

Abstract
Rationale: DNA-based microbiological studies are moving beyond studying healthy human microbiota to investigate diverse infectious diseases, including chronic respiratory infections, such as those in the airways of people with cystic fibrosis (CF) and non-CF bronchiectasis. The species identified in the respiratory secretion microbiota from such patients can be classified into those that are common and abundant among similar subjects (core) versus those that are infrequent and rare (satellite). This categorization provides a vital foundation for investigating disease pathogenesis and improving therapy. However, whether the core microbiota of people with different respiratory diseases, which are traditionally associated with specific culturable pathogens, are unique or shared with other chronic infections of the lower airways is not well studied. Little is also known about how these chronic infection microbiota change from childhood to adulthood.
Objectives: We sought to compare the core microbiota in respiratory specimens from children and adults with different chronic lung infections.
Methods: We used bacterial 16S rRNA gene pyrosequencing, phylogenetic analysis, and ecological statistical tools to compare the core microbiota in respiratory samples from three cohorts of symptomatic children with clinically distinct airway diseases (protracted bacterial bronchitis, bronchiectasis, CF), and from four healthy children. We then compared the core pediatric respiratory microbiota with those in samples from adults with bronchiectasis and CF.
Measurements and Main Results: All three pediatric disease cohorts shared strikingly similar core respiratory microbiota that differed from adult CF and bronchiectasis microbiota. The most common species in pediatric disease cohort samples were also detected in those from healthy children. The adult CF and bronchiectasis microbiota also differed from each other, suggesting common early infection airway microbiota that diverge by adulthood. The shared core pediatric microbiota included both traditional pathogens and many species not routinely identified by standard culture.
Conclusions: Our results indicate that these clinically distinct chronic airway infections share common early core microbiota, which are likely shaped by natural aspiration and impaired clearance of the same airway microbes, but that disease-specific characteristics select for divergent microbiota by adulthood. Longitudinal and interventional studies will be required to define the relationships between microbiota, treatments, and disease progression.
Keywords: core microbiota, cystic fibrosis, bronchiectasis, protracted bacterial bronchitis
A growing body of evidence indicates that the airways of even healthy people contain detectable microbes (1–4), and that these microbiota are altered in many chronic lung diseases. For example, culture-independent analyses of cystic fibrosis (CF) respiratory samples have revealed remarkably diverse microbial communities compared with culture results. Among the bacteria detected most frequently, and at highest abundance, in those CF samples were anaerobic species not usually detected by standard laboratory methods (5). Both cross-sectional and longitudinal studies indicate that CF airway microbiota generally decrease in diversity over time, concurrent with a decrease in average lung function (6–9). Whether these ecological changes are the cause of lung function decline, or whether they are the result of increasing antibiotic exposure or changing properties of the infected airways, are not understood (9). Furthermore, exactly where the species that comprise CF secretion microbiota come from, and whether features unique to the CF airway or its secretions select for CF-specific microbiota, are unknown.
Several other childhood lung diseases also involve chronic airway infection, including non-CF bronchiectasis and protracted bacterial bronchitis (10). At least some underlying factors that predispose to infection in CF, bronchiectasis, and protracted bacterial bronchitis may be shared (e.g., mucus hypersecretion or stasis, impaired airway clearance), whereas others vary (e.g., inflammatory response, intensity of antibiotic use, physicochemical mucus characteristics), and the associated infectious microbiota could therefore differ at the beginning or throughout these diseases. In support of this concept, specific microbes have been associated with the pathogenesis of each of these diseases in culture-based studies. For example, Staphylococcus aureus and Pseudomonas aeruginosa are traditionally associated with CF lung disease (11). In contrast, Streptococcus pneumoniae, Moraxella catarrhalis, and Haemophilus influenzae are traditionally associated with both protracted bacterial bronchitis and early bronchiectasis (10), with P. aeruginosa associated with more advanced bronchiectasis disease (12). Treatment regimens, therefore, tend to differ for each disease, designed to target specific pathogens. Moreover, it has been suggested that these bacteria play primary roles in initiating or driving associated symptoms (13), which could imply disease-specific infection pathogenesis. However, it is now known that children with CF have lung disease without detectable infection by P. aeruginosa or S. aureus (14, 15). The identification of diverse microbiota in even healthy airways further complicates the task of delineating “infected” and/or “pathogenic” from “normal” conditions. Therefore, comparing the earliest stages of chronic infection for diverse airways diseases could be helpful in understanding whether infection begins (and drives disease) in distinct or similar ways in different conditions.
An analytical approach that has been particularly useful in understanding environmental and infectious microbiota involves partitioning the constituent microbes into core and satellite species (7). In cross-sectional studies, core species are those that are detected in the majority of samples from subjects with a specific disease, whereas satellite species are those that are infrequent. Although this categorization does not necessarily identify which species are pathogens, it provides a useful framework within which to compare microbiota from different subject groups, and the resulting similarities and differences have important implications for infection pathogenesis. Here, we compared the core and satellite microbiota in three very different cohorts of children with protracted bacterial bronchitis, bronchiectasis, and CF using next-generation sequencing and ecological analytical approaches. Because these study subjects were children, and therefore at relatively early stages of disease, we hypothesized that these cohorts would have similar, rather than disease-specific, early core respiratory sample microbiota. We then compared pediatric and adult CF and bronchiectasis respiratory metacommunities, using data from two of our recent adult studies (16, 17), to identify evidence of disease-specific changes in microbiota as patients with each disease progress to adulthood.
Methods
Patients and Clinical Samples
All children in this study were participants in previous studies of CF, protracted bacterial bronchitis, and bronchiectasis, respectively (18–20). Inclusion and exclusion criteria and study details are listed in the online supplement. Sputum and bronchoalveolar lavage (BAL) fluid, as well as clinical data, were collected during those studies, as previously described (18–20). The study was approved by the institutional review boards at Seattle Children’s Hospital (Seattle, Washington) for the CF samples, and at the Royal Children’s Hospital (Brisbane, QLD, Australia) for the protracted bacterial bronchitis, bronchiectasis and control pediatric samples. Data from two adult bronchiectasis (n = 38 samples, age 37–74 yr) and CF (n = 30, 18–55 yr) cohorts from previous studies were used for metacommunity comparisons with the microbiota from the pediatric cohorts (16, 17).
DNA Extraction and Quantitative PCR
Full details of DNA extraction from BAL and sputum samples, and of the use of quantitative PCR to determine total bacterial abundance using universal bacterial primers, are in the online supplement.
Bacterial Tag-Encoded FLX Amplicon Pyrosequencing and Sequence Analyses
The bacterial taxonomy of each sample was evaluated using bacterial tag-encoded FLX-titanium pyrosequencing of a fragment of the 16S rRNA gene covering the V1–V3 variable regions, as described previously (21). Details of amplification, sequencing, and sequence processing and analysis are in the online supplement.
Statistical Analyses
To avoid potential biases in comparisons of diversity between local communities due to varying number of sequences per sample, a randomized resampling method, using three indices of diversity (S*, H’, and 1-D), were employed as previously described (7). Bacterial species within each metacommunity were partitioned into core and satellite species groups using the Poisson distribution test as previously described (7) (see the online supplement).
Results
Pediatric Study Cohorts
We analyzed the microbiota in respiratory samples (sputum and BAL fluid) from three cohorts of children with bronchiectasis, protracted bacterial bronchitis, and CF, respectively. All of these children either underwent bronchoscopy for chronic wet cough or were expectorating, indicative of symptomatic disease. As shown in Table 1, the cohorts were substantially different in terms of demographic and treatment features that could play important roles in determining the airway microbiota, including age, antibiotic exposure, and P. aeruginosa culture positivity, which have each been shown to correlate inversely with airway microbiota diversity in CF, and FEV1 % predicted, which correlates directly with diversity (9). The children with CF also had higher rates of inhaled corticosteroid use, a treatment that could conceivably alter airway microbiota constituency (2). The CF cohort originated entirely from a single hospital in the United States, whereas all children with bronchiectasis and protracted bacterial bronchitis were from a single hospital in Australia. Each of these differences, if they were to influence airway microbiota, would be unlikely to bias our analyses toward finding microbial similarities between the three cohorts.
Table 1.
Demographic characteristics of the pediatric study groups
| Clinical Characteristic | BE* | PBB | CF |
|---|---|---|---|
| Subjects, n | 19 | 12 | 25 |
| Geographic origin | Australia (Brisbane) | Australia (Brisbane) | United States (Seattle) |
| Sample type | 9 BAL and 10 sputum | All BAL | All sputum |
| Age, yr (mean ± SD, range) | 8.9 ± 4.7, 1.8–16.3† | 2.3 ± 1.7, 0.6–5.5‡,§ | 12.5 ± 3.5, 2.3–17.7 |
| Female sex, % | 36 | 25 | 68 |
| FEV1 % predicted (mean ± SD, range, [no. of subjects with measurements]) | 85.7 ± 21, 41–120 (15) | 117 ± 1.4, 116 and 118 (2)‡,§ | 77.9 ± 24, 22.2–116.2 (21) |
| Subjects on antibiotics at sampling, n (%)|| | 8/19 (42) | 1/12 (8) | 7/25 (28) |
| Subjects on corticosteroids at sampling, n (%) | 6/19 (32), 1 oral, 5 inhaled | 2/12 (16), all inhaled | 11/25 (48), all inhaled |
| Culture positive for P. aeruginosa, n (%)¶ | 0 | 0 | 8 (34) |
| Culture positive for S. aureus, n (%)¶ | 2 (11) | 2 (16) | 22 (96) |
| Culture positive for H. influenzae, n (%)¶ | 4 (19) | 6 (50) | 7 (30) |
Definition of abbreviations: BAL = bronchoalveolar lavage; BE = bronchiectasis; CF = cystic fibrosis; PBB = protracted bacterial bronchitis.
Of the 10 children with bronchiectasis with additional diagnoses available, 5/10 (50%) had idiopathic bronchiectasis, 1 had previously had an airway foreign body, 1 had primary ciliary dyskinesia, 1 had chronic aspiration due to a tracheoesophageal fistula, 1 had Mounier-Kuhn syndrome, and 1 had common variable immunodeficiency.
P < 0.01 compared with CF.
P < 0.001 compared with CF.
P < 0.001 compared with bronchiectasis.
Antibiotics for children with CF included tobramycin (inhaled), linezolid, levofloxacin, ciprofloxacin, trimethoprim-sulfamethoxazole, ticarcillin-clavulanate, and amoxicillin-clavulanate. Antibiotics for children with protracted bacterial bronchitis included only erythromycin. Antibiotics for children with bronchiectasis included erythromycin, azithromycin, roxithromycin, clarithromycin, amoxicillin-clavulanate, cefotaxime, ceftriaxone, and ticarcillin-clavulanate.
Of 23 subjects with CF with culture data available; all subjects with protracted bacterial bronchitis and bronchiectasis had available culture data.
Pediatric Community Diversity and Composition
Bacterial pyrosequencing data were used to assess and compare diversity and composition of microbiota from respiratory samples within and across the 3 pediatric cohorts. A total of 408,335 bacterial sequence reads (mean ± SD per sample, 5,041 ± 3,557), identifying 104 genera and 225 distinct operational taxonomic units classified to species level (see Table E1 in the online supplement), were generated from all samples combined. The average numbers of bacterial sequence reads per sample were similar among the three cohorts (mean ± SD: bronchiectasis, 4,284 ± 3476; protracted bacterial bronchitis, 4,471 ± 3718; and CF, 4,785 ± 2,702). Estimates of total bacterial abundances were also similar when quantified by quantitative PCR (for all samples, including sputum and BAL cell pellets, from which sufficient DNA remained after sequencing analysis, mean cfu/ml equivalents/sample ± SD: bronchiectasis, 1.47 × 107 ± 2.32 × 107, n = 18 samples; protracted bacterial bronchitis, 9.89 × 106 ± 1.38 × 107, n = 10; and CF, 5.82 × 107 ± 6.28 × 107, n = 21).
Bacterial diversity within and between cohorts was compared using three indices of diversity: species richness (S*, the total number of species), Shannon-Wiener (H’, a metric that accounts for both number and relative abundance of species), and Simpson’s (1-D, a measure of the probability that two species randomly selected from a sample will differ). Bacterial diversity between bronchiectasis sputum and BAL samples was compared using all three measures and found not to be significantly different (P > 0.05, Figure E1), and hence those samples were combined for subsequent analyses. S* was found to vary substantially within cohorts, with means (±SD) of 30.6 (±10.5), 30.9 (±11.5), and 19.5 (±10.0) for the bronchiectasis, protracted bacterial bronchitis, and CF cohorts, respectively (Figure 1); similar results were obtained for H’ and 1-D (Figure 1 and Table E2). Diversity between pediatric cohorts was not significantly different (P > 0.05) in all instances, except for one measure (H’) between the protracted bacterial bronchitis and CF cohorts (Table E2).
Figure 1.
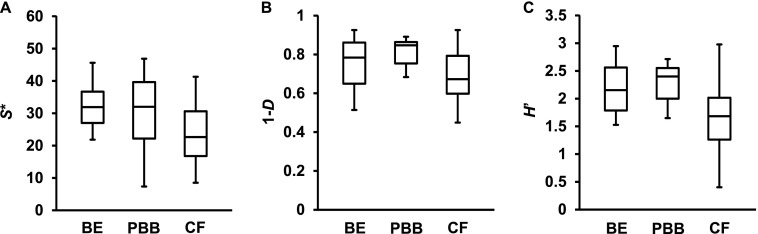
Box plot comparisons of bacterial diversity between the bronchiectasis (BE), protracted bacterial bronchitis (PBB), and cystic fibrosis (CF) cohorts. Given are three measures of diversity: species richness (S*), Simpson’s complement index (1-D), and Shannon-Wiener index (H’). The top and bottom boundaries of each box indicate the 75th and 25th quartile values, respectively, and lines within each box represent the 50th quartile (median) values. Ends of whiskers mark the lowest and highest diversity values in each instance. One-way ANOVA summary statistics are given in Table E2. The only significant difference found was for H’ between the CF and PBB cohorts (P = 0.027, Table E2).
Similarities and differences in community membership and structure for microbiota samples both within and between disease cohorts were assayed with two different indices: Sørensen (SSOR; which accounts for the number of species present in each community and those that are shared) and Bray-Curtis quantitative (SBC; similar to SSOR, but also accounts for abundance of each species). Each index varies in value from 0 to 1, with higher values indicating greater similarity. It has been previously observed that community composition is highly variable between patients with CF (7, 22). Here, we found that that observation held regardless of underlying pediatric lung disease, with mean (±SD) similarities in bacterial community membership taken pairwise for samples within pediatric cohorts of: SSOR = 0.44 ± 0.14 for bronchiectasis (n = 171 pairwise comparisons); SSOR = 0.51 ± 0.12 for protracted bacterial bronchitis (n = 66); and SSOR = 0.50 ± 0.12 for CF (n = 300), with a mean similarity of the entire pool of samples of SSOR = 0.46 ± 0.13 (n = 1,540) (Figure 2). For SBC, the mean between all samples was SBC = 0.19 ± 0.15 (n = 1,540), whereas that within cohorts was: SBC = 0.18 ± 0.14 for bronchiectasis (n = 171); SBC = 0.27 ± 0.12 for protracted bacterial bronchitis (n = 66); and SBC = 0.27 ± 0.17 for CF (n = 300). Therefore, individual sample microbiota differed within each pediatric disease group to a similar degree, and the microbiota identified within each pediatric cohort’s samples were pooled together into disease-specific metacommunities (defined as the set of all microbiota identified in samples from a given cohort) for subsequent comparisons.
Figure 2.

Dendrograms of bacterial community membership for all patients from the bronchiectasis (BE), protracted bacterial bronchitis (PBB), and cystic fibrosis (CF) cohorts. Black, gray, and white shaded boxes are given to show positions of BE, PBB, and CF samples, respectively. Patient species profiles were compared using the Sørensen index of similarity and unweighted pair-group method using arithmetic mean (UPGMA).
Microbiota within a Metacommunity Framework
Previously, we established that the categorization of component species in CF microbiota into core and satellite species revealed important aspects of species-abundance distributions within a metacommunity that would be neglected without such a distinction (7). A coherent metacommunity could be expected to exhibit a direct relationship between prevalence and abundance of individual species within the constituent communities. Consistent with this prediction, the abundance of species in each pediatric study cohort was significantly correlated with the number of individual respiratory sample communities that those species occupied (Figure 3A).
Figure 3.

Distribution and dispersal of bacterial species among bronchiectasis (BE), protracted bacterial bronchitis (PBB), and cystic fibrosis (CF) pediatric cohorts. (A) The number of samples for which each detected bacterial species (open circles) was observed, plotted against the abundance (log10 scale) of that species among all samples within each cohort (BE, r2 = 0.64, F1,175 = 311.5, P < 0.0001; PBB, r2 = 0.72, F1,128 = 321.1, P < 0.0001; and CF, r2 = 0.75, F1,141 = 418.4, P < 0.0001). (B) A dispersal plot to identify which bacterial species are randomly distributed within each cohort, a measure used to assign core versus satellite status. Index of dispersion was calculated as the ratio of variance to mean of abundance for each species within each cohort and plotted for each sample. The line depicts the 2.5% confidence limit for the χ2 distribution. Species that fall below this line are randomly distributed and were considered satellite species, whereas those that are above the line are nonrandomly distributed and were considered core species. The 97.5% confidence limit was not plotted, as no species fell below that line.
The species identified in each pediatric cohort metacommunity were then classified as core or satellite based upon their distributions (Figure 3B). Of the 177 species that comprised the bronchiectasis metacommunity, 88 were core and 89 were satellite species. The protracted bacterial bronchitis metacommunity (130 species) was comprised of 63 core and 67 satellite species, and the CF metacommunity (143 species) was comprised of 66 core and 77 satellite species.
Similarities between the three pediatric metacommunities were assayed using SSOR and SBC indices for all species, and for the core and satellite groups. The resulting cluster analysis revealed community membership to be relatively well conserved between cohorts for all species (SSOR = 0.74 ± 0.02 [n = 3 pairwise comparisons]), but much more highly conserved between the core groups (SSOR = 0.93 ± 0.02) (Figure 4). The satellite groups, comprised of randomly distributed species, were highly divergent (SSOR = 0.35 ± 0.01). Results from Bray-Curtis quantitative analyses were similar: all, SBC = 0.50 ± 0.10; core, SBC = 0.51 ± 0.10; and satellite, SBC = 0.10 ± 0.02. These results indicate that the high level of similarity between the three pediatric cohort metacommunities is attributable to core species.
Figure 4.

Dendrograms of community membership similarity between the pediatric bronchiectasis (BE), protracted bacterial bronchitis (PBB), and cystic fibrosis (CF) bacterial metacommunities and compared with adult CF and BE metacommunities. Given are whole, core and satellite microbiota. Metacommunity profiles were compared using the Sørensen index of similarity and unweighted pair-group method using arithmetic mean (UPGMA). Similarities between the microbiota from different cohorts are read as the location of the horizontal line connecting those cohorts (the “node”) on the y axis; for example, the similarity between pediatric core microbiota was 90% or greater, whereas that between adult core microbiota was 40% or greater.
To determine whether disease-specific microbiota emerge with disease progression into adulthood for the two chronic diseases, bronchiectasis and CF (protracted bacterial bronchitis usually improves long term with antibiotic treatment [10]), we compared the microbiota from our previous studies of adult CF (16) and bronchiectasis (17) cohorts with the pediatric cohort microbiota. The adult bronchiectasis cohort metacommunity (n = 38 patients) was comprised of 86 core and 54 satellite species, whereas the adult CF cohort (n = 30) metacommunity was comprised of 68 core and 81 satellite species. The resulting analyses revealed adult community membership to be highly divergent in all cases (whole microbiota, SSOR = 0.36 ± 0.03; core, SSOR = 0.46 ± 0.06; and satellite, SSOR = 0.09 ± 0.03 [n = 6 pairwise comparisons in all cases]; Figure 4). When relative abundances of constituent taxa were included to examine differences in community structure, an even more pronounced divergence was observed than for membership (whole, SBC = 0.07 ± 0.03; core, SBC = 0.07 ± 0.04; and satellite, SBC = 0.01 ± 0.01 [n = 6 in all cases]). In addition, comparisons between the adult CF and bronchiectasis metacommunities also revealed a high degree of divergence both in terms of membership (whole, SSOR = 0.38; core, SSOR = 0.48; and satellite, SSOR = 0.16; Figure 4) and structure (whole, SBC = 0.05; core, SBC = 0.05; and satellite, SBC = 0.02). Therefore, the adult CF and bronchiectasis microbiota differed substantially from each other, contrasting with the similarities among the pediatric cohort microbiota.
To test whether the adult and pediatric metacommunities were significantly similar or dissimilar, the whole, core, and satellite microbiota were compared between cohorts using the Raup and Crick probability-based similarity index (SRC) to determine whether compositional similarities in the microbiota were more or less significantly similar than expected by chance (Figure 5). The resulting cluster analysis revealed the pediatric whole, core, and satellite microbiota to be significantly dissimilar (SRC < 0.05) from the corresponding adult CF and bronchiectasis microbiota. Likewise, the whole and core microbiota from the pediatric cohorts were significantly similar (SRC > 0.95), adding further weight to the observations of pediatric community membership (SSOR) and structure (SBC) (Figure 5). In contrast, the pediatric satellite microbiota were not significantly similar to or dissimilar from each other (SRC > 0.05 and < 0.95), which would be expected, given that satellite microbiota in a given cohort represent rare and randomly distributed species. Furthermore, the adult CF and bronchiectasis microbiota were significantly dissimilar from each other (SRC < 0.05; Figure 5).
Figure 5.

Dendrograms of Raup and Crick probability-based similarity index (SRC) between the pediatric bronchiectasis (BE), protracted bacterial bronchitis (PBB), and cystic fibrosis (CF) bacterial metacommunities and compared with adult CF and BE metacommunities. Given are the whole, core, and satellite microbiota. SRC < 0.95 and SRC > 0.05 denote similarity no greater than expected by chance. SRC < 0.05 denotes significant dissimilarity, and SRC > 0.95 significant similarity. The 0.05 and 0.95 thresholds are depicted with dashed lines in each instance. Dendrograms were constructed using unweighted pair-group method using arithmetic mean (UPGMA).
Similarity of percentages (SIMPER) analysis of the metacommunities was used to identify those species that contributed most to the observed similarity between the three pediatric cohorts. These species, all core group members, are listed in decreasing order of contribution in Table 2. H. influenzae contributed the greatest amount to the observed similarity between pediatric samples. Most of the major contributors have previously been associated with respiratory tract infections and other opportunistic infections, and many are known inhabitants of the oral cavity, including both aerobic and anaerobic species (Table E1). These findings contrasted with the SIMPER analysis of the adult CF cohort, wherein P. aeruginosa contributed over 83% to the observed similarity between samples (Table E3a) in addition to three other species: S. pneumoniae, Prevotella melaninogenica, and Veillonella parvula. Within the adult bronchiectasis cohort, the main contributors to the overall similarity between samples were H. influenzae (50.4%), P. aeruginosa (8.62%), and V. dispar (8.11%) (Table E3b), along with S. pneumoniae and, as for the pediatric cohorts, other species associated with respiratory infections, many of which are known oral microbiota members. As also observed for the pediatric cohorts, the species identified from the SIMPER analyses for both adult cohorts were all core species in their respective metacommunities. Although there were some species identified in common for the adult and pediatric cohorts by the SIMPER analyses, these similarities were greatly outweighed by the differences in species content, accounting for the observed differences in whole and core microbiota within each adult–pediatric disease cohort pair.
Table 2.
Similarity of percentages analysis of bacterial community similarity (Bray-Curtis) between pediatric metacommunities
| Occupancy* |
Mean Abundance | ||||||
|---|---|---|---|---|---|---|---|
| Species | BE | PBB | CF | (%) | Mean Contribution | % Contribution | Cumulative % |
| Haemophilus influenzae | 12 | 11 | 19 | 23.22 | 18.79 | 37.44 | 37.44 |
| Streptococcus mitis | 18 | 12 | 25 | 14.59 | 8.45 | 16.84 | 54.28 |
| Prevotella melaninogenica | 16 | 11 | 24 | 9.16 | 4.59 | 9.15 | 63.43 |
| Veillonella dispar | 19 | 12 | 24 | 4.48 | 3.90 | 7.77 | 71.20 |
| Fusobacterium nucleatum | 17 | 11 | 21 | 2.63 | 1.99 | 3.97 | 75.17 |
| Neisseria flavescens | 12 | 11 | 19 | 5.04 | 1.73 | 3.44 | 78.61 |
| Porphyromonas catoniae | 17 | 11 | 18 | 1.66 | 1.05 | 2.09 | 80.70 |
| Haemophilus parainfluenzae | 15 | 10 | 16 | 1.30 | 1.04 | 2.06 | 82.76 |
| Porphyromonas gingivalis | 17 | 9 | 17 | 1.60 | 0.97 | 1.92 | 84.69 |
| Prevotella nanceiensis | 13 | 11 | 15 | 0.85 | 0.54 | 1.08 | 85.77 |
| Prevotella histicola | 12 | 7 | 19 | 0.81 | 0.46 | 0.92 | 86.69 |
| Granulicatella adiacens | 15 | 10 | 19 | 0.53 | 0.44 | 0.87 | 87.57 |
| Prevotella oris | 10 | 4 | 13 | 1.07 | 0.37 | 0.73 | 88.30 |
| Sphingomonas echinoides | 9 | 7 | 3 | 0.73 | 0.36 | 0.71 | 89.02 |
| Gemella morbillorum | 13 | 11 | 21 | 0.86 | 0.35 | 0.70 | 89.71 |
| Prevotella pallens | 16 | 8 | 13 | 0.63 | 0.32 | 0.63 | 90.35 |
Definition of abbreviations: BE = bronchiectasis; CF = cystic fibrosis; PBB = protracted bacterial bronchitis.
Given is occupancy, or the number of patients for each disease group in which a given species was detected. Next is mean % abundance of sequences for a species across the samples it was observed to occupy. Mean contribution represents the average contribution of a given species to the average similarity between samples (overall mean = 50.2%). Percentage contribution is the mean contribution divided by mean similarity across samples. The list of species is not exhaustive, so cumulative % value does not sum to 100%. Species-level identities of detected taxa are reported here. However, given the length of the ribosomal sequences analyzed, these identities should be considered putative.
Total n for each pediatric disease cohort: BE, 19; PBB, 12; CF, 25.
To investigate whether the core microbiota shared between the three pediatric disease groups were unique to children with airways disease, we identified the microbiota in BAL fluid from four children who underwent bronchoscopy for reasons other than suspected infection or chronic cough (described in the online supplement and Ref. 18). Of the 10 shared core species that contributed the most to similarity between disease cohorts from Table 2, 6 were detected in all four “control” samples, and the other 4 species were detected in 3 of the 4 samples, suggesting that the shared core microbiota are commonly found in the airways of children without chronic cough or infection (Table E4), consistent with prior airway microbiota studies that included samples from healthy subjects (3, 23).
Discussion
We found that cohorts of symptomatic children with three clinically distinct airways diseases—CF, bronchiectasis, and protracted bacterial bronchitis—shared remarkably similar respiratory sample core microbiota, the most common of which were also found in airway samples from children without clinical evidence of infection. The shared core microbiota included both traditional pathogens and many bacteria that are either not identified by routine clinical laboratory methods or are generally classified as “oral flora,” and are routinely ignored with respect to pathophysiology or treatment. In contrast, the respiratory sample microbiota from adults with CF and bronchiectasis differed significantly from each other and from those of children with the same disease. These results indicate that the core microbes in different chronic respiratory infection types likely begin similarly, yet are divergent by adulthood.
Although these observations provide valuable insight into the microbiota in diseased airways, they also identify next steps to take in understanding the pathogenesis of diverse airway infections. Given the high abundance and prevalence of the core species among pediatric patients, one interpretation of these results could be that the core species represent those that contribute the most to airway inflammation and disease progression. Alternatively, it could be that the satellite species play key pathogenic roles in specific contexts or stages of infection; for example, infection at any stage with specific low-abundance or infrequent bacteria could lead to increased inflammation, airway damage, mucus hypersecretion, and/or clinical deterioration. Although these questions are not fully answered by the current study, the relative rarity of any specific species in each satellite group may indicate a consistent, general role for the core groups in pathogenesis. Future studies will be required to longitudinally compare the changes over time in airway microbiota in health and disease to better understand the microbial contribution to pathogenesis, and the best therapeutic approach to infection.
These findings provide valuable perspective for results from studies of many respiratory diseases, usually at more advanced stages (2). A study using a relatively low-depth sequencing technique, 16S rRNA gene clone sequencing, comparing microbiota in explanted lungs from adults with end-stage bronchiectasis and CF identified diverse communities often dominated by Pseudomonas in both sample sets (24). Likewise, two studies using the same technique applied to sputa from patients with CF and bronchiectasis found some similarities and some differences between the subject groups, but no formal comparisons were made (25, 26). In support of our results (17), a recent pyrosequencing study of 21 older adults with non-CF bronchiectasis showed their sputum to contain diverse microbiota (27). Studies of sputum, BAL fluid, and explanted lung tissue from patients at various stages of disease due to chronic obstructive pulmonary disease and asthma, sometimes comparing with healthy subjects, have yielded varying and often conflicting results, in some cases demonstrating disease-specific microbiota, and, in others, identifying substantial overlap (1, 2, 23, 28, 29). In all of these studies, the effects of therapies that could conceivably impact airway microbiota, such as intubation, steroids, and antibiotics, were difficult to control for. Our results suggest that the microbiota observed in those older patients, at later stages of disease, likely had similar (or identical) beginnings and diverged over time. Whether those changes resulted from features of the underlying disease or from differences in treatment are not yet clear. Although the differences in antibiotic use among study cohorts support the idea that antibiotics did not have a profound, lasting impact on airway microbiota (similar to observations from CF [8, 30]), and there was no evidence that differences in steroid use resulted in differences in microbiota in these children, chronic or repetitive drug exposure could have gradual effects (as indicated by longitudinal studies in CF [9]). Regardless, these findings suggest that factors intrinsic to airways predominate in establishing the early microbiota in these chronic respiratory infections.
Several observations support these conclusions. Culture-based studies of children with CF demonstrated that radiographic lung disease, respiratory symptoms, and inflammation all precede the detection of “standard CF pathogens” (14, 15, 31, 32). All of the shared pediatric core microbes identified in the current study, many of which would traditionally be considered “oral flora,” were also commonly identified in oropharyngeal swabs from infants with CF by pyrosequencing (33). The identification of such “oral flora” has been associated with both inflammation (32) and structural lung disease (15) in children with CF. Similarly, piglets with engineered CF transmembrane conductance regulator mutations (34) concurrently develop diverse airway microbiota and lung disease similar to those observed in children with CF. The cultured microbiota of CF pigs were similar in constituency to those of wild-type pigs, but more persistent and higher in abundance. Therefore, the requirement for traditional pathogens for CF lung disease or symptom development is questionable.
Both cross-sectional and longitudinal studies of CF respiratory samples using molecular methods have demonstrated correlations between decreasing microbiota diversity, age, and disease progression (3, 8, 9, 30). Interestingly, some organisms traditionally associated with chronic airways disease were classified as core species in some disease cohorts, but not others in our study (for example, P. aeruginosa, S. aureus, and several species each of Veillonella and Prevotella were core members in both the CF and bronchiectasis metacommunities, but satellite for protracted bacterial bronchitis; Table E1). Collectively, these results could indicate that particular microbes are associated with and/or accelerate specific lung diseases. Alternatively, changes over time in airway microbiota, including the gradual decrease in community diversity observed in CF and in chronic obstructive pulmonary disease (23), and the gradual identification of P. aeruginosa in people with CF and non-CF bronchiectasis (24), could all result from intensified antibiotic treatment and/or underlying disease, but this concept would not require the microbes that eventually dominate to be particularly pathogenic. Similarly, changes in the respiratory microbiota could result from evolution (therapy driven or otherwise) in another reservoir that seeds the airways, such as the oropharynx (as suggested by abundance in our study of microbes traditionally associated with this space [35]) or the gastrointestinal tract (which a recent longitudinal study of infants with CF suggested could be a source for respiratory microbiota [33]).
Together with the above earlier studies, our results support a unified model of early airway infections, in which a defect in the clearance of otherwise normal airway microbiota (for example, due to early airway injury, altered mucus, defective ciliary motility, mechanical obstruction, or immunodeficiency) contributes to progressive inflammation and airway damage. In this model, an extension of the “vicious cycle hypothesis” (13), either medical treatment or the physical effects of underlying disease (such as diseased tissue or accumulated secretions) could lead to progressive change in the microbiota. These microbiological changes (including eventual selection for P. aeruginosa in CF or bronchiectasis) could, in turn, accelerate lung disease, or they could be consequences rather than causes of this progression. Future studies, either interventional or longitudinal, may be able to refine this model.
Antibiotics play important roles in the management of all three of these symptomatic chronic infections. For example, inhaled and oral antibiotics are commonly used in CF and bronchiectasis, both for maintenance (11, 36) and exacerbation treatments (11, 13). For protracted bacterial bronchitis, a prolonged course of antibiotics usually leads to resolution of symptoms (18). These observations underscore the importance of bacteria in the pathogenesis of each disease; the current findings provide a rationale for re-examining and comparing the culture-based antibiotic approaches to each of these airway infections, particularly during early stages.
Our study was limited by the small size of each subject group. The groups were not matched for age, treatment, disease severity, or demographics, limiting our ability to determine relationships between microbiota and these factors. However, the similarity in microbiota among demographically and clinically divergent pediatric groups could be interpreted to further support the concept of a common early pediatric respiratory microbiota; additional study with demographically well matched cohorts would be required to test this idea. Because the four “control” children in this study underwent bronchoscopy for respiratory symptoms (usually stridor [18]), they cannot be considered to be truly healthy, but they offered convenient comparison samples, because they had no clinical evidence of infection. We did not distinguish between viable and nonviable cells in the current analysis, due to the small volumes of many of the samples analyzed, and thus the detection of a microbe cannot definitively be said to indicate its persistence (although even transient or nonviable bacterial cells could potentially contribute to airway inflammation). Similarly, it could be proposed that the microbial diversity found in our study and others is due to contamination by upper airway bacteria during sample collection. However, the significant similarity in microbiota among sputum and BAL samples from the children with bronchiectasis in our study, as observed in studies of children with CF (5, 22), as well as the high abundance of microbes in our samples, argue against this issue significantly altering our findings.
In conclusion, children with early chronic lung infections due to three distinct diseases shared strikingly similar core airway microbiota that included many bacteria not generally identified by clinical laboratory culture. These similarities were independent of underlying diagnosis or disease severity, geography, antibiotic and steroid use, and age. These results indicate that different chronic airway infections begin similarly, remaining similar even after symptoms have begun, but diverging by adulthood. Our findings provide a baseline to compare with in future, longitudinal microbiota studies of diverse chronic airway infections, which would provide vital insight into the microbial determinants of chronic lung disease progression, and could lead to improved treatments.
Acknowledgments
Acknowledgments
The authors thank the participating patients and their families, B. van Yserloo and W. Osborne for technical assistance, and R. Gibson and H. Smith-Vaughan for helpful discussions.
Footnotes
Supported by Cystic Fibrosis Foundation grant HOFFMA07P0, American Thoracic Society grant CF-07-003, National Institutes of Health grant K02HL105543 (L.R.H.), the United Kingdom Natural Environment Research Council grant NE/H019456/1 (C.J.v.d.G.), and Australia’s National Health and Medical Research Council grants 1042601, 1040830, 1,019,834 and 1,034,703 (A.B.C.).
The sequence data reported in this paper have been submitted to the National Center for Biotechnology Information Short Read Archive database (Bioproject accession no. PRJNA200702).
Author Contributions: C.J.v.d.G. designed and performed the study and wrote the manuscript; L.C., G.B.R., C.P., and R.L.M. performed analyses and wrote the manuscript; K.D.B. and G.J.R. designed the study and wrote the manuscript; A.B.C. and L.R.H. designed the study, contributed specimens, performed the study, and wrote the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han MK, Huang YJ, Lipuma JJ, Boushey HA, Boucher RC, Cookson WO, Curtis JL, Erb-Downward J, Lynch SV, Sethi S, et al. Significance of the microbiome in obstructive lung disease. Thorax. 2012;67:456–463. doi: 10.1136/thoraxjnl-2011-201183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blainey PC, Milla CE, Cornfield DN, Quake SR. Quantitative analysis of the human airway microbial ecology reveals a pervasive signature for cystic fibrosis. Sci Transl Med. 2012;4:153ra130. doi: 10.1126/scitranslmed.3004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L, et al. Lung HIV Microbiome Project. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers GB, Carroll MP, Serisier DJ, Hockey PM, Jones G, Bruce KD. Characterization of bacterial community diversity in cystic fibrosis lung infections by use of 16s ribosomal DNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol. 2004;42:5176–5183. doi: 10.1128/JCM.42.11.5176-5183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox MJ, Allgaier M, Taylor B, Baek MS, Huang YJ, Daly RA, Karaoz U, Andersen GL, Brown R, Fujimura KE, et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS ONE. 2010;5:e11044. doi: 10.1371/journal.pone.0011044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Gast CJ, Walker AW, Stressmann FA, Rogers GB, Scott P, Daniels TW, Carroll MP, Parkhill J, Bruce KD. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2011;5:780–791. doi: 10.1038/ismej.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stressmann FA, Rogers GB, van der Gast CJ, Marsh P, Vermeer LS, Carroll MP, Hoffman L, Daniels TWV, Patel N, Forbes B, et al. Long-term cultivation-independent microbial diversity analysis demonstrates that bacterial communities infecting the adult cystic fibrosis lung show stability and resilience. Thorax. 2012;67:867–873. doi: 10.1136/thoraxjnl-2011-200932. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci USA. 2012;109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang AB, Redding GJ, Everard ML. Chronic wet cough: protracted bronchitis, chronic suppurative lung disease and bronchiectasis. Pediatr Pulmonol. 2008;43:519–531. doi: 10.1002/ppul.20821. [DOI] [PubMed] [Google Scholar]
- 11.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 12.Redding GJ. Bronchiectasis in children. Pediatr Clin North Am. 2009;56:157–171, xi. doi: 10.1016/j.pcl.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Chalmers JD, Smith MP, McHugh BJ, Doherty C, Govan JR, Hill AT. Short- and long-term antibiotic treatment reduces airway and systemic inflammation in non–cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2012;186:657–665. doi: 10.1164/rccm.201203-0487OC. [DOI] [PubMed] [Google Scholar]
- 14.Sagel SD, Gibson RL, Emerson J, McNamara S, Burns JL, Wagener JS, Ramsey BW Inhaled Tobramycin in Young Children Study Group; Cystic Fibrosis Foundation Therapeutics Development Network. Impact of Pseudomonas and Staphylococcus infection on inflammation and clinical status in young children with cystic fibrosis. J Pediatr. 2009;154:183–188. doi: 10.1016/j.jpeds.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mott LS, Park J, Murray CP, Gangell CL, de Klerk NH, Robinson PJ, Robertson CF, Ranganathan SC, Sly PD, Stick SM AREST CF. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax. 2012;67:509–516. doi: 10.1136/thoraxjnl-2011-200912. [DOI] [PubMed] [Google Scholar]
- 16.Rogers GB, Cuthbertson L, Hoffman LR, Wing PAC, Pope C, Hooftman DAP, Lilley AK, Oliver A, Carroll MP, Bruce KD, et al. Reducing bias in bacterial community analysis of lower respiratory infections. ISME J. 2013;7:697–706. doi: 10.1038/ismej.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogers GB, van der Gast CJ, Cuthbertson L, Thomson SK, Bruce KD, Martin ML, Serisier DJ. Clinical measures of disease in adult non-CF bronchiectasis correlate with airway microbiota composition. Thorax. 2013;68:731–737. doi: 10.1136/thoraxjnl-2012-203105. [DOI] [PubMed] [Google Scholar]
- 18.Chang AB, Yerkovich ST, Gibson PG, Anderson-James S, Petsky HL, Carroll ML, Masters IB, Marchant JM, Wurzel D, Upham JW.Pulmonary innate immunity in children with protracted bacterial bronchitis J Pediatr 2012161621–625.e1 [DOI] [PubMed] [Google Scholar]
- 19.Kapur N, Grimwood K, Masters IB, Morris PS, Chang AB. Lower airway microbiology and cellularity in children with newly diagnosed non-CF bronchiectasis. Pediatr Pulmonol. 2012;47:300–307. doi: 10.1002/ppul.21550. [DOI] [PubMed] [Google Scholar]
- 20.Wolter DJ, Emerson JC, McNamara S, Buccat AM, Qin X, Cochrane E, Houston LS, Rogers GB, Marsh P, Prehar K, et al. Staphylococcus aureus small-colony variants are independently associated with worse lung disease in children with cystic fibrosis. Clin Infect Dis. 2013;57:384–391. doi: 10.1093/cid/cit270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP) PLoS ONE. 2008;3:e3326. doi: 10.1371/journal.pone.0003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris JK, De Groote MA, Sagel SD, Zemanick ET, Kapsner R, Penvari C, Kaess H, Deterding RR, Accurso FJ, Pace NR. Molecular identification of bacteria in bronchoalveolar lavage fluid from children with cystic fibrosis. Proc Natl Acad Sci USA. 2007;104:20529–20533. doi: 10.1073/pnas.0709804104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maughan H, Cunningham KS, Wang PW, Zhang Y, Cypel M, Chaparro C, Tullis DE, Waddell TK, Keshavjee S, Liu M, et al. Pulmonary bacterial communities in surgically resected noncystic fibrosis bronchiectasis lungs are similar to those in cystic fibrosis. Pulm Med. 2012;2012:746358. doi: 10.1155/2012/746358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bittar F, Richet H, Dubus J-C, Reynaud-Gaubert M, Stremler N, Sarles J, Raoult D, Rolain J-M. Molecular detection of multiple emerging pathogens in sputa from cystic fibrosis patients. PLoS One. 2008;3:e2908. doi: 10.1371/journal.pone.0002908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duff RM, Simmonds NJ, Davies JC, Wilson R, Alton EW, Pantelidis P, Cox MJ, Cookson WOCM, Bilton D, Moffatt MF. A molecular comparison of microbial communities in bronchiectasis and cystic fibrosis. Eur Respir J. 2013;41:991–993. doi: 10.1183/09031936.00052712. [DOI] [PubMed] [Google Scholar]
- 26.Tunney MM, Einarsson GG, Wei L, Drain M, Klem ER, Cardwell C, Ennis M, Boucher RC, Wolfgang MC, Elborn JS. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respir Crit Care Med. 2013;187:1118–1126. doi: 10.1164/rccm.201210-1937OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, Woyke T, Allgaier M, Bristow J, Wiener-Kronish JP, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma J Allergy Clin Immunol 2011127372–381.e1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, Cooper J, Sin DD, Mohn WW, Hogg JC. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, Tunney MM, Wolfgang MC. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One. 2012;7:e45001. doi: 10.1371/journal.pone.0045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zemanick ET, Wagner BD, Sagel SD, Stevens MJ, Accurso FJ, Harris JK. Reliability of quantitative real-time PCR for bacterial detection in cystic fibrosis airway specimens. PLoS One. 2010;5:e15101. doi: 10.1371/journal.pone.0015101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gangell C, Gard S, Douglas T, Park J, de Klerk N, Keil T, Brennan S, Ranganathan S, Robins-Browne R, Sly PD AREST CF. Inflammatory responses to individual microorganisms in the lungs of children with cystic fibrosis. Clin Infect Dis. 2011;53:425–432. doi: 10.1093/cid/cir399. [DOI] [PubMed] [Google Scholar]
- 33.Madan JC, Koestler DC, Stanton BA, Davidson L, Moulton LA, Housman ML, Moore JH, Guill MF, Morrison HG, Sogin ML, et al. Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: interaction between intestinal and respiratory tracts and impact of nutritional exposures. MBio. 2012;3:e00251–12. doi: 10.1128/mBio.00251-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cardenas PA, Cooper PJ, Cox MJ, Chico M, Arias C, Moffatt MF, Cookson WO. Upper airways microbiota in antibiotic-naïve wheezing and healthy infants from the tropics of rural Ecuador. PLoS One. 2012;7:e46803. doi: 10.1371/journal.pone.0046803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barker AF, Couch L, Fiel SB, Gotfried MH, Ilowite J, Meyer KC, O’Donnell A, Sahn SA, Smith LJ, Stewart JO, et al. Tobramycin solution for inhalation reduces sputum Pseudomonas aeruginosa density in bronchiectasis. Am J Respir Crit Care Med. 2000;162:481–485. doi: 10.1164/ajrccm.162.2.9910086. [DOI] [PubMed] [Google Scholar]