

Abstract
In type 1 diabetes, proinflammatory cytokines secreted by infiltrating immune cells activate the unfolded protein response (UPR) in islet β-cells, which leads to attenuation of global mRNA translation. Under such conditions, privileged mRNAs required for adaptation to the prevailing stress are maintained in an actively translated state. Pdx1 is a β-cell transcription factor that is required for the adaptive UPR, but it is not known how translation of its mRNA is maintained under these conditions. To study translation, we established conditions in vitro with MIN6 cells and mouse islets and a mixture of proinflammatory cytokines (IL-1β, TNF-α, and IFN-γ) that mimicked the UPR conditions seen in type 1 diabetes. Cell extracts were then subjected to polyribosome profiling to monitor changes to mRNA occupancy by ribosomes. Similar to other privileged mRNAs (Atf4 and Chop), Pdx1 mRNA remained partitioned in actively translating polyribosomes under the UPR, whereas the mRNA encoding a proinsulin-processing enzyme (Cpe) and others partitioned into inactively translating monoribosomes. Bicistronic luciferase reporter analyses revealed that the distal portion of the 5′-untranslated region of mouse Pdx1 (between bp −105 to −280) contained elements that promoted translation under both normal and UPR conditions, and this region exhibited conserved sequences and secondary structure similar to those of other known internal ribosome entry sites. Our findings suggest that Pdx1 protein levels are maintained in the setting of the UPR, in part, through elements in the 5′-untranslated region that confer privileged mRNA translation in a 5′-7-methylguanylate cap–independent manner.
Type 1 diabetes (T1D) is characterized by loss of immune tolerance to insulin-producing β-cells, leading early to impaired insulin production and later to β-cell destruction and frank hyperglycemia (1, 2). Whereas many studies have focused on the immune system as the primary trigger of T1D, emerging data have pointed to a potentially equally important role of the β-cell itself in T1D pathogenesis (3). Recent studies of prediabetic nonobese diabetic (NOD) mice (a model of T1D) have shown that the accumulation of unfolded proteins in the endoplasmic reticulum (ER) leads to ER stress (4, 5). It has been hypothesized that in β-cells, ER stress is a major driver of antigen and neoantigen exposure and thereby a trigger for autoimmunity. In support of this hypothesis, NOD mice treated with chemical protein folding chaperones have more robust β-cell function and a significantly reduced incidence of diabetes (6). Further, islets from individuals with T1D show increased levels of ER stress markers (6, 7).
ER stress initially triggers an adaptive response known as the unfolded protein response (UPR). The UPR activates 3 ER membrane-resident signaling proteins: inositol-requiring 1 (IRE1), activating transcription factor 6 (ATF6), and PRKR-like endoplasmic reticulum kinase (PERK), which collectively function to remediate ER stress by promoting protein folding and suppressing new protein production (8). Whereas many studies have elucidated the transcriptional responses of the UPR (downstream of IRE1 and ATF6), fewer studies have investigated the translational responses of the UPR (downstream of PERK). During the UPR, phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) by PERK causes suppression of general protein synthesis (generally, those proteins encoded by capped mRNAs) (9, 10). However, several “privileged” mRNAs escape translational suppression during this process, and the proteins encoded by these mRNAs are typically required for adaptation to ER stress, including activating transcription factor 4 (ATF4) (11). The mechanisms that promote translation of these privileged mRNAs are varied, but generally appear to involve elements in the 5′-untranslated region (UTR), such as repressing and activating upstream open reading frames (uORFs) and internal ribosomal entry sites (IRESs) (12).
Pdx1 is a homeodomain transcription factor that is critical for pancreas development and the postnatal function of islet β-cells (13–15). Whereas mutation of the Pdx1 gene in both mice and humans results in pancreatic agenesis, haploinsufficiency of Pdx1 leads to a maturity-onset diabetes of the young (16). Pdx1+/− mice fed a high-fat diet show heightened susceptibility to ER stress owing to failure in the UPR (17). Pdx1 plays a key role in the UPR by directly promoting transcription of Atf4, Wfs1, and Ero1b, among others (17). These findings suggest that maintenance of Pdx1 mRNA translation in the setting of ER stress is important for remediation of stress. We hypothesized that Pdx1 mRNA translation is heightened during ER stress. To test this hypothesis, we established a cell culture system in vitro that recapitulates the early translational changes seen during T1D. Our findings reveal that the 5′-UTR of the Pdx1 mRNA contains DNA elements that confer relative resistance to translational repression in the setting of the UPR, thereby establishing for the first time a mechanism that maintains Pdx1 protein production during proinflammatory signaling.
Materials and Methods
Antibodies
Antibodies against β-actin, ATF4, CAAT/enhancer binding protein homologous protein (CHOP), eIF2α, and phospho-eIF2α were detailed previously (18). Other antibodies used in these studies include anti-Pdx1 (07-696; Millipore), anti-carboxypeptidase E (CPE) (11044; Abcam), anti-puromycin (EQ001; Kerafast), anti-phospho-eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) (9455; Cell Signaling Technology), and anti-4EBP1 (sc-6024; Santa Cruz Biotechnology). Fluorophore-labeled secondary antibodies IRDye 800 and IRDye 700 were purchased from Li-Cor Biosciences.
Animals, islets, and cell lines
Mice (C57BL/6J) were purchased from The Jackson Laboratory and maintained at the Indiana University Laboratory Animal Resource Center under pathogen-free conditions according to protocols approved by the institutional animal care and use committee. The mouse insulinoma cell line MIN6 was maintained in culture conditions as described previously (19). Mouse islets were isolated from collagenase-perfused pancreata and cultured in RPMI 1640 medium as described previously (20). Islets were picked by hand and counted and then were allowed to recover overnight before experimentation. Islet or MIN6 cell cultures were treated with a cytokine cocktail containing 5 ng/mL IL-1β, 10 ng/mL TNF-α, and 100 ng/mL IFN-γ or with 1 μM thapsigargin for the indicated times.
Transient transfections and luciferase assays
Transient transfections were performed using 70% confluent MIN6 cells in 6-well tissue culture plates. For each transfection, 2 μg of reporter plasmid (C49 constructs) was combined with 6 μL of Metafectene reagent (Biontex Laboratories) and added to cells in serum-free medium for 6 hours, transfection medium was removed, and culture medium was returned to the cultures. Transfected cells were harvested 24 hours later, and RNA was isolated or protein was assayed for luciferase activity using a commercially available dual-luciferase activity kit (Promega) and luminometer (Turner BioSystems).
Immunoblot analysis
Whole-cell extracts from cell lines and islets were prepared and subjected to immunoblot analysis as described previously (21). For immunoblotting of puromycin incorporation into protein, puromycin was added to the culture medium at 1 μg/mL during the final 15 minutes of incubation, and then cells were washed twice with cold PBS and lysed as described previously (20). Immunoblot analyses of MIN6 and islet extracts were performed after separation of protein extracts on a 4% to 20% gradient SDS-polyacrylamide gel. Immunoblots were visualized using fluorescently labeled secondary antibodies (Li-Cor Biosciences) and quantified using Li-Cor software.
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA from MIN6 cells and islets was recovered using an RNeasy kit (QIAGEN), reverse transcribed, and subjected to qRT-PCR. Data were normalized to Actb message levels, except in the case of RNA from polyribosomal profiling experiments, which are reported as the percentage of total recovered RNA. All qRT-PCR data represent the means of triplicate determinations from at least 3 independent experiments. Primer sequences were previously described for Xbp1s, Chop, Atf4, and Actb (22–25). For Pdx1, the following primers were used: 5′-CGGACATCTCCCCATACGAAG-3′ (forward) and 5′-CCCCAGTCTCGGTTCCATTC-3′ (reverse). For Cpe, the following primers were used: 5′-GCTCAGGTAATTGAAGTCTT-3′ (forward) and 5′-TACTGCTCACGAATACAGTT-3′ (reverse). For firefly luciferase, the following primers were used: 5′-AGAGGATGGAACCGCTGGAGAG-3′ (forward) and 5′-GCTTCTGCCAACCGAACGGAC-3′ (reverse). For Renilla luciferase, the following primers were used: 5′-CAAAGAGAAAGGTGAAGTTCGTCG-3′ (forward) and 5′-TGGAAAAGAATCCTGGGTCCG-3′ (reverse).
Polyribosomal profile (PRP) experiments
A total of 500 islets or 5 × 106 MIN6 cells were treated with or without cytokines or thapsigargin and then were washed twice with cold PBS containing 50 μg/mL cycloheximide and harvested in 500 μL of lysis buffer containing 20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 100 mM NaCl, 1% Triton X-100, 50 U/mL ribonuclease inhibitor (Promega), and 50 μg/mL cycloheximide. Lysates were clarified by centrifugation at 13 000g for 10 minutes at 4°C. A portion of the lysate supernatant was preserved as the input sample to determine total mRNA levels. The remainder of the lysate was then applied to a 10% to 40% sucrose gradient solution containing 20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 100 mM NaCl, and 50 μg/mL cycloheximide. The sucrose gradients were subjected to centrifugation at 4°C in a Beckman SW-41Ti rotor at 270 000g for 2 hours. A piston gradient fractionator (BioComp Instruments) was used to fractionate the gradients, and absorbance of RNA at 254 nm was recorded using an in-line UV monitor. The eluate was collected using a fraction collector, and total RNA from the fractions was purified, reverse transcribed, and subjected to qRT-PCR. Monoribosome-associated RNA was collected from fractions 1 to 5, and polyribosome-associated RNA was collected from fractions 6 to 10.
Recombinant DNA and mutagenesis
The C49 bicistronic vector construct was described previously (26). 5′-UTR fragments were PCR-amplified from genomic mouse or human DNA and subcloned into the EcoRI site of the C49 vector. PCR amplifications were performed using the following primers: for human PDX1 5′-UTR, 5′-AAAGCGAGCAGGGTGGCG-3′ (forward) and 5′-GGCTGCGGCCCGGGATT-3′ (reverse); for mouse Pdx1 5′-UTR, 5′-AAAATTGAAACAAGTGCAGGTG-3′ (forward) and 5′-GGTGGCAGCCGGCACTTG-3′ (reverse); and for mouse Cpe 5′-UTR, 5′-GTGAGGCGAGAGGAGGCTGGTGCTG-3′ (forward) and 5′-CGCGTCCCCGCGAGCTGCACTGCC-3′ (reverse). Vectors containing the 5′-UTR insert were verified by automated sequencing. Deletion mutations were introduced in the respective C49 constructs using a site-directed mutagenesis kit (Agilent).
Statistics
All data are presented as means ± SEM. One-way ANOVA (followed by a Dunnett posttest) was used for comparisons in which 2 or more conditions were compared with a single control, and a Student t test was performed when 2 conditions were compared. Prism (version 5.0; GraphPad Software) was used for all statistical analyses. Statistical significance was defined as a value of P < .05.
Results
Proinflammatory cytokines activate the early UPR in MIN6 β-cells and mouse islets
To study mRNA translation responses as seen in T1D, we first established physiologic cell culture conditions that mimic the UPR. Previously, it was reported that the UPR is observed upon incubation of β-cells and islets with a mixture of proinflammatory cytokines for 24 hours (27, 28). To correlate more precisely the changes to mRNA and protein levels of key ER stress–responsive markers, we incubated MIN6 β-cells and primary mouse islets with a mixture of proinflammatory cytokines (IL-1β, TNF-α, and IFN-γ) at concentrations thought to approximate the T1D milieu (21) and then performed qRT-PCR and immunoblot analyses. As a positive control for overt ER stress and robust UPR activation, cells were also treated with 1 μM thapsigargin (an inhibitor of the sarcoendoplasmic reticulum Ca2+ ATPase [SERCA] pump) for 4 hours. As shown in Figure 1A, treatment of both MIN6 cells and mouse islets with thapsigargin led to statistically significant increases in the ER stress markers Atf4, Chop, and Xbp1s, with increases in corresponding protein levels of ATF4 and CHOP (Figure 1B).
Figure 1.

Proinflammatory cytokines activate the early UPR in mouse MIN6 cells and islets. A, MIN6 cells (top panel) or mouse islets (bottom panel) were untreated (CTL), treated with cytokines (Cyto, 5 ng/mL IL-1β, 10 ng/mL TNF-α, and 100 ng/mL IFN-γ) for 24 hours, or treated with 1 μM thapsigargin (Tg) for 4 hours. cDNA from total cell lysates was subjected to qRT-PCR for the genes indicated. B, Total cell lysates from MIN6 cells (top panel) or mouse islets (bottom panel) were subjected to immunoblot analyses after 24 hours of cytokine mix or 4 hours of thapsigargin treatment, and results were quantified and displayed in the bar graphs. Data represent means ± SEM (n = 6–8). *, P < .05 compared with the control.
Similar to the effect of thapsigargin, treatment of MIN6 cells and mouse islets with cytokines for 24 hours led to significant increases in Atf4, Chop, and Xbp1s mRNAs. In addition, we observed a significant increase in the protein levels of ATF4 in both MIN6 cells and islets (but not of CHOP, a protein that increases later in the course of ER stress) (Figure 1B). These data are consistent with the early effect of the UPR to enhance Atf4 mRNA translation (11). We next examined the mRNA and protein levels of a factor thought to be suppressed by the UPR. The levels of the mRNA encoding the insulin-processing enzyme carboxypeptidase E (Cpe) were unchanged in MIN6 cells and islets (Figure 1A) at 24 hours of cytokine incubation, yet its protein levels were reduced (Figure 1B), in agreement with the known UPR-mediated suppression of this protein (29). Together, these data indicate that proinflammatory cytokines induce the UPR in MIN6 β-cells and mouse islets.
Proinflammatory cytokines regulate mRNA translation in MIN6 β-cells and mouse islets
In the setting of ER stress, the UPR attempts to mitigate new protein production partly via inhibition of translation. The phosphorylation of eIF2α by PERK inhibits the initiation phase of translation of most cellular mRNAs, but paradoxically causes an increase in the initiation of select “privileged” mRNAs that are required for adaptation to ER stress (9, 11). As shown in Figure 2, A and B (and in agreement with prior studies [28]), cytokine treatment of MIN6 cells and islets led to an increase in eIF2α phosphorylation (similar to thapsigargin). We also investigated the abundance and phosphorylation of 4EBP1, which regulates translation initiation through association with eIF4E, a component of the mRNA 5′-7-methylguanylate cap. Neither cytokine nor thapsigargin treatment resulted in changes in 4EBP1 abundance or phosphorylation at the time points evaluated (Figure 2, A and B). To assess the effects of cytokines and thapsigargin on mRNA translation, we subjected total RNA from MIN6 cells and islets to PRP experiments. Figure 2, A and B, shows typical PRPs for MIN6 cells and mouse islets treated with and without cytokines or thapsigargin, indicating the positions of the 80S monoribosome-associated (initiating or inactively translating) RNAs and the polyribosome-associated (actively translating) RNAs. In MIN6 cells, thapsigargin treatment resulted in the relative depletion of polyribosome-associated RNA compared to monoribosome-associated RNA (reflected as a reduced polyribosome/monoribosome [P/M] ratio of 0.866 ± 0.03 vs 0.578 ± 0.09, P < .05). This finding is consistent with the known effects of the UPR, because a block in translation initiation causes relative retention of monoribosomes, as polyribosomes elongate and “run off” of RNA transcripts (30). Upon treatment with cytokines for 24 hours, MIN6 cells showed no change in the P/M ratio. However, prolonged cytokine treatment (72 hours) revealed a loss of polyribosome-associated mRNA in MIN6 cells, consistent with the UPR (P/M ratio of 0.866 ± 0.03 vs 0.741 ± 0.02, P < .05) (Figure 2A). Interestingly, in the case of mouse islets, we did not observe alterations in polyribosome-associated RNA after extended cytokine or thapsigargin treatment (Figure 2B), a result that may reflect the relative insensitivity of the PRP technique to the prevailing effects of the UPR in primary islet tissue. To assess total protein synthesis under these conditions, we next incubated MIN6 cells with puromycin after cytokine and thapsigargin treatment. As shown in Figure 2C, puromycin incorporation into protein is reduced in response to both cytokines and thapsigargin, consistent with the reduction in protein synthesis that is a hallmark of the UPR.
Figure 2.

Effect of proinflammatory cytokines on mRNA translation in MIN6 cells and mouse islets. A, MIN6 β cells were untreated (CTL), treated with cytokines (Cyto) for the indicated time, or treated with 1 μM thapsigargin (Tg) for 4 hours and subjected to immunoblot (upper panel) or PRP analysis (lower panel). B, Mouse islets were untreated (CTL), treated with cytokines for the indicated time, or treated with 1 μM thapsigargin for 24 hours and subjected to immunoblot (upper panel) or PRP analysis (lower panel). C, Immunoblot of MIN6 cells for puromycin after treatment as indicated. Representative data from at least 3 independent experiments are shown for each panel.
We surmised that the PRPs (which reflect total RNA engagement at ribosomes) may be relatively insensitive to the translational events induced by cytokines at the 24-hour time point and that alterations in engagement of specific mRNAs with polyribosomes may be more evident. To examine specific mRNA engagement with mono- and polyribosomes, we next performed qRT-PCR from individual PRP fractions from MIN6 cells to quantitate changes in ribosome association. We found that Atf4 mRNA was shifted rightward toward greater occupancy by polyribosomes after cytokine treatment, similar to thapsigargin treatment, suggesting increased ribosome engagement and mRNA translation under these conditions (Figure 3A). This shift toward polyribosomes is in agreement with the increased protein levels of ATF4 observed (Figure 1B). Similarly, we observed a rightward shift of Chop mRNA toward polyribosomes with cytokine and thapsigargin treatments (Figure 3B). In contrast, Cpe mRNA showed a leftward shift toward monoribosomes under both cytokine and thapsigargin conditions (Figure 3C), consistent with reduced engagement of translating ribosomes and reduced CPE levels (Figure 1B). As shown in Figure 4, a similar shift (or trend to shift) into monoribosomes was also observed for several other transcripts, including Actb, Tbp, and Ccnd1 in response to cytokines and thapsigargin, whereas no change was observed for Bip, an mRNA with an previously characterized IRES (31, 32). These data suggest that proinflammatory cytokines, similar to thapsigargin, induce differential effects on mRNA translation, depending on the nature of the transcript.
Figure 3.

Effect of proinflammatory cytokines and thapsigargin on ribosomal occupancy of Atf4, Chop, and Cpe. MIN6 β cells were untreated (CTL), treated with proinflammatory cytokines (Cyto) for 24 hours, or treated with thapsigargin (Tg) for 4 hours and then were subjected to PRP analysis with fractionation of the sedimentation gradient. A, Analysis of Atf4 mRNA in PRP fractions. B, Analysis of Chop mRNA in PRP fractions. C, Analysis of Cpe mRNA in PRP fractions. Representative data are shown on the left in A, B, and C, and quantitative data on the percentage of total mRNA residing in polyribosomes (fractions 6–10) are shown on the right. Data represent means ± SEM (n = 3–5). *, P < .05 compared with the control.
Figure 4.

Effect of proinflammatory cytokines and thapsigargin on ribosomal occupancy of Actb, Tbp, Ccnd1, and Bip. MIN6 β cells were untreated (CTL), treated with proinflammatory cytokines (Cyto) for 24 hours, or treated with thapsigargin (Tg) for 4 hours and then were subjected to PRP analysis with fractionation of the sedimentation gradient. The percentage of each mRNA species (indicated above each bar graph) residing in polyribosomes is shown. Data represent means ± SEM (n = 3–5). *, P < .05 compared to the control.
Pdx1 mRNA 5′-UTR enhances mRNA translation in the setting of the UPR
Pdx1 was recently shown to activate genes necessary for the adaptive UPR (17). We therefore hypothesized that Pdx1 mRNA translation must be maintained, at least initially, as a requisite for the adaptive UPR. As shown in Figure 5, A and B, neither Pdx1 mRNA nor Pdx1 protein levels changed after 24 hours of treatment with cytokines or 4 hours of treatment with thapsigargin in MIN6 cells. Notwithstanding the phosphorylation of eIF2α under these conditions, the association of Pdx1 mRNA with polyribosomes was unchanged after either cytokine or thapsigargin treatment (Figure 5C). These data suggested to us that Pdx1 mRNA translation was maintained by mechanisms that are distinctly different from those that suppress translation of Cpe or those that enhance translation of Atf4 and Chop (since the latter 2 shift from relative monoribosome occupancy to polyribosome occupancy) (Figure 3).
Figure 5.

Pdx1 retains ribosomal occupancy in the setting of UPR activation. A, MIN6 β-cells were untreated (CTL), treated with cytokines (Cyto) for 24 hours, or treated with thapsigargin (Tg) for 4 hours and then were subjected to qRT-PCR, immunoblot analysis, or PRP analysis with fractionation of the sedimentation gradient. A, qRT-PCR analysis in whole-cell extracts. B, Immunoblot analysis (top panel) with corresponding quantitation (n = 3–5, lower panel). C, qRT-PCR analysis of Pdx1 mRNA in PRP fractions (left panel) and corresponding quantitation of the percentage of total mRNA in polyribosomes. Data represent means ± SEM (n = 3–6).
Many mRNAs containing a 5′-7-methylguanylate “cap” are targeted for eIF2α-dependent translation initiation and are translationally repressed when eIF2α is phosphorylated during the UPR (12). Some privileged mRNAs contain elements in the 5′-UTR that aid in ribosome recruitment and initiation under conditions of stress (12). To investigate whether the maintenance of Pdx1 mRNA translation during the UPR is governed by its 5′-UTR, we cloned the 5′-UTR of the mouse Pdx1 gene into a bicistronic luciferase reporter vector (C49) between the Renilla and firefly luciferase genes (26). This vector allows the expression of the first gene (Renilla luciferase) to be driven by conventional, cap-dependent translation initiation, whereas expression of the second gene (firefly luciferase) would be driven by cap-independent translation initiation mediated by the cloned 5′-UTR element (Figure 6A). Upon transfection of the bicistronic reporter constructs into cell lines, the ratio of the firefly/Renilla luciferase activity is a gauge of the intrinsic ribosome initiation activity (26).
Figure 6.
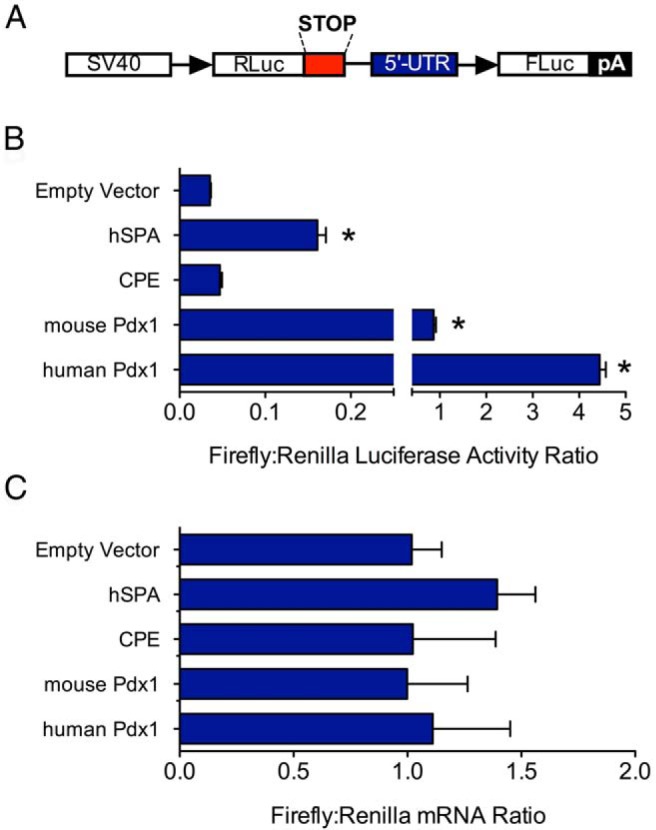
Pdx1 5′-UTR allows cap-independent translation initiation of downstream ORFs. A, Schematic diagram of the bicistronic reporter vector, showing positions of the SV40 promoter, Renilla luciferase gene (RLuc), 5′-UTR, and firefly luciferase gene (FLuc). B, MIN6 cells were transfected with bicistronic vector containing no insert (Empty vector), 5′UTR of hSPA, 5′-UTR of Cpe, and 5′-UTRs of mouse and human Pdx1 genes and then were subjected to luciferase assay. C, Same as in panel B, except that total RNA from each condition was subjected to qRT-PCR for luciferase and Renilla mRNAs (normalized to Actb mRNA). Data represent means ± SEM (n = 3–8). *, P < .05 compared with the empty vector control.
As shown in Figure 6B, the mouse Pdx1 mRNA 5′-UTR showed ∼15-fold enhancement of the firefly/Renilla activity ratio in MIN6 cells compared with that for an empty vector control or a bicistronic vector containing the Cpe 5′-UTR (Figure 6B). Mouse Pdx1 mRNA 5′-UTR also displayed a ∼5-fold enhancement of firefly/Renilla activity ratio compared with that for the human surfactant protein A (hSPA) 5′-UTR, which contains a putative IRES (26). To determine whether the human PDX1 gene also directs cap-independent translation, we inserted its 5′-UTR into the bicistronic reporter vector. The human PDX1 5′-UTR showed a striking ∼65-fold enhancement of the firefly/Renilla luciferase activity ratio in MIN6 β-cells compared with that for the empty vector control (Figure 6B). Importantly, the increased firefly/Renilla activity ratios with the human and mouse 5′-UTRs were not caused by a cryptic promoter element in this region, because levels of Renilla and firefly luciferase mRNAs by real-time PCR were identical for all constructs tested (Figure 6C). Next, we performed these experiments under cytokine- and thapsigargin-treated conditions to determine whether translational enhancement persists under UPR conditions. As shown in Figure 7, the Pdx1 5′-UTR firefly/Renilla activity was diminished under conditions of cytokine and thapsigargin stress, but still exhibited absolute activity that was comparable to or greater than that of hSPA, which contains a characterized IRES element. Collectively, the data in Figures 5 to 7 indicate that the Pdx1 5′-UTR drives cap-independent translation of a downstream open reading frame (ORF) and that its absolute activity is comparable to that of known IRESs under conditions of ER stress.
Figure 7.
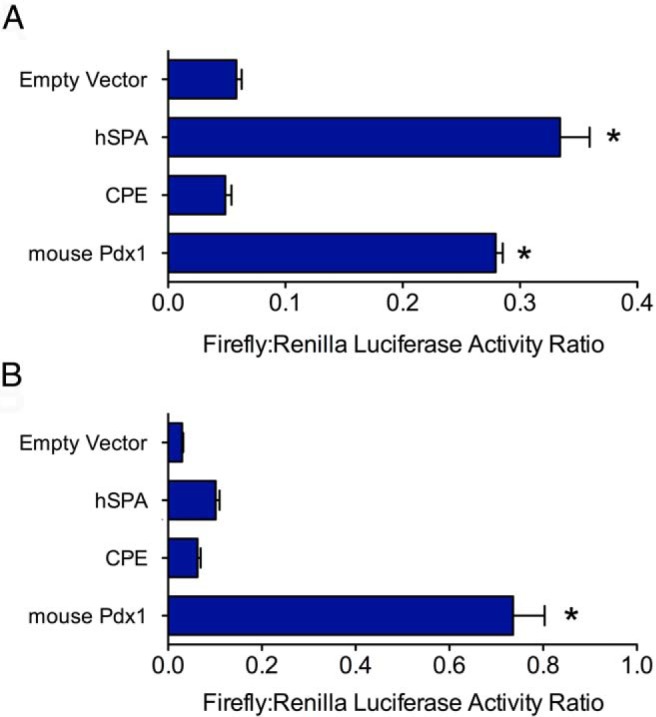
Pdx1 5′-UTR augments cap-independent translation in the setting of cytokines and thapsigargin. MIN6 cells were transfected with the bicistronic reporter vectors containing no insert (Empty vector), hSPA, Cpe, or mouse Pdx1 5′-UTRs, treated with cytokines for 24 hours or thapsigargin for 4 hours and then were subjected to dual-luciferase assay. A, Results after cytokine treatment. B, Results after thapsigargin treatment. Data represent means ± SEM (n = 3–8). *, P < .05 compared with the empty vector control.
Distal portion of the 5′-UTR of Pdx1 is sufficient to drive translational activity
Several mechanisms have been proposed for privileged mRNA translation during stress, including (but not limited to) uORFs and IRESs. As shown in Figure 8A, a comparison of mouse and human Pdx1 5′-UTRs revealed 70% identity and the existence of a conserved uORF start site beginning at bp −171 (mouse) and at bp −187 (human). To interrogate potential structural features within the 5′-UTR that drive enhanced translation, we generated deletion mutations of the 5′-UTR, as shown in Figure 8B, and subjected them to analysis in the bicistronic vector. A deletion of the proximal mouse 5′-UTR (bp −1 to −104) that included a portion of the uORF (DM1) did not affect the firefly/Renilla activity ratio, suggesting that an intact putative uORF and structural elements within this region are not required for translational augmentation (Figure 8C). In contrast, deletion of the distal region of the 5′-UTR (bp −105 to −280) abrogated the firefly/Renilla activity ratio almost completely. The cognate distal deletion of the human PDX1 5′-UTR showed a similar loss in activity (Figure 8C). Notably, these alterations in firefly/Renilla activity ratios were not caused by changes in firefly or Renilla luciferase mRNA levels (Figure 8D). These data indicate that the distal portion of the Pdx1 mRNA 5′-UTR is required for full translational activity and that the presence of an intact putative uORF is not required.
Figure 8.

Deletional analysis of the Pdx1 5′UTR. A, Comparison of mouse and human Pdx1 5′-UTRs, showing in gray highlighting the positions of the putative uORFs. B, Schematic representation of deletion mutants of the mouse and human Pdx1 5′-UTRs that were inserted into the bicistronic vector between the Renilla luciferase (RLuc) and firefly luciferase (FLuc) genes. C, Results of dual-luciferase assays after transfection of the constructs in panel B into MIN6 cells. D, qRT-PCR from total RNA for the Renilla luciferase and firefly luciferase mRNAs (normalized to Actb mRNA). Data represent means ± SEM (n = 3–8). *, P < .05 compared with the empty vector control.
Discussion
In T1D, protein unfolding and ER stress in the β-cell are thought to arise when local or generalized inflammation (arising from innate or adaptive immune processes and proinflammatory cytokines) triggers pathways that lead to nitric oxide production, oxidative stress, and/or reductions in SERCA2 levels (33). Under these conditions, the UPR induces generalized reductions in mRNA translation initiation, along with a simultaneous increase in or maintenance of translation of specific mRNAs needed for stress remediation (12). In this study, we examined the differential translational responses to proinflammatory cytokines of key stress-responsive mRNAs in the β-cell. Pdx1 mRNA exhibits unique properties under stress that contribute to its ongoing translation.
The translational regulation of mRNAs occurs predominately at the levels of initiation and elongation, where factors such as eIF2α, eIF4E, and the eIF4F complex control initiation, and eucaryotic translation elongation factor 2 (eEF2) controls elongation (34, 35). Under conditions of fuel surfeit, pathways such as the mammalian target of rapamycin (mTOR) promote eIF4E and eEF2 activity to enhance initiation and elongation (36). In contrast, under conditions of stress, such as during fuel (amino acid) deficiency or ER stress, translation of many transcripts is repressed at the level of initiation by phosphorylation of eIF2α (12). This repression is also mediated by the availability and assembly of cap-binding translation factors. In the β-cell, it was recently shown that continued translation of mouse Cpe mRNA is dependent on the cap component eIF4G1 and is repressed under conditions of ER stress (37). In contrast, other mRNAs are translationally activated during stress. The Atf4 and Chop mRNAs each contain inhibitory uORFs that repress translation of the downstream coding ORF under normal conditions, but, during the UPR, initiation at these uORFs is repressed, whereas translation from the coding ORF predominates (38, 39).
Our studies demonstrate that both activation and repression of translation are pertinent to β-cell physiology during the UPR and that proinflammatory cytokines are sufficient to induce such translational control. Interestingly, whereas thapsigargin causes rapid shutdown of global translation initiation, we observed that cytokines cause changes to the ribosomal occupancy of Atf4, Chop, and Cpe before changes in global translation (as assessed by PRP analysis) are evident. This finding indicates that physiological stress, such as that induced by proinflammatory cytokines, regulates translation of specific transcripts before a global shutdown of translation initiation and that such responses may be critical to an appropriate β-cell stress response.
Pdx1 is a protein of critical importance to the β-cell, because it is necessary for β-cell development and function. Pdx1 has also been shown to play a key role in β-cell ER stress remediation, because it activates the genes necessary for ER homeostasis, including Atf4, Wfs1, and Ero1lb. Haploinsufficiency of Pdx1 in mice predisposes animals to ER stress and β-cell apoptosis (17). Previously, we showed that in the setting of T1D in the NOD mouse, the reduction in Pdx1 protein levels in the β-cell may be a contributing factor to the increasing ER stress and β-cell dysfunction seen in that model (5). Our studies here are the first to provide a potential mechanism whereby the Pdx1 5′-UTR maintains translation at relatively heightened levels during ER stress, despite global suppression of translation initiation. We point out that cytokine treatment (and thapsigargin to a lesser extent) caused a relative reduction in the translational activity mediated by the Pdx1 5′-UTR. Nevertheless, the absolute translational activity of this 5′-UTR element was greater than that seen with many other 5′-UTR elements, including that of hSPA.
Our studies indicated that a discrete region of the 5′-UTR of the mouse Pdx1 mRNA has properties that enhance translation of downstream coding regions. Using a bicistronic reporter plasmid, we showed that the region between −105 and −280 independently drives reporter gene translation, whereas the region between −1 and −104 and the 5′-UTR of Cpe is unable to do so. The homologous region of the human PDX1 gene retained similar properties, suggesting that the phenomenon may be applicable to humans. Although we investigated the 5′-UTR of Pdx1 based on its conservation across species, we acknowledge that other mechanisms may also contribute to translational regulation, including elements within the 3′-UTR.
The mechanisms driving translation of mRNAs during stress have been studied for a number of genes and include phenomena such as IRESs and uORFs. uORFs have been estimated to occur in up to 40% of transcripts (35), but whether such elements function in a regulatory manner requires experimental verification. We identified a conserved uORF in the mouse and human Pdx1 5′-UTR. Because (1) translation of uORFs typically requires 5′-end–dependent ribosome scanning in a manner similar to traditional cap-dependent translation initiation (35) and (2) deletion of the distal portion of this uORF in the mouse gene did not affect translational activity, we do not believe that the uORF mechanism is applicable to the translational activity of the Pdx1 5′-UTR.
IRESs are a heterogeneous group of elements found in the 5′-UTR of viral and cellular mRNAs that allow 5′ cap-independent recruitment of ribosomes for translation initiation (40). Although the existence of an IRES is largely determined experimentally, it is believed that specific forms of secondary structure in IRESs may allow for identification of such elements (41). In this regard, the Pdx1 5′-UTR contains ∼75% G-C content and is predicted to contain secondary structural elements that may mimic those of other IRESs. Nevertheless, the existence of IRESs in mammalian mRNAs continues to remain controversial, but our data suggest that the Pdx1 5′-UTR may harbor such an element, particularly because it can drive translation of a downstream ORF without the need for a 5′ cap.
Taken together, our findings shed new light on mechanisms that maintain Pdx1 protein synthesis under conditions of stress. Although not directly tested in this study, it is conceivable that regulation of Pdx1 mRNA translation by its 5′-UTR may be important during pancreas development, because cellular differentiation might also be considered a form of cellular stress. A better understanding of the mechanisms involved in Pdx1 translation, such as the factors that may promote Pdx1 mRNA translation via its 5′-UTR, could lead to therapies aimed to preserve Pdx1 expression in diabetic and prediabetic states.
Acknowledgments
We acknowledge N. Stull and K. Benninger in the Indiana University Diabetes Research Center Islet Core for isolation of islets and Dr P. Silveyra from Pennsylvania State University for providing the bicistronic reporter vector.
This work was supported by the National Institutes of Health (Grants R01 DK060581 and R01 DK083583, the Ball Bros. Foundation and the George and Francis Ball Foundation (grants to R.G.M.), the American Diabetes Association (Junior Faculty Award to S.A.T.), the Manpei Suzuki Diabetes Foundation (postdoctoral fellowship grant to M.H.), and the American Heart Association (predoctoral fellowship grant to A.T.T.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ATF4
- activating transcription factor 4
- ATF6
- activating transcription factor 6
- CHOP
- CAAT/enhancer binding protein homologous protein
- EEF2
- eucaryotic translation elongation factor 2
- eIF2α
- eukaryotic translation initiation factor 2α
- 4EBP1
- eukaryotic translation initiation factor 4E-binding protein 1
- ER
- endoplasmic reticulum
- hSPA
- human surfactant protein A
- IRE1
- inositol-requiring 1
- IRES
- internal ribosomal entry site
- NOD
- nonobese diabetic
- ORF
- open reading frame
- PERK
- PRKR-like endoplasmic reticulum kinase
- P/M
- polyribosome/monoribosome
- PRP
- polyribosomal profile
- qRT-PCR
- quantitative real-time RT-PCR
- SERCA
- sarcoendoplasmic reticulum Ca2+ ATPase
- TID
- type I diabetes
- uORF
- upstream open reading frame
- UPR
- unfolded protein response
- UTR
- untranslated region.
References
- 1. Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10:501–513 [DOI] [PubMed] [Google Scholar]
- 2. Mathis D, Vence L, Benoist C. β-Cell death during progression to diabetes. Nature. 2001;414:792–798 [DOI] [PubMed] [Google Scholar]
- 3. Maganti A, Evans-Molina C, Mirmira RG. From immunobiology to β-cell biology: The changing perspective on type 1 diabetes [published online ahead of print April 29, 2014]. Islets. doi: 10.4161/isl.28778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Sullivan-Murphy B, Urano F. ER stress as a trigger for β-cell dysfunction and autoimmunity in type 1 diabetes. Diabetes. 2012;61:780–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tersey SA, Nishiki Y, Templin AT, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Engin F, Yermalovich A, Nguyen T, et al. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci Transl Med. 2013;5:211ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marhfour I, Lopez XM, Lefkaditis D, et al. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia. 2012;55:2417–2420 [DOI] [PubMed] [Google Scholar]
- 8. Kaufman RJ, Back SH, Song B, Han J, Hassler J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in β-cells. Diabetes Obes Metab. 2010;12(suppl 2):99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Back SH, Scheuner D, Han J, et al. Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in β cells. Cell Metab. 2009;10:13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang HY, Wek RC. Phosphorylation of the α-subunit of the eukaryotic initiation factor-2 (eIF2α) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem. 2005;280:14189–14202 [DOI] [PubMed] [Google Scholar]
- 11. Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108 [DOI] [PubMed] [Google Scholar]
- 12. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. β-Cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the β-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12:1763–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609 [DOI] [PubMed] [Google Scholar]
- 15. Offield MF, Jetton TL, Labosky PA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–995 [DOI] [PubMed] [Google Scholar]
- 16. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17:138–139 [DOI] [PubMed] [Google Scholar]
- 17. Sachdeva MM, Claiborn KC, Khoo C, et al. Pdx1 (MODY4) regulates pancreatic β cell susceptibility to ER stress. Proc Natl Acad Sci USA. 2009;106:19090–19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robbins RD, Tersey SA, Ogihara T, et al. Inhibition of deoxyhypusine synthase enhances islet β cell function and survival in the setting of endoplasmic reticulum stress and type 2 diabetes. J Biol Chem. 2010;285:39943–39952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyazaki J, Araki K, Yamato E, et al. Establishment of a pancreatic β cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132 [DOI] [PubMed] [Google Scholar]
- 20. Stull ND, Breite A, McCarthy RC, Tersey SA, Mirmira RG. Mouse islet of Langerhans isolation using a combination of purified collagenase and neutral protease. J Vis Exp. 2012;67:e4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maier B, Ogihara T, Trace AP, et al. The unique hypusine modification of eIF5A promotes islet β cell inflammation and dysfunction in mice. J Clin Invest. 2010;120:2156–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evans-Molina C, Robbins RD, Kono T, et al. Peroxisome proliferator-activated receptor γ activation restores islet function in diabetic mice through reduction of endoplasmic reticulum stress and maintenance of euchromatin structure. Mol Cell Biol. 2009;29:2053–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iype T, Francis J, Garmey JC, et al. Mechanism of insulin gene regulation by the pancreatic transcription factor Pdx-1: application of pre-mRNA analysis and chromatin immunoprecipitation to assess formation of functional transcriptional complexes. J Biol Chem. 2005;280:16798–16807 [DOI] [PubMed] [Google Scholar]
- 24. Kulkarni RN, Roper MG, Dahlgren G, et al. Islet secretory defect in insulin receptor substrate 1 null mice is linked with reduced calcium signaling and expression of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2b and -3. Diabetes. 2004;53:1517–1525 [DOI] [PubMed] [Google Scholar]
- 25. Lipson KL, Fonseca SG, Ishigaki S, et al. Regulation of insulin biosynthesis in pancreatic β cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254 [DOI] [PubMed] [Google Scholar]
- 26. Wang G, Guo X, Silveyra P, Kimball SR, Floros J. Cap-independent translation of human SP-A 5′-UTR variants: a double-loop structure and cis-element contribution. AJP Lung Cell Mol Physiol. 2009;296:L635–L647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cardozo AK, Ortis F, Storling J, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005;54:452–461 [DOI] [PubMed] [Google Scholar]
- 28. Chambers KT, Unverferth JA, Weber SM, Wek RC, Urano F, Corbett JA. The role of nitric oxide and the unfolded protein response in cytokine-induced β-cell death. Diabetes. 2008;57:124–132 [DOI] [PubMed] [Google Scholar]
- 29. Jeffrey KD, Alejandro EU, Luciani DS, et al. Carboxypeptidase E mediates palmitate-induced β-cell ER stress and apoptosis. Proc Natl Acad Sci USA. 2008;105:8452–8457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Teske BF, Baird TD, Wek RC. Methods for analyzing eIF2 kinases and translational control in the unfolded protein response. Methods Enzymol. 2011;490:333–356 [DOI] [PubMed] [Google Scholar]
- 31. Cho S, Park SM, Kim TD, Kim JH, Kim K-T, Jang SK. BiP internal ribosomal entry site activity is controlled by heat-induced interaction of NSAP1. Mol Cell Biol. 2007;27:368–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim YK, Jang SK. Continuous heat shock enhances translational initiation directed by internal ribosomal entry site. Biochem Biophys Res Commun. 2002;297:224–231 [DOI] [PubMed] [Google Scholar]
- 33. Evans-Molina C, Hatanaka M, Mirmira RG. Lost in translation: endoplasmic reticulum stress and the decline of β-cell health in diabetes mellitus. Diabetes Obes Metab. 2013;15(suppl 3):159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dever TE, Green R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol. 2012;4:a013706–a013706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Somers J, Pöyry T, Willis AE. A perspective on mammalian upstream open reading frame function. Int J Biochem Cell Biol. 2013;45:1690–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945 [DOI] [PubMed] [Google Scholar]
- 37. Liew CW, Assmann A, Templin AT, et al. Insulin regulates carboxypeptidase E by modulating translation initiation scaffolding protein eIF4G1 in pancreatic β cells. Proc Natl Acad Sci USA. 2014;111:E2319–E2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA. 2004;101:11269–11274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Palam LR, Baird TD, Wek RC. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem. 2011;286:10939–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Filbin ME, Kieft JS. Toward a structural understanding of IRES RNA function. Curr Opin Struct Biol. 2009;19:267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]