen-host interactome opens avenues for and imposes burdens on computational modeling. For Salmonella typhimurium, only 62 interactions with human proteins are reported to date, and the computational modeling based on such a small training data is prone to yield model overfitting. In this work, we propose a multi-instance transfer learning method to reconstruct the proteome-wide Salmonella-human PPI networks, wherein the training data is augmented by homolog knowledge transfer in the form of independent homolog instances. We use AdaBoost instance reweighting to counteract the noise from homolog instances, and deliberately design three experimental settings to validate the assumption that the homolog instances are effective to address the problems of data scarcity and data unavailability. The experimental results show that the proposed method outperforms the existing models and some predictions are validated by the findings from recent literature. Lastly, we conduct gene ontology based clustering analysis of the predicted networks to provide insights into the pathogenesis of Salmonella.
Introduction
Pathogen-host protein-protein interaction (PPI) plays an important role in revealing the molecular-level dynamic mechanism of microbial pathogenesis. Fast and accurate reconstruction of proteome-wide pathogen-host PPI networks is essential to reveal the host cellular processes that pathogen proteins may interfere with. In recent years, high throughput experimental techniques have drastically accumulated much knowledge about intra-species PPI networks, though noisy and far incomplete [1], [2]. Accordingly the majority of computational methods have been developed as the complement of labor-intensive biological experiments for intra-species PPI network reconstruction, e.g. yeast PPI network [3], Arabidopsis thaliana PPI network [4], human PPI network [5], etc. However, the current host-pathogen PPI networks are comparatively much smaller. The latest HIV-human PPI database [6] contains about 3,638 interactions, the P.falciparum-H.sapiens PPI dataset [7] contains about 1,112 interactions, and the small Salmonella-human PPI data [8] contains only 62 interactions. At present there are very few computational methods developed for pathogen-host PPI networks reconstruction, e.g. HIV-human PPI prediction [9]–[13], P.falciparum-H.sapiens PPI prediction [14] and Salmonella-human PPI prediction [15]–[17]. To improve the predictive performance, most of the reported methods simultaneously leverage a catalog of biological feature information (see Table 1).
Table 1. Summary of feature information extracted from literature.
| Integration of feature information | Literature |
| sequence k-mer, interlog, gene ontology, metabolic pathways | [7] |
| binding motif, gene expression profile, gene ontology, sequence similarity, post-translational modification, tissue distribution, PPI network topology | [9], [10] |
| protein domain profile, sequence k-mer | [11] |
| structural similarity | [12] |
| protein domain profile, gene expression, gene ontology, gene co-expression | [14] |
Salmonella typhimurium is a facultative intracellular pathogen that causes a variety of diseases from acute gastroenteritis to systemic infection. After invasion into the lumen of host small intestine, Salmonella secretes effectors that interact with the host cellular proteins to ensure its survival in the host cellular environment and gain control of the host immune response [18]. To gain more insight into the inflammation/immune signaling pathways that Salmonella induced or interfered with, we need to fast and accurately reconstruct the complete Salmonella-human PPI networks. Unfortunately, the current experimentally derived Salmonella-human PPI network contains only 62 interactions [8], much smaller than the HIV-human PPI network [6] and the P.falciparum-H.sapiens PPI network [7]. As a fast complement to experimental techniques, computational modeling can accelerate the reconstruction of Salmonella-human PPI networks at low cost. To our knowledge, only a few computational methods have been developed to date for Salmonella-human PPI prediction [15]–[17]. Schlekera et al. [15] used protein sequence similarity and protein domain similarity to predict Salmonella-human PPIs. Kshirsagar et al. proposed two machine learning methods to predict Salmonella-human PPIs [16], [17], wherein the random forest method [16] imputed the missing feature information of gene ontology and gene expression, and the multi-task learning method [17] proposed DC optimization to integrate the feature information of gene ontology, gene expression and pathways. Except the similarity based method [15], the other two methods both adopt data integration to improve the model performance. Data integration is a popular method to enrich the abundance of feature information, but it has two major disadvantages: (1) aggregating more features without augmenting the size of training data is prone to increase the risk of model overfitting on small data; (2) integration of multiple aspects of feature information poses more demanding data constraints on the computational modeling. If the required feature information is not available for the proteins to be predicted, the data integration methods [8]–[11], [14], [16]–[17] will fail to work. Even for those methods that exploit only one type of feature information, e.g. protein structural similarity [12], gene ontology [14], etc., the problem of data unavailability should also be properly addressed. Most of the types of effective feature information listed in Table 1 are derived from costly experiments and are likely to be not available for some proteins. Thus we need to deliberately consider effective substitution of missing feature information and design proper experimental setting to validate the feature substitution. Kshirsagar et al. [16] conducted explicit substitution for the missing feature information of gene ontology and gene expression, but did not explicitly estimate the model performance in the case of feature information substitution. As compared to non-sequence feature information, protein sequence is cheap to obtain and imposes the least demanding data constraints on computational modeling. However, it has been reported that protein sequence alone is not sufficient to train a satisfactory model for PPI prediction [19].
In this work, we propose a multi-instance transfer learning method to reconstruct the proteome-wide Salmonella-human PPI networks. In the method, gene ontology (GO) is used as discriminative features to represent proteins. Due to the incompleteness and scarcity of gene ontology knowledge, we treat the homolog GO information (the aggregated GO information from the homologs) as independent homolog instance to augment or substitute for the target instances (the GO information from the protein itself). The potential noise from homologs is counteracted by AdaBoost instances reweighting algorithm [20], [21]. To validate the effectiveness of the method, we design the following three experimental settings: (1) Single Instance Learning as the baseline model that conducts no homolog knowledge transfer; (2) Multi-instance Learning Novel where the training data are represented with target instances and homolog instances, while the test data are represented with homolog instances only; (3) Multi-instance Learning where both the training data and the test data are represented with target instances and homolog instances. Last, we use the proposed method to reconstruct the proteome-wide Salmonella-human PPI networks, based on which we further conduct gene ontology based clustering analysis to provide valuable cues for further biomedical research.
Materials and Methods
Data and materials
Schleker et al. [8] experimentally derived 62 interactions between 25 Salmonella proteins and 51 human proteins, based on which Kshirsagar et al. [16], [17] developed two machine learning methods for Salmonella-human PPI prediction. PPI prediction is generally treated as a problem of two-class classification where the PPIs are treated as positive data and a negative data is needed for computational modeling. At present the experimental negative data is hardly available and the common practice to generate negative data is random sampling. Random sampling is based on the assumption that the expected number of negatives (non-interacting protein pairs) is several orders of magnitude higher than the number of positives (interacting protein pairs) [22], such that the negative space is randomly sampled with larger probability than the positive space. The human proteins for negative data sampling are taken from the latest SwissProt database [23]. Besides the way of negative data sampling, the second problem is to determine the ratio of positive data to negative data. Here we adopt 1∶1 ratio instead of highly skewed ratio like 1∶100 [16], [17] based on the two points: (1) it is hard to simulate the true ratio of positive data to negative data and simulation of the ratio makes little sense to computational modeling; (2) simply pooling so large a negative data is prone to yield an extremely unbalanced training data and thus yields a highly biased model.
To validate how well the proposed model generalizes to unseen data, we further need to construct a validation set from recent literature. We find 18 Salmonella-human PPIs in [18] and two novel interactions (SspH2, SGT1) & (SspH2, Nod1) in [24]. After excluding the non-protein interactions (e.g. cholesterol, inositol phospates) and the interactions that have been collected in [8], we obtain 7 novel interactions as validation set.
Transfer learning
As compared to traditional supervised learning, transfer learning focuses on useful knowledge/information transfer across related domains that are heterogeneously subjected to distinct statistical distributions [25]. One major merit of transfer learning is that there is no need to make the assumption of independent and identical distribution (iid) between target domain and auxiliary domain. Such the relaxation opens up wide avenues for transfer learning in the field of biological data analysis. In recent years, many sophisticated machine learning methods have been developed to exploit the auxiliary data for useful biological information transfer [26], [27], [28].
In this work, the homolog knowledge is exploited to make up for data scarcity as well as to address the concern of data unavailability. Unlike computing individual kernel matrices in [26], [27] and training individual classifiers in [28], the homolog knowledge transfer is conducted here by means of independent homolog instances under AdaBoost learning framework [20], [21]. The merit is that the independent homolog instance is used to augment and enhance the target instance, and especially substitute for the target instance when the required feature information is not available. Meanwhile the potential of negative knowledge transfer by homolog instances can be attenuated by AdaBoost instance reweighting algorithm[20], [21].
GO feature construction
The homologs for each protein are extracted from SwissProt 57.3 database [23] using PSI-BLast
[29]. We choose default PSI-BLast parameters setting (E-value = 10) to enlarge the coverage of homologs and GO terms. The GO terms are extracted from GOA database [30] (114 Release, as of 28 November, 2012). For each protein i, there are two sets of GO terms, one set contains the GO terms from homologs denoted as homolog set
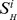 , and the other set contains the GO terms from the protein itself denoted as target set
, and the other set contains the GO terms from the protein itself denoted as target set
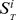 . Here the term target is used to denote the protein itself (comparative to homolog) instead of the pathogen targeted protein. Based on the denotations, two feature vectors for each PPI pair (
. Here the term target is used to denote the protein itself (comparative to homolog) instead of the pathogen targeted protein. Based on the denotations, two feature vectors for each PPI pair (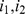 ) are formally defined as follows:
) are formally defined as follows:
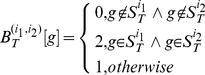 |
(1) |
 |
(2) |
where  denotes component g of the target instance
denotes component g of the target instance
 and
and  denotes component g of the homolog instance
denotes component g of the homolog instance
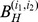 . Formula (1) and formula (2) mean that if the interacting proteins pair shares the same GO term g, the corresponding component in the feature vector
. Formula (1) and formula (2) mean that if the interacting proteins pair shares the same GO term g, the corresponding component in the feature vector 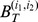 or
or 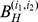 is set 2; if neither protein in the protein pair possesses the GO term g, the value is set 0; otherwise the value is set 1. The definition is symmetrical, i.e., the protein pair (
is set 2; if neither protein in the protein pair possesses the GO term g, the value is set 0; otherwise the value is set 1. The definition is symmetrical, i.e., the protein pair (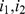 ) and the protein pair (
) and the protein pair (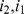 ) have identical feature representation.
) have identical feature representation.
Multi-instance transfer learning
In the scenario of traditional supervised learning, each data point is represented by only one instance, but only one instance may not be sufficient to depict a complex object in most cases. For example, as biological macromolecules protein molecule constantly changes spatial conformations and DNA molecule temporally changes expression levels. Full depiction of the temporal and spatial information needs more than one instance. For another example, evolutionary information is usually used for us to understand the molecular functions of novel proteins or the interactions between orthologs (interlog) [7]. In the case that we can not mingle the information of the protein itself with the information of the homologs, multi-instance representation is a good choice.
In this work, we depict each protein with two instances, the target instance and the homolog instance, The target instance is used to represent the GO information of the protein itself and the homolog instance is used to capture the evolutionary information of the target protein. Besides enriching the feature information of target instance, the homolog instance serves the second purpose of substituting the target instance when the target GO information is not available. We can see that the homolog knowledge transfer by means of independent homolog instances is a novel way to simultaneously solve the problems of data scarcity and data unavailability. Despite the merits, homolog instances may carry a certain level of noise from evolutionary divergence and the noise probably does harm to the model performance. For the reason, we need to choose noise-resistant machine learning methods to counteract the noise contained in the homolog instances. To our knowledge, AdaBoost is an empirically established and theoretically proven machine learning method that boosts an ensemble of weak learners by instances reweighting [20], [21], [31]. It has been theoretically proven that by means of regularization technique, multiple rounds of instances reweighting help AdaBoost to achieve maximum margin between two-class hyper-planes [31]. The regularization technique penalizes the noise/outlier at the cost of high training error to achieve low generalization error. In this work, we adopt the latest variant Modest AdaBoost [21] that softens the weight distributions between easy-to-classify instances and the hard-to-classify instances. For completeness, Modest AdaBoost [21] is briefly described as follows:
Given training instances
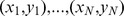 , initialize instance weights
, initialize instance weights 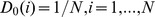 ;
;-
For m = 1,…, M and while
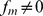
-
Use distribution
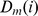 and weighted least squares to train weak classifier:
and weighted least squares to train weak classifier: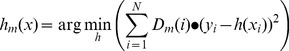
(3) -
Compute the inverted distribution
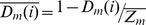
(4) -
Compute
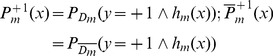
(5) 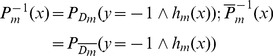
(6) -
Set
Update the weight distributions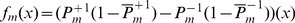
(7) 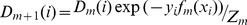
(8)
-
-
Construct the final classifier
where M denotes the number of iterations in the training,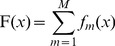
(9) 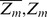 are normalizing coefficients that are chosen to satisfy
are normalizing coefficients that are chosen to satisfy 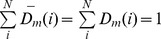 ,
, 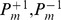 denote the probability that the weak classifier
denote the probability that the weak classifier 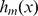 assigns label
assigns label 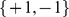 to the instance
to the instance  under the weight distribution
under the weight distribution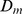 , and similarly
, and similarly 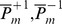 denote the probability under the inverted weight distribution
denote the probability under the inverted weight distribution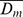 . Formula (8) suggests that the weight increases (
. Formula (8) suggests that the weight increases (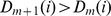 ) for those misclassified instances (
) for those misclassified instances (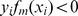 ). The weight distribution
). The weight distribution 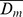 inverted from
inverted from 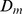 conversely assigns higher weights to those correctly-classified instances. We can see that
conversely assigns higher weights to those correctly-classified instances. We can see that 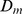 pays more attention to those easy-to-classify instances while
pays more attention to those easy-to-classify instances while 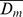 pays more attention to those hard-to-classify instances. Modest AdaBoost made a compromise between the two weight distributions to make soft the decision function
pays more attention to those hard-to-classify instances. Modest AdaBoost made a compromise between the two weight distributions to make soft the decision function 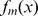 (see formula (7)).
(see formula (7)).
In the test phase, each test protein pair (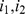 ) is represented by two instances, the target instance
) is represented by two instances, the target instance
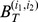 and the homolog instance
and the homolog instance
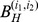 . The decision committee
. The decision committee 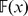 as defined in Formula (9) yields two outputs
as defined in Formula (9) yields two outputs 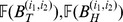 for the two instances (
for the two instances (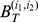 ,
,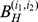 ). In multi-instance AdaBoost, the final decision value for the protein pair (
). In multi-instance AdaBoost, the final decision value for the protein pair (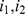 ) is defined as follows:
) is defined as follows:
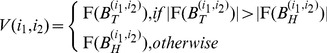 |
(10) |
where  denotes the absolute value. The final class label for the protein pair (
denotes the absolute value. The final class label for the protein pair (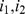 ) is defined as follows:
) is defined as follows:
| (11) |
For those positive predictions, the decision values are further normalized to measure the confidence level of prediction:
| (12) |
where 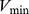 denotes the minimum decision value and
denotes the minimum decision value and 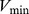 denotes the maximum decision value.
denotes the maximum decision value.
Model evaluation and model selection
To validate the effectiveness of homolog knowledge transfer by means of independent homolog instances, we design three experimental settings: (1) Single Instance Learning that does not consider homolog knowledge transfer; (2) Multi-instance Learning Novel where the training data point is represented with target instance and homolog instance, while the test data point is represented with the homolog instance only.; (3) Multi-instance Learning where both the training data point and the test data point is represented with target instance and homolog instance. The experimental setting (2) is explicitly designed to estimate the model robustness against data unavailability, and the experimental setting (3) is designed to validate the assumption that the homolog instance is effective to augment the training data and thus to solve the problem of data scarcity for Salmonella-human PPI prediction.
As regards with Modest AdaBoost [21], there are two hyper-parameters to be empirically determined, one parameter M is the rounds of training, and the other parameter is the base learner. Here M is chosen within {50, 100, 150, 200, 250, 300, 350} and the base learner is a decision tree with the number of tree splits chosen within {1, 2, 3, 4, 5, 6, 7, 8, 9, 10}. The model performance is estimated by 10-fold cross validation using the following performance metrics: ROC-AUC (AUC of Receiver Operating Characteristic), PR-AUC (AUC of Precision recall curve), SP (Specificity), SE (Sensitivity) and MCC (Matthews correlation coefficient). We first derive several intermediate variables from confusion matrix M as defined in formula (13), and then we calculate SP, SE and MCC for each label (SPl, SEl and MCCl) as defined in formula (14), based on which to further calculate the overall accuracy (Acc) and the overall MCC (MCC) as defined in formula (15).
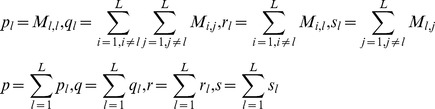 |
(13) |
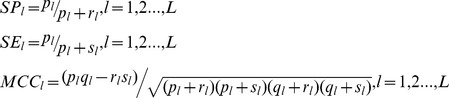 |
(14) |
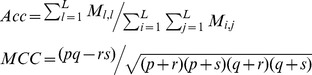 |
(15) |
where the confusion matrix 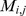 records the counts that class
records the counts that class  are classified to class
are classified to class 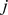 and L denotes the number of class labels. The AUC metric is calculated on the basis of the decision values defined by formula (10). For comparison with the existing methods, we also report the F1 score defined as follows:
and L denotes the number of class labels. The AUC metric is calculated on the basis of the decision values defined by formula (10). For comparison with the existing methods, we also report the F1 score defined as follows:
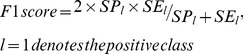 |
(16) |
Results
Model performance
Cross validation performance evaluation on benchmark data
We conduct 10-fold cross validation to validate the effectiveness of homolog knowledge transfer by means of independent homolog instances. The ROC curves for the three experimental settings are illustrated in Figure 1. From Figure 1, we can see that both the experimental setting Multi-instance Learning and the experimental setting Multi-instance Learning Novel outperform the baseline experimental setting Single Instance Learning, with ROC-AUC scores equal to 0.8335, 0.8176 and 0.8003, respectively. Besides ROC curve, precision-recall curve (PR curve) is another performance metric that is often used to measure the performance of two-class classification, especially in the scenario of highly skewed (extremely unbalanced) training data [32]. For comprehensive study, the PR curves for the three experimental settings are plot in Figure 2, with PR-AUC scores equal to 0.8678, 0.8369 and 0.8325, respectively. From Figure 1 and Figure 2, we can see that the homolog knowledge transfer by means of independent homolog instances does improve the model performance (Multi-instance Learning versus Single Instance Learning), and the homolog instances can be treated as effective substitute when the required feature information is not available (Multi-instance Learning Novel versus Single Instance Learning).
Figure 1. ROC curves for the three experimental settings (Multi-instance Learning, Multi-instance Learning Novel, Single Instance Learning).

Figure 2. Precision-Recall curves for the three experimental settings (Multi-instance Learning, Multi-instance Learning Novel, Single Instance Learning).

The ROC Curve and the PR Curve focus on the positive class (interaction), paying little attention to the negative class (non-interaction). In this work, we comprehensively survey the performance on both he positive class and the negative class, and use MCC to measure the predictive bias. The performance metrics in terms of SP, SE and MCC are given in Table 2. From Table 2, we can see that the SE, SP and MCC values show no significant variance between the positive class and the negative class in the three experimental settings, except the SE values (positive class: 0.8065, negative class: 0.6935) in the setting Multi-instance Learning Novel. The MCC values on the two classes suggest that there is little predictive bias. As compared to ROC-AUC scores and PR-AUC scores, the performance metrics of overall accuracy and overall MCC shows obvious performance increase between Multi-instance Learning and Single Instance Learning (overall accuracy 78.23% versus 70.97%; overall MCC 0.6306 versus 0.5260), so does it between Multi-instance Learning Novel and Single Instance Learning (overall accuracy 75% versus 70.97%; overall MCC 0.5833 versus 0.5260). The results once again suggest that the homolog instances are effective to augment the training data and substitute for the missing target GO information. The very small performance difference between Multi-instance Learning and Multi-instance Learning Novel further verifies the effectiveness of homolog instances.
Table 2. Model performance estimation by 10-fold cross validation.
| Multi-instance transfer learning | Multi-instance Learning | Multi-instance Learning Novel | Single Instance Learning | ||||||||
| SP | SE | MCC | SP | SE | MCC | SP | SE | MCC | |||
| Positive | 0.7692 | 0.8065 | 0.6338 | 0.7246 | 0.8065 | 0.5936 | 0.7031 | 0.7258 | 0.5290 | ||
| Negative | 0.7966 | 0.7581 | 0.6284 | 0.7818 | 0.6935 | 0.5780 | 0.7167 | 0.6935 | 0.5234 | ||
| Accuracy | 78.23% | 75% | 70.97% | ||||||||
| MCC | 0.6306 | 0.5833 | 0.5260 | ||||||||
| ROC-AUC | 0.8335 | 0.8176 | 0.8003 | ||||||||
| PR-AUC | 0.8678 | 0.8369 | 0.8325 | ||||||||
| F1 Score | 0.80 | 0.76 | 0.71 | ||||||||
| *Random forest [16] | SP | SE | |||||||||
| Positive | 0.817 | 0.407 | |||||||||
| F1 Score | 0.52 | ||||||||||
| *Multi-task learning [17] | F1 Score | 0.758 | |||||||||
Note: * denotes the existing models.
The F1 score defined in formula (16) takes into account SP and SE on the positive class. As shown in Table 2, the three experimental settings Multi-instance Learning, Multi-instance Learning Novel and Single Instance Learning achieve F1 scores 0.80, 0.76 and 0.71, respectively. The relatively significant increase of F1 scores also demonstrates the effectiveness of homolog knowledge transfer by means of independent homolog instances.
Performance comparison with existing models
To further validate the model merits, we compare our proposed multi-instance transfer learning method with two reported machine learning methods, one is Random Forest [16], and the other is multi-task learning method [17]. As shown in Table 3, Random Forest [16] achieved rather low SE value (0.407) on the positive class, much lower than our method (SE = 0.8065), though a little higher SP values than our method (SP 0.817 versus 0.7692). Such a low SE value (0.407) suggests that Random Forest [16] is likely to be highly biased towards the negative class. In addition, Random Forest [16] also achieved much lower F1 score than our method (F1 score 0.52 versus 0.80).
Table 3. Model validation on data from recent literatures.
| Salmonella effector proteins | Targeted human proteins | Confidence level |
| AvrA | MTOR | 0.3271 |
| SipA | F-actin caspase-3 | 0.5157 |
| SipC | F actin | 0.5185 |
| PibB2 | Kinesin-1 | 0.2943 |
| SseI | F-actin | 0.5185 |
| SspH2 | SGT1 | 0.1059 |
| SspH2 | Nod1 | 0.5410 |
The multi-task learning method [17] simultaneously exploited multiple PPI networks from the bacteria S.typhi, B.anthracis, F.tularensis and Y.pestis. The method reported the F1 score only (0.758). Model evaluation with inadequate performance metric is one of major weaknesses to both the methods [16], [17]. As compared to the Random Forest method [16] and the multi-task learning method [17], our proposed multi-instance transfer learning method has the following merits (1) we design the experimental setting Single Instance Learning as the baseline to verify the performance gain and robustness of information substitution by the homolog instances. Unfortunately, the Random Forest method [16] should have designed the baseline experimental setting of no imputing missing values, and the multi-task learning method [17] should have designed the single-task learning of only S.typhi without information from B.anthracis, F.tularensis and Y.pestis, as the baseline experimental setting. Without the baseline performance, we can not gain knowledge about the performance increase by imputing missing values [16] and optimizing multiple sub-tasks learning [17]; (2) Besides the baseline experimental setting, we also design an extreme experimental setting Multi-instance Learning Novel where the feature information of the test proteins is completely not available (e.g. novel proteins). Both the methods [16], [17] did not test the model robustness against data unavailability; (3) Besides gene ontology, the two methods [16], [17] also need other feature information such as gene expression, RNAi expression, conserved pathways, Pfam interactions, etc., which may also be not available. In our methods, only gene ontology is needed and thus the data constraint is much less demanding. Of course, the above-mentioned comparison is rough in a sense. The randomness introduced by data partition of cross validation and negative data sampling makes it hard to conduct strictly fair comparison between different computational methods. Furthermore, the performance increase of novel method against the existing methods benefits to a certain degree from the update of data (e.g. GOA update). Nevertheless, a relatively rough comparison helps us to gain some knowledge about the reliability of novel models. For the existing models, it is necessary to frequently update the model training using up-to-date data.
Schlekera et al. [15] used protein sequence similarity (sequence identity was 21%) to predict interlogs, achieving 49% recognition rate (i.e. SE = 0.49) on the dataset [8]. The SE value is equivalent to the Random Forest [16] (SE = 0.407) but much lower than the proposed multi-instance transfer learning method (Multi-instance Learning SE = 0.8065; Multi-instance Learning Novel SE = 0.8065; Single Instance Learning SE = 0.7258).
Model validation on data from recent literature
To further validate the model performance, we conduct independent test using the data from recent literature. The recent experimental findings are generally scarce and scattered in massive biomedical literature, and it is hard to manually collect adequate data for model validation. By manual search, we find 18 Salmonella-human PPIs in [18]. Excluding the non-protein interactions (e.g. cholesterol, inositol phospates) and the interactions that have been collected in [8], we obtain 5 novel interactions, together with two novel interactions (SspH2, SGT1) & (SspH2, Nod1) [24], we obtain 7 novel interactions as validation set. The predictions on the validation set are shown in Table 3, where the normalized decision values in the third column measure the confidence level of the predictions. All the 7 novel PPIs are correctly recognized by the proposed method. Interestingly, the two proteins O43765 and O95905 both are related to the gene SGT1, but only O43765 is predicted to interact with SspH2. According to gene ontology annotations, O95905 is annotated as “Novel regulator of p53 stability and function” (http://www.uniprot.org/uniprot/O95905) and O43765 is annotated as “Co-chaperone that binds directly to HSC70 and HSP70 and regulates their ATPase activity” (http://www.uniprot.org/uniprot/O43765). It has been reported in [24] that SGT1 interacting with SspH2 was restricted to those SGT1 proteins that have NLR co-chaperone function. O43765 fulfils co-chaperone function while O95905 does not, O43765 (SGT1) is predicted to interact with SspH2 while O95905 is not. The details of the predicted interactions between SspH2 and SGT1 are consistent with the experimental findings in [24].
Reconstruction of proteome-wide Salmonella-human PPI networks
Proteome-wide PPI predictions
To reconstruct the proteome-wide Salmonella-human PPI networks, the 25 Salmonella proteins in the 62 interactions [8] are used as pathogen proteins and the human proteins are taken from the file uniprot_sprot_human.dat.gz available at ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/taxonomic_divisions/. Excluding those uncurated proteins and those proteins that already appear in the 62 interactions [8], we totally obtain 20,334 reviewed human proteins. Hence the prediction space contains 508,350 (25×20,334) protein pairs. The predictions are given in File S1. Among the 20,334 human proteins, 6271 human proteins are predicted to interact with the 25 Salmonella proteins and 75,381 novel interactions are detected by our method. Comparatively, the Random Forest method [16] predicted 190,868 novel interactions between 22,653 human proteins and 486 Salmonella proteins, of which 461 Salmonella proteins were not included in the 62 PPIs [8]. After excluding the 461 Salmonella proteins and the related predictions, the Random Forest method [16] predicted 134,339 interactions, much larger than 75,381 interactions predicted by our method, suggesting a larger coverage of true interactions and meanwhile a higher risk of false positive predictions.
Overlap with existing models
The overlap of predictions between different computational methods is generally small. For instance, the overlap between the predictions by the three methods [9], [12], [13] contains only 4 HIV-human PPIs. Such a low overlap largely results from the two points: (1) the overlap analysis is not based on proteome-wide predictions; (2) the threshold of the final predictions is generally chosen to be much higher than the threshold for model estimation [10], [16], [17], and thus narrows down the scale of the predicted PPI networks. As regards with Salmonella-human PPI prediction, the Random Forest method [16] yielded 134,339 interactions, our method yields 75,381 interactions and there are 23,159 overlapped predicted PPIs (see File S2). The overlap rate between Random Forest [16] and our method is 12.41%. Here the overlap rate among K methods is formally defined as 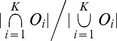 , where
, where 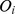 denoted the predictions yielded by the i-th method and
denoted the predictions yielded by the i-th method and 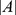 denotes the cardinality of set A. The 23,159 overlapped PPIs are supposed to be more reliable. As compared to the low overlap rate among the three methods [9], [12], [13], the large overlap rate between Random Forest [16] and our method is largely due to the proteome-wide prediction space. Besides, as compared to large training data, the small training data is prone to yield a relatively larger portion of false positive predictions, which may also contribute to the large overlap rate. It is worth noting that if we further define as positives those predictions with
denotes the cardinality of set A. The 23,159 overlapped PPIs are supposed to be more reliable. As compared to the low overlap rate among the three methods [9], [12], [13], the large overlap rate between Random Forest [16] and our method is largely due to the proteome-wide prediction space. Besides, as compared to large training data, the small training data is prone to yield a relatively larger portion of false positive predictions, which may also contribute to the large overlap rate. It is worth noting that if we further define as positives those predictions with 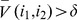 (
(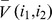 is defined in formula (12)), the predicted interactions are supposed to be more reliable.
is defined in formula (12)), the predicted interactions are supposed to be more reliable.
Schlekera et al. [15] used the 62 Salmonella-human interactions to derive interlogs, i.e. the interacting pairs between Salmonella protein orthologs and human protein orthologs. Unlike the Random Forest method [16] and our method, Schlekera et al. [15] did not predict the interactions between the 25 Salmonella proteins contained in the benchmark data with other human proteins, so we do not conduct analysis of network overlap with the method.
Discussions
Reconstruction of pathogen-host PPI networks is of importance to reveal the underlying mechanism of pathogen infection and host defence. At present, it is still a challenging task for labor-intensive experimental techniques to accurately map the proteome-wide pathogen-host protein interactome. Computational modeling is a good complement to experimental methods for fast proteome-wide interactome mapping at low cost. Unfortunately, the current experimentally derived pathogen-host PPI networks are rather small, for instance, there are only 62 known PPIs between Salmonella typhimurium and human, such a small network would yield a computational model that does not generalize well. On the other hand, computational modeling need deliberate dwelling on the problem that the knowledge in biological databases (e.g. gene ontology) is currently not complete.
In this work, we conduct homolog knowledge transfer by means of independent homolog instances to serve the purposes: (1) augmenting the training data to reduce the risk of model overfitting; (2) enriching the feature information to improve the model performance; (3) substituting for the missing feature information to enhance the model robustness against data unavailability. The multi-instance transfer learning method is implemented under the framework of AdaBoost, where the gene ontology knowledge from the homologs is treated as independent homolog instance to augment the training data. The noise from the homolog instances could be counteracted by AdaBoost instance reweighting algorithm.
To validate the assumptions that the independent homolog instances are effective to solve the problems of data scarcity and data unavailability for Salmonella-human PPI prediction, we deliberately design three experimental settings: Multi-instance Learning, Multi-instance Learning Novel and Single Instance Learning and comprehensively survey the model performance by multiple performance metrics: SP, SE, Accuracy, MCC, ROC-AUC and PR-AUC. The results of 10-fold cross validation experiments show that the homolog knowledge transfer by means of independent homolog instances does improve the model performance and helps the model work properly in the extreme case that the gene ontology knowledge is completely not available. The experimental results also show that the proposed multi-instance transfer learning method significantly outperforms the existing machine learning methods with less demanding data constraints. To further validate how well the proposed model generalizes to unseen data, we collect 7 experimentally derived interactions from the recent literatures. Al78 interactions can be correctly recognized by our method. Interestingly, for the two proteins {O43765, O95905} that are related to gene SGT1, only the protein O43765 that fulfils the molecular function of co-chaperone is predicted to interact with SspH2, which is consistent with the recent findings.
Noteworthily, there are two concerns that need to be addressed, one concern is about the feasibility of training model on the small Salmonella-human PPI data, and the other concern is about false positive rate. Although the proposed model demonstrates sound performance from the point of view of cross validation and independent test experiments, the smallness of data may not convince us of the validity of the predictions as the large HIV data does [6], [10], [28]. The worry comes from whether or not it is feasible to use so small a data to train a satisfactory model. Actually, some machine learning methods like SVM (support vector machine) are derived from small-example statistical learning theory [35], where only a small number of support vectors (SV) are needed to define the two-class decision hyper-planes and a large number of non-SVs are discarded. The gracefulness of sparseness is often used to reduce the computational complexity [28], [36], [37]. It has been theoretically proven that the regularization technique helps AdaBoost to achieve large margin between two-class hyperplanes and sparseness like SVM [31], implying that it is still feasible to train a satisfactory model on such a small data for Salmonella-human PPI prediction. We note that the multi-instance transfer learning method is specifically proposed to augment very small data. For large data like HIV, the proposed method will increase the computational complexity and thus seems not to be a proper solution. As regards with the second concern, both biological experiments and computational predictions are supposed to yield a certain level of false positives. For biological experiments, different experimental techniques are generally specific to particular types of interactions, for instance, Y2H as an in vivo technique is highly effective at detecting transient interactions and can be readily applied to screen large genome-wide libraries, but is limited by its biases toward non-specific interactions [38]. Likewise computational method is also prone to yield false positive predictions, especially on small training data, and the 7 correctly recognized interactions (see Table 3) may be worried to result from high false positive rate. Actually, our proposed method greatly reduces the risk of false positive predictions as compared to the Random Forest [16] (predicted 75,381 interactions versus predicted 134,339 interactions). To ensure the validity of the predictions, we can increase the decision threshold as 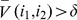 (
(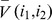 is defined in formula (12)).
is defined in formula (12)).
The ultimate goal of model development and model estimation is to reconstruct reliable proteome-wide protein interaction networks between Salmonella typhimurium and Homo sapiens. To gain insights into the patterns of Salmonella infection and host response, the predicted PPI networks are further subjected to gene ontology based clustering analysis. We simply cluster together the Salmonella targeted human proteins on the basis of gene ontology terms. To provide valuable cues about the host protein complexes, molecular functions and signaling pathways that Salmonella proteins may interfere with, we assign to the same cluster the human partners that possess the same GO term, with each cluster corresponding to one PPI sub-network. All the GO terms of human proteins are classified into thee major classes, i.e., biological processes (P), molecular functions (F) and cellular compartments (C). For each major class, we further discuss the two cases: (1) all the 25 Salmonella proteins are involved in the PPI sun-network, denoted as P1, F1 and C1, respectively; (2) NOT all the 25 Salmonella proteins are involved in the PPI sun-network, denoted as P2, F2 and C2, respectively. P1, F1 and C1 are given in File S3, File S4 and File S5, respectively. P2, F2 and C2 are given in File S6, File S7 and File S8, respectively. For the sake of quite a large number of predicted PPI sub-networks, we only demonstrate several PPI sun-networks as examples, interested readers are referred to File S3∼File S8 for insightful biological cues.
PPI sub-network GO: 0032862 - activation of Rho GTPase activity
It has been reported in [33] that Salmonella typhimurium injects toxins SopE, SopE2 and SptP to change the GTP/GDP loading state of Rho GTPases by transient interactions, wherein SopE and SopE2 mimic eukaryotic G-nucleotide exchange factors (GEF) and thereby activate Rho GTPase signaling pathways in the infected host cells. In this work, the Salmonella proteins are predicted with 7 human proteins that are annotated with GO term GO: 0032862 (activation of Rho GTPase activity). The predicted PPI sub-network GO: 0032862 is illustrated in Figure 3. The two Salmonella SopE proteins (O52623, Q7CQD4) are predicted to interact with 5 human proteins {Q9H8V3, Q6ZW31, Q5VT97, Q92574, P21709}, suggesting that the SopE activates Rho GTPase signaling pathways by directly mimicking the molecular functions of the host proteins or indirectly interacting with the host proteins. Among the 7 human proteins, the two human proteins {Q9H8V3, Q92574} are predicted to be targeted by all the 25 Salmonella proteins while the two proteins {Q8IYL9, P32248} are predicted to be targeted by only one Salmonella protein.
Figure 3. The predicted Salmonella-human PPI sub-network GO: 0032862 (biological process: activation of Rho GTPase activity).
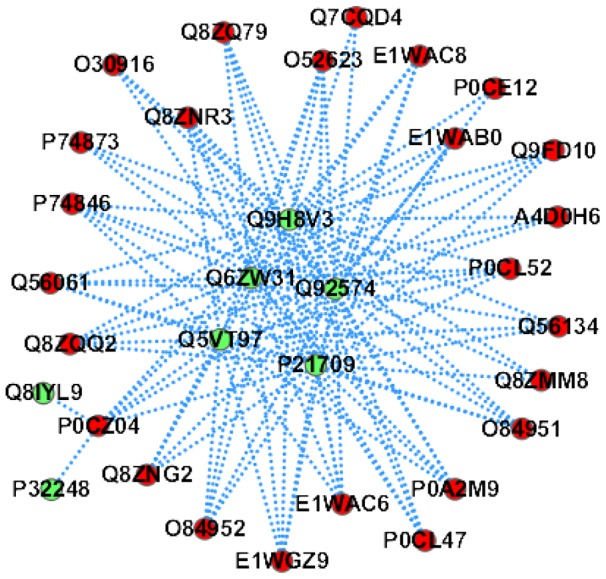
The red node denotes Salmonella protein and the green node denotes human protein.
PPI sub-network GO: 0008234 - cysteine-type peptidase activity
The predicted PPI sub-network is illustrated in Figure 4. In Figure 4, the Salmonella proteins are predicted to interact with 70 human proteins that fulfil the molecular function “cysteine-type peptidase activity” (GO: 0008234), wherein the Salmonella protein Q8ZQQ2 is a hub protein that is predicted to target 63 human proteins. The Salmonella protein slrP (Q8ZQQ2) is an effector protein that functions to alter the host cell physiology and promote bacterial survival in the host tissues. This protein is an E3 ubiquitin ligase that interferes with the host ubiquitination pathway and leads to significant decrease of thioredoxin activity and increase of the host cell death (http://www.uniprot.org/uniprot/Q8ZQQ2). The six human proteins {P5521, Q14790, P42575, O00303, P55210, P55211} are predicted to be targeted by all the 25 Salmonella proteins, wherein P5521 is involved in the activation cascade of caspases responsible for apoptosis execution, whose over-expression promotes programmed cell death. The predictions suggest the Salmonella proteins may also interfere with the host apoptotic signaling pathways.
Figure 4. The predicted Salmonella-human PPI sub-network GO: 0008234 (molecular function: cysteine-type peptidase activity).

The red node denotes Salmonella protein and the green node denotes human protein.
PPI sub-network GO: 0007205 - activation of protein kinase C activity by G-protein coupled receptor protein signaling pathway
The predicted PPI sub-network is illustrated in Figure 5. As shown in Figure 5, all the 20 predicted human partners except {P46663, P17677} are densely connected with the 25 Salmonella proteins. Salmonella protein spiC (P0CZ04) is predicted to target all the 20 human proteins and the human proteins {Q07954, P49619, Q86XP1, Q9NRD5, Q16760, P52824, Q9Y6T7, Q9P212, Q8TEW0, Q5KSL6, P23743} are predicted to be targeted by all the 25 Salmonella proteins. Salmonella protein spiC (P0CZ04) is a virulence protein that plays a central role in mammalian macrophage infection by inhibiting phagosome-lysosome fusion and cellular trafficking (http://www.uniprot.org/uniprot/P0CZ04). The human protein Q07954 is an endocytic receptor involved in endocytosis and in phagocytosis of apoptotic cells, and may modulate the cellular events such as APP metabolism, kinase-dependent intracellular signaling, neuronal calcium signaling as well as neurotransmission (http://www.uniprot.org/uniprot/Q07954). The molecular function annotations of the two proteins suggest that Salmonella protein spiC (P0CZ04) is highly likely to interact with the human protein Q07954.
Figure 5. The predicted Salmonella-human PPI sub-network GO: 0007205 (biological process: activation of protein kinase C activity by G-protein coupled receptor protein signaling pathway).

The red node denotes Salmonella protein and the green node denotes human protein.
PPI sub-network GO: 0003777 - microtubule motor activity
In [34], it is reported that Salmonella typhimurium grows within the host cells in a permissive compartment termed Salmonella-containing vacuole (SCV), and some SPI-2 effectors modulate microtubule motor activity on the SCV. In this work, the Salmonella proteins are predicted with 74 human proteins that are annotated with the GO term GO: 0003777 (microtubule motor activity) (see Figure 6), and the SPI-2 effector sseJ (Q9FD10) is predicted to interact with 26 human proteins that are involved in the molecular functions “microtubule motor activity”. From Figure 6, we can see the predicted PPI sub-network GO: 0003777 is densely connected and the 10 human proteins {Q9H0B6, Q8NCM8, Q8NCM8, Q9GZS0, Q12840, P33176, Q02224, Q9NS87, O95239, O14576} are predicted to be targeted by all the 25 Salmonella proteins. The human protein Q9H0B6 (Kinesin light chain 2) is a light chain of Kinesin (a microtubule-associated force-producing protein that may play a role in organelle transport), and the light chain may function in coupling of cargo to the heavy chain or in the modulation of its ATPase activity (http://www.uniprot.org/uniprot/Q9H0B6). The prediction suggests that the Salmonella proteins may interfere with the host organelle transport and ATPase activity.
Figure 6. The predicted Salmonella-human PPI sub-network GO: 0003777 (molecular function: microtubule motor activity).

The red node denotes Salmonella protein and the green node denotes human protein.
Supporting Information
XML file contains the proteome-wide predicted Salmonella-human PPIs.
(XML)
XML file contains the overlapped interactions between our method and Random Forest [16].
(XML)
XML file contains the clusters of interacting human partners that participate in specific biological processes. All the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
XML file contains the clusters of interacting human partners that fulfil specific molecular functions. All the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
Text file contains the clusters of interacting human partners that are localized in specific cellular compartments. All the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
XML file contains the clusters of interacting human partners that participate in specific biological processes. NOT all the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
XML file contains the clusters of interacting human partners that fulfil specific molecular functions. NOT all the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
XML file contains the clusters of interacting human partners that are localized in specific cellular compartments. NOT all the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
Acknowledgments
Thanks for the helpful comments from the anonymous reviewers.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
The work is partly supported by China Postdoctoral Science Foundation Funded Projects (No. 2013M531869, No. 2014T70821). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. von Mering C, Krause R, Snel B, Cornell M, Oliver SG, et al. (2002) Comparative assessment of large-scale datasets of protein-protein interactions. Nature 417: 399–403. [DOI] [PubMed] [Google Scholar]
- 2. Edwards A, Kus B, Jansen R, Greenbaum D, Greenblatt J, et al. (2002) Bridging structural biology and genomics: assessing protein interaction data with known complexes. Trends Genet 18: 529–536. [DOI] [PubMed] [Google Scholar]
- 3. Wu X, Zhu L, Guo J, Zhang D, Lin K (2006) Prediction of yeast protein-protein interaction network: insights from the Gene Ontology and annotations. Nucleic Acids Res 34: 2137–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeBodt S, Proost S, Vandepoele K, Rouzé P, Peer Y, et al. (2009) Predicting protein-protein interactions in Arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genomics 10: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shen J, Zhang J, Luo X, Zhu W, Yu K, et al. (2007) Predicting protein–protein interactions based only on sequences information. Proc Natl Acad Sci USA 104: 4337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fu W, Sanders-Beer BE, Katz KS, Maglott DR, Pruitt KD, et al. (2009) Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res (Database Issue) 37: D417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wuchty S (2011) Computational prediction of host-parasite protein interactions between P. falciparum and H. sapiens. PLoS ONE 6: e26960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schleker S, Sun J, Raghavan B, Srnec M, Müller N, et al. (2012) The current Salmonella-host interactome. Proteomics Clin Appl 6: 117–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tastan O, Qi Y, Carbonell J, Klein-Seetharaman J (2009) Prediction of interactions between HIV- 1 and human proteins by information integration. In Proceedings of the Pacific Symposium on Biocomputing (PSB-2009) pp 516–527. [PMC free article] [PubMed] [Google Scholar]
- 10. Qi Y, Tastan O, Carbone J, Klein-Seetharaman, Weston J, et al. (2010) Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins. Bioinformatics 26: i645–i652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dyer M, Muralib T, Sobrala B (2011) Supervised learning and prediction of physical interactions between human and HIV proteins. Infect Genet Evol 11: 917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doolittle J, Gomez S (2010) Structural similarity-based predictions of protein interactions between HIV-1 and Homo sapiens. Virology J 7: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mukhopadhyay A, Maulik U, Bandyopadhyay S (2012) A novel biclustering approach to association rule mining for predicting HIV-1-human protein interactions. PLoS One 7: e32289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dyer M, Muralib T, Sobrala B (2007) Computational prediction of host-pathogen protein-protein interactions. Bioinformatics 23: i159–i166. [DOI] [PubMed] [Google Scholar]
- 15. Schleker S, Garcia-Garcia J, Klein-Seetharaman J, Oliva B (2012) Prediction and comparison of Salmonella-human and Salmonella-Arabidopsis interactomes. Chem Biodivers 9: 991–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kshirsagar M, Carbonell J, Judith K (2012) Techniques to cope with missing data in host–pathogen protein interaction prediction. Bioinformatics 28: i466–i472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kshirsagar M, Carbonell J, Judith K (2013) Multitask learning for host-pathogen protein interactions. Bioinformatics 29: i217–i226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Srikanth CV, Regino ML, Hallstrom K, McCormick B (2011) Salmonella effector proteins and host-cell responses. Cell Mol Life Sci 68: 3687–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu J, Guo M, Needham C, Huang Y, Cai L, et al. (2010) Simple sequence-based kernels do not predict protein-protein interactions. Bioinformatics 26: 2610–2614. [DOI] [PubMed] [Google Scholar]
- 20. Freund Y, Schapire RE (1997) A decision-theoretic generalization of on-line learning and an application to boosting. J Comput Syst Sci 55: 119–139. [Google Scholar]
- 21. Vezhnevets A, Vezhnevets V (2005) Modest AdaBoost – teaching AdaBoost to generalize better. Graphicon 12: 987–997. [Google Scholar]
- 22. Jansen R, Gerstein M (2004) Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Curr Opin Microbiol 7: 535–545. [DOI] [PubMed] [Google Scholar]
- 23. Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, et al. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31: 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhavsar A, Brown N, Stoepe J, Wiermer M, Martin D, et al. (2013) The Salmonella type III effector SspH2 specifically exploits the NLR co-chaperone activity of SGT1 to subvert immunity. PLoS Pathog 9: e1003518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pan S, Yang Q (2010) A Survey on Transfer Learning. IEEE Trans Knowl Data Eng 22: 1345–1359. [Google Scholar]
- 26. Mei S, Wang F, Zhou S (2011) Gene ontology based transfer learning for protein subcellular localization. BMC Bioinformatics 12: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mei S (2012) Multi-label multi-kernel transfer learning for human protein subcellular localization. PLoS ONE 7: e37716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mei S (2013) Probability weighted ensemble transfer learning for predicting interactions between HIV-1 and human proteins. PLoS ONE 8: e79606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barrell D, Dimmer E, Huntley R, Binns D, O'Donovan C, et al. (2009) The GOA database in 2009–an integrated Gene Ontology Annotation resource. Nucleic Acids Res (Database Issue) 37: D396–D403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meir R, Ratsch G (2003) An introduction to boosting and leveraging. Lect Notes Artif Int 2600: 118–183. [Google Scholar]
- 32. Davis J, Goadrich M (2006) The relationship between precision-recall and ROC curves. In Proceedings of the 23rd International Conference on Machine Learning. [Google Scholar]
- 33. Schlumberger MC, Hardt WD (2005) Triggered phagocytosis by Salmonella: bacterial molecular mimicry of RhoGTPase activation/deactivation. Curr Top Microbiol Immunol 291: 29–42. [DOI] [PubMed] [Google Scholar]
- 34. Wasylnka JA, Bakowski MA, Szeto J, Ohlson MB, Trimble WS, et al. (2008) Role for myosin II in regulating positioning of Salmonella-containing vacuoles and intracellular replication. Infect Immun 76: 2722–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vapnik V (1999) An Overview of Statistical Learning Theory. IEEE Trans Neural Netw 10: 988–999. [DOI] [PubMed] [Google Scholar]
- 36. Mei S (2014) SVM ensemble based transfer learning for large-scale membrane proteins discrimination. J Theor Biol 340: 105–110. [DOI] [PubMed] [Google Scholar]
- 37. Dong J, Adam K, Ching Y (2005) Fast SVM Training Algorithm with Decomposition on Very Large Data Sets. IEEE Trans Pattern Anal Mach Intell 27: 603–18. [DOI] [PubMed] [Google Scholar]
- 38. Gonzalez MW, Kann MG (2012) Chapter 4: Protein Interactions and Disease. PLoS Comput Biol 8: e1002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
XML file contains the proteome-wide predicted Salmonella-human PPIs.
(XML)
XML file contains the overlapped interactions between our method and Random Forest [16].
(XML)
XML file contains the clusters of interacting human partners that participate in specific biological processes. All the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
XML file contains the clusters of interacting human partners that fulfil specific molecular functions. All the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
Text file contains the clusters of interacting human partners that are localized in specific cellular compartments. All the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
XML file contains the clusters of interacting human partners that participate in specific biological processes. NOT all the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
XML file contains the clusters of interacting human partners that fulfil specific molecular functions. NOT all the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
XML file contains the clusters of interacting human partners that are localized in specific cellular compartments. NOT all the 25 Salmonella proteins are involved in the predicted PPI sub-networks.
(XML)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.