

Abstract
Rationale: Alveolar liquid clearance is regulated by Na+ uptake through the apically expressed epithelial sodium channel (ENaC) and basolaterally localized Na+-K+-ATPase in type II alveolar epithelial cells. Dysfunction of these Na+ transporters during pulmonary inflammation can contribute to pulmonary edema.
Objectives: In this study, we sought to determine the precise mechanism by which the TIP peptide, mimicking the lectin-like domain of tumor necrosis factor (TNF), stimulates Na+ uptake in a homologous cell system in the presence or absence of the bacterial toxin pneumolysin (PLY).
Methods: We used a combined biochemical, electrophysiological, and molecular biological in vitro approach and assessed the physiological relevance of the lectin-like domain of TNF in alveolar liquid clearance in vivo by generating triple-mutant TNF knock-in mice that express a mutant TNF with deficient Na+ uptake stimulatory activity.
Measurements and Main Results: TIP peptide directly activates ENaC, but not the Na+-K+-ATPase, upon binding to the carboxy-terminal domain of the α subunit of the channel. In the presence of PLY, a mediator of pneumococcal-induced pulmonary edema, this binding stabilizes the ENaC-PIP2-MARCKS complex, which is necessary for the open probability conformation of the channel and preserves ENaC-α protein expression, by means of blunting the protein kinase C-α pathway. Triple-mutant TNF knock-in mice are more prone than wild-type mice to develop edema with low-dose intratracheal PLY, correlating with reduced pulmonary ENaC-α subunit expression.
Conclusions: These results demonstrate a novel TNF-mediated mechanism of direct ENaC activation and indicate a physiological role for the lectin-like domain of TNF in the resolution of alveolar edema during inflammation.
Keywords: epithelial sodium channel, pneumonia, protein kinase C-α, pulmonary edema, tumor necrosis factor
At a Glance Commentary
Scientific Knowledge on the Subject
The lectin-like domain of TNF, mimicked by the TIP peptide, improves alveolar liquid clearance (ALC) in multiple animal models of both hydrostatic and permeability edema, which has led to interest in its potential clinical use as a treatment for pulmonary edema. The precise mechanism of action of the peptide that leads to increased Na+ uptake in a homologous lung epithelial system has not been determined.
What This Study Adds to the Field
In a combined in vitro–in vivo study, we show that the TIP peptide directly binds to the intracellular carboxy-terminal domain of the α subunit of apical epithelial Na+ channel (ENaC). This binding maintains expression of the α subunit of ENaC and stabilizes the channel’s complex formation with myristoylated alanine-rich C kinase substrate and phosphatidylinositol 4,5-bisphosphate, which are essential for the open conformation, in the presence of pneumococcal pneumolysin (PLY), an important mediator of permeability edema. Knock-in mice expressing a TNF mutant lacking a functional ALC-stimulatory domain are more prone to develop edema than their wild-type counterparts after instillation of a low dose of PLY, which does not induce significant barrier dysfunction. Taken together, these data demonstrate, for the first time to our knowledge, a novel mechanism of ENaC activation and suggest a physiological role for the lectin-like domain of TNF in ALC.
Alveolar liquid clearance (ALC) is critical to preventing excess fluid accumulation in the alveoli (alveolar edema), which compromises gas exchange (1–4). Dysfunctional ALC correlates with morbidity and mortality in ARDS patients (5). ALC is mediated mainly by vectorial Na+ transport through the apical epithelial sodium channel (ENaC) and the basolateral Na+-K+-ATPase in type II alveolar epithelial cells (AECs). The magnitude of ENaC-mediated Na+ uptake in type II AEC depends on (1) proteolytic cleavage-induced excision of inhibitory tracts within proximal regions of the extracellular domains of the α and γ subunits, (2) the average open probability (Po) time of the channel, and (3) the expression level of functional channels. Functional ENaC channels typically consist of α, β, and γ subunits (6–8), with the α subunit being crucial for neonatal and adult lung liquid clearance (9). ENaC subunit expression is regulated by clathrin-mediated endocytosis (10), as well as by Nedd-4-1- or -2-mediated ubiquitinylation and degradation (11). The open conformation of ENaC is stabilized by its direct binding to phosphatidylinositol 4,5-bisphosphate (PIP2) in complex with myristoylated alanine-rich C kinase substrate (MARCKS) (12–14). MARCKS has been demonstrated to be a target of both the α and δ isoforms of protein kinase C (PKC-α and PKC-δ, respectively) (15, 16).
Proinflammatory cytokines, including IL-1β, transforming growth factor β, and tumor necrosis factor (TNF) can significantly impair ENaC activity, in a p38 MAP kinase– or PKC-dependent manner (17–20). TNF has a dual role in ENaC regulation (21). Activation of TNF receptor 1 mediates transcriptional inhibition of all three ENaC subunits—α, β, and γ—and induces PKC-dependent post-translational inhibition of ENaC-α subunit expression in vitro and in damaged lungs in vivo (19). By contrast, TNF increases edema reabsorption in a rat pneumonia model (22) and stimulates Na+ uptake in A549 cells in a catecholamine-independent manner (23). These activities are most likely mediated by the lectin-like domain of the cytokine (24), which is spatially distinct from the receptor binding sites (25, 26). The lectin-like domain of TNF is mimicked by the 17–amino acid circular TIP peptide (26), which increases amiloride-sensitive Na+ uptake in type II AECs (27, 28). The TIP peptide has been shown to activate ALC in several animal models of hydrostatic and permeability edema (21, 29–31). In a recently performed, placebo-controlled, double-blind Phase I clinical trial, TIP peptide (also named AP301) was observed not to induce serious adverse effects upon inhalation (32)
Bacterial and viral toxins have been shown to induce significant changes in ENaC Po and/or expression, leading to impaired ALC (33–36). It is therefore of high clinical relevance to identify novel mechanisms that increase ENaC function during pulmonary edema associated with ARDS and pneumonia, for which no standard therapy exists to date. Preliminary data from a Phase IIa interventional, randomized, double-blind, placebo-controlled, parallel-group study of patients with acute lung injury indicated that oral inhalation of TIP peptide caused earlier onset and more pronounced activation of pulmonary edema clearance, as compared to placebo-treated patients (Department of Anesthesia and Intensive Care Medicine, Medical University Vienna; ClinicalTrials.gov Identifier: NCT01627613). As such, the TIP peptide represents a therapeutic candidate for the treatment of pulmonary edema, but its mechanism of action remains elusive. This study was designed to determine how the TIP peptide, mimicking the lectin-like domain of TNF, activates ENaC in the presence or absence of pneumolysin (PLY). We also investigated whether the lectin-like domain of TNF plays a physiological role in ALC during PLY-induced edema formation. Some of the results of these studies have been previously reported in the form of an abstract (37).
Methods
Methodological details other than those described below are available in the online supplement.
Generation of Triple-Mutant TNF Knock-in Mice
C57BL/6 triple-mutant TNF knock-in (TKI) mice were generated upon substitution of nucleotides encoding for three crucial amino acids for the Na+ uptake stimulatory activity of TNF (Thr104, Glu106, and Glu109) by Ala-encoding nucleotides. To isolate the genomic TNF gene fragment, a hybridization probe of 300 bp was generated by PCR amplification using the primers mTNF 5′(atc ggt acc tta cac ggc gat ctt tcc gcc c) and mTNF 3′(atc ggt acc tta cac ggc gat ctt tcc gcc c) in combination with genomic C57BL/6 mouse DNA as the template. To isolate relevant fragments for homologous recombination, hybridization library screening (Incyte Genomics, St. Louis, MO) was performed and positive clones for the TNF gene locus were validated by restriction analysis. To improve the efficacy of the mutation procedure, a smaller PstI/HindIII fragment, containing the TIP domain, located in the first exon, was subcloned into pBluescript II KS (Stratagene, La Jolla, CA), as such generating the pBluescript mTNF subconstruct. Desired point mutations were inserted making use of the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) in combination with appropriate primers. Molecular characterization of positive embryonic stem (ES) cell clones required insertion of a Leu110Ser mutation in the third round, to generate an additional NheI restriction site. Next, the modified PstI/HindIII fragment was reintegrated into the sequence for native TNF. A neomycin resistance/thymidine kinase (neo/TK) selection cassette, flanked by loxP sites (floxed), was inserted into the mutated TNF construct to select transformed mouse ES cells. The sequence of the selection cassette was isolated from the vector construct and was integrated at nucleotide position 9,552 into an intron region of the TNF gene. This integration of the triple-mutated gene into the mouse genome was performed at the Roche Centre for Medical Genomics in the laboratories of Dr. Horst Bluethmann (F. Hoffmann-La Roche AG, Basel, Switzerland). The targeting vector was first introduced by electroporation into a culture of C57BL/6-derived ES cells. Cells in which the mutated gene, including the selection cassette, was integrated into the genome upon homologous recombination were selected for resistance to the neomycin-like drug G418. Subsequently, cells were transfected with Cre recombinase, an enzyme recognizing loxP sites, which excises the intervening DNA. Cre/lox recombination caused removal of the selection cassette from the mutated gene. Cells were then injected into mouse blastocysts. Reimplantation of these blastocysts into the uterus of pseudo-pregnant dams resulted in the birth of 27 chimeric mice, 5 of which (4 female, 1 male) were intercrossed with C57BL/6 mice. Genotyping demonstrated that the first generation of offspring from the chimeric mice included heterozygous individuals for the mutated gene, implying that the ES cells had entered the germline of the chimeric animals.
Results
The Primary Target of the TIP Peptide Is ENaC
Because both Na+-K+-ATPase (29) and ENaC (27, 28, 38) can be activated by the TIP peptide, we investigated in more detail which of these Na+ transporters is the primary target in a homologous cell system and whether the peptide has a direct or indirect effect on their activity.
As shown in Figure 1A, TIP peptide (50 μg/ml), when applied in the cell bath, significantly increased inward currents in H441 cells, a model of human Na+ absorptive airway epithelia (39), using a whole-cell, voltage-clamped, patch-clamp protocol with voltage steps ranging from −140 to +60 mV. The recorded current was completely inhibited by the ENaC inhibitor amiloride (10 μM). Deglycosylation of H441 cells with 100 U of N-glycosidase F 5 minutes before adding TIP peptide blunted the peptide’s effects on Na+ uptake (Figure 1A, inset), suggesting a crucial interaction of the peptide with glycosylated plasma membrane components, in correspondence with our previously obtained data in a heterologous cellular ENaC expression system (38).
Figure 1.
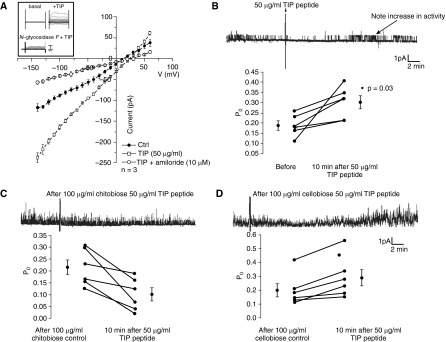
(A) Whole-cell, voltage-clamped patch-clamp current measurements (pA) of H441 cells treated or not with TIP peptide (50 μg/ml) in the cell bath in the presence or absence of amiloride (10 μM, n = 3/group). Inset: Effect of N-glycosidase F pretreatment (100 U, 5 min) of H441 cells on TIP peptide–mediated increase in Na+ uptake. (B–D) Single-channel patch-clamp measurements of 2F3 cells treated for 10 minutes with TIP peptide (50 μg/ml, n = 5) (B) or with TIP peptide pretreated with N,N′-diacetylchitobiose (100 μg/ml, n = 6) (C) or cellobiose (100 μg/ml, n = 6) (D).
To further test the effect of the TIP peptide on amiloride-sensitive ENaC activity, we performed single-channel analysis in membrane patches from Xenopus 2F3 cells (in cell-attached mode). As shown in Figure 1B, Po of ENaC increased from 0.18 ± 0.03 (before addition of TIP peptide) to 0.30 ± 0.04 (10 minutes after addition of TIP peptide into the pipette; P < 0.03; n = 5). Addition of the mutant TIP peptide, in which three residues (one Thr and two Glu) crucial for the Na+ uptake activation were exchanged against Ala (27), had no stimulatory effect (10 minutes after addition of mutant TIP: Po = 0.15 ± 0.04; n = 5; data not shown). Preincubation of TIP peptide with a sugar specifically binding to the lectin-like domain of TNF (N,N′-diacetylchitobiose (100 μg/ml)) (25, 26) decreased Po from 0.22 ± 0.04 to 0.10 ± 0.03 (10 minutes after addition of TIP; n = 6) (Figure 1C), whereas the control sugar cellobiose (100 μg/ml) had no inhibitory effect (Po increased from 0.20 ± 0.03 [before addition] to 0.29 ± 0.04 [10 minutes after addition] of TIP; n = 6) [Figure 1D]).
Pull-down experiments in which we used biotinylated human TNF (hTNF) revealed direct binding of human TNF (10 ng/ml) to recombinant ENaC-α, but not to the β and γ subunits (Figure 2A). A 2,000-fold molar excess of a PEGylated, soluble TNF receptor 1 complex over hTNF (20 μg/ml; Amgen, Thousand Oaks, CA), which does not interfere with the ALC-activating effect of TNF (21), only moderately blunted binding. By contrast, N,N′-diacetylchitobiose (100 μg/ml), but not cellobiose at the same concentration, completely inhibited the interaction (Figure 2A). In support of these results, there was strong binding activity of the TIP peptide, mimicking the lectin-like domain of TNF, to recombinant ENaC-α, with much weaker binding to the β and γ subunits (Figure 2B). As demonstrated in Figure 2C, biotinylated TIP or mutant TIP peptides, coupled to Dynabeads MyOne Streptavidin T1 magnetic beads (Invitrogen, Carlsbad, CA), bind to ENaC-α in lysates from control or ENaC-α-overexpressing H441 cells, with the signal being significantly lower for the mutant than for the native peptide. N,N′-diacetylchitobiose significantly reduced binding, whereas preincubation with cellobiose had no effect.
Figure 2.
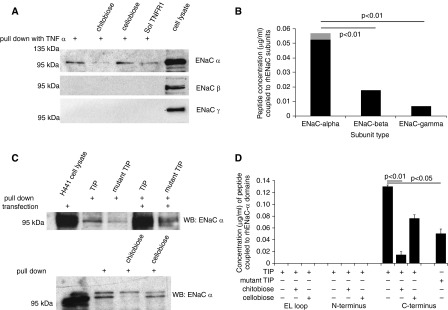
(A) Representative pull-down experiment assessing binding of biotinylated human TNF (10 ng/ml) to the apical epithelial Na+ channel (ENaC)-α, -β, and -γ subunits in H441 cell lysates. (B) Measurement of binding of biotinylated TIP peptide to recombinant ENaC-α, -β, and -γ subunits (n = 5). (C) Upper panel: Representative pull-down experiment assessing binding of biotinylated TIP or mutant TIP peptide to endogenous or overexpressed ENaC-α in H441 cell lysates. Lower panel: Influence of preincubation of TIP peptide with either N,N′-diacetylchitobiose or cellobiose (both in 10-fold molar excess over TIP peptide) upon binding to ENaC-α. (D) Binding activity of biotinylated TIP or mutant TIP peptide to glutathione-S-transferase–coupled domains of ENaC-α. EL loop = extracellular loop.
These results correspond to data from a study of molecular docking between TNF and N,N′-diacetylchitobiose, indicating that oligosaccharide binding to TNF sterically hinders its association with residues involved in the Na+ uptake stimulating effect of the cytokine (40).
As shown in Table 1, the TIP peptide (50 μg/ml) and the mutant TIP peptide failed to increase the enzymatic activity or the Na+-binding affinity of the Na+-K+-ATPase at either pH 7.5 or pH 6.5. These data indicate that TIP peptide concentrations previously shown to activate Na+ uptake (27) do not directly modify Na+-K+-ATPase function.
Table 1.
Effect of TIP and Mutant TIP Peptides (50 μg/ml) on Na+-K+-ATPase Activity and Na+ Affinity (N = 5)
| Enzymatic Activity (%) |
Na+ Affinity (K1/2/mM) | ||
|---|---|---|---|
| pH 7.6 | pH 6.5 | ||
| Control | 100 | 100 | 0.41 ± 0.02 |
| TIP 50 μg/ml | 95 | 99 | 0.36 ± 0.02 |
| Mutant TIP 50 μg/ml | 99 | 98 | 0.33 ± 0.02 |
Experiments were performed with 2.1 μg/ml Na+,K+-ATPase (enzymatic activity of 1,506 μmol Pi per milligram of protein and hour at 37°C).
TIP Peptide Directly Binds to the Carboxy-Terminal Domain of ENaC-α
To determine to which subdomain of ENaC-α the lectin-like domain of TNF binds, we used glutathione-S-transferase–tagged recombinant proteins of extracellular loop, amino-terminal, and carboxy-terminal domains of ENaC-α (14). Proteins were coupled to glutathione-S-transferase SpinTrap columns (GE Healthcare, Piscataway, NJ) and incubated with biotinylated or mutant TIP peptide. After elution of the complexes, biotinylated TIP peptide was detected only in the eluent of the carboxy-terminal domain–containing column (Figure 2D). Molecular docking studies carried out with the homology modeling approach and using the structure of the ENaC-related, acid-sensing ion channel 1 (ASIC-1) as a template (41) predicted a preferential binding of the TIP peptide, but not of the mutant TIP peptide, to the carboxy-terminal domain of ENaC-α, in correlation with our experimental data (Figure E1 in the online supplement). The potential interaction domain between the lectin-like domain of TNF and ENaC-α was previously proposed to be involved in gating (42).
The observation that the TIP peptide interacts with the carboxy-terminal intracellular domain of ENaC-α suggests it has to be internalized. Figures 3A and 3B demonstrate binding and uptake of rhodamine-labeled TIP peptide in H441 cells, reaching a maximal uptake level around 60 minutes, as quantified in Figure 3C. This uptake was completely blunted upon depletion of cholesterol in the H441 cells by 1 mM methyl-β-cyclodextrin (Figures 3A–3C). Cholesterol depletion has been shown not to affect ENaC expression, but rather to reduce its Po time and to induce its redistribution from low-density, cholesterol-rich membrane rafts to higher-density regions of the membrane (43, 44). As such, these data indicate that the TIP peptide has to be taken up in cholesterol-rich lipid rafts in the vicinity of ENaC.
Figure 3.

(A and B) Binding and uptake of rhodamine-labeled TIP peptide (red) in H441 cells pretreated or not with β-methylcyclodextrin (1 mM). (C) Quantification of uptake of rhodamine-labeled TIP peptide in H441 cells, pretreated or not with β-methylcyclodextrin (1 mM) and subsequently treated with trypsin to remove all extracellularly bound peptide (n = 20/group, mean [black bars] ± SD [gray bars]).
The Lectin-like Domain of TNF Blunts PLY-induced Down-regulation of ENaC-α Expression
Toxins of bacterial and viral origin, such as LPS, PLY, and influenza A virus M2 protein, can significantly impair ENaC activity in a p38 MAP kinase– or PKC-dependent manner (33–36). We have recently shown that the pneumococcal toxin PLY impairs ENaC function (34). As shown in Figure 4A, PLY (60 ng/ml) reduces ENaC-α protein expression in H441 cells after 24-hour incubation, an effect blunted by coincubation of the cells with TIP peptide (50 μg/ml). Interactions between PY motifs in the carboxy-terminal domain of human ENaC subunits with WW domains of the E3 ubiquitin ligases Nedd-4-1 and Nedd-4-2 are facilitated by extracellular signal-regulated protein kinases 1 and 2 (ERK1/2)–mediated phosphorylation of T residues in the vicinity of the PY motif, which initiates its degradation (11, 45). ERK1/2 is in turn activated by the phospholipase C (PLC)/PKC pathway (46). The protective effect of the peptide on ENaC-α expression likely occurs by means of inhibiting the PLC/PKC/ERK1/2 pathway. Indeed, the TIP peptide blunts PLY (125 ng/ml)–mediated activation of PLC (Figure 4F), PKC-α (Figures 4B and 4C) and ERK1/2 (Figures 4D and 4E). The PKC-α-specific inhibitor Ro32-0432 (10 nM) blunted the PLY-mediated suppression of ENaC-α protein expression (Figure 4G). This finding suggests an important role for this PKC isoform in PLY-mediated ENaC dysfunction. In the presence of PLY, the PKC-α inhibitor Ro32-0432 induces activation of PKC-δ (Figure 4H), an effect which does not occur with the TIP peptide (Figure 4I). A plausible explanation for the differential effects of a PKC-α inhibitor versus the TIP peptide on the status of PKC-δ in PLY-treated cells is that the peptide blunts PLC activation, which is upstream from both PKC-α and PKC-δ. The ENaC inhibitor amiloride (10 μM) partially abolished the protective activity of the TIP peptide on PLY-mediated PKC-α activation, which indicates cross-talk between ENaC and PKC-α, as previously proposed by others (47; see also Figures 4B and 4C).
Figure 4.

(A) Effect of pneumolysin (PLY) (60 ng/ml, 24 h) treatment on apical epithelial Na+ channel (ENaC) expression in H441 cells, assessed in a representative Western blot (WB). (B) Representative WB used to demonstrate protein kinase C-α (PKC-α) activation, measured as the ratio of phosphorylated (phospho)Ser657/total PKC-α expression in the plasma membrane in H441 cells treated for 1 hour with PLY (60 ng/ml). Cells were pretreated for 30 minutes with TIP peptide (50 μg/ml), either in combination with amiloride (10 μM) or not. (C) Quantification of the phospho/total PKC-α ratio in H441 cells treated with PLY in the presence or absence of TIP peptide (50 μg/ml) and amiloride (10 μM) (mean ± SD data from three independent experiments in triplicates). (D) Representative WB used to assess activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) in H441 cells treated for 1.5 hours with PLY (125 ng/ml) upon pretreatment or not with TIP peptide (50 μg/ml, 30 min). (E) Quantification of the phospho/total ERK1/2 ratio in H441 cells treated with PLY in the presence or absence of TIP peptide (means ± SD of three independent experiments in triplicates). (F) Effect of preincubation with the phosphatidylcholine-specific phospholipase C (PC-PLC) inhibitor D609 (30 μM) or with TIP peptide (50 μg/ml) upon PC-PLC activation in basal or PLY-treated H441 cells. (G) Ro32-0432 (10 nM) preserves ENaC-α expression in the presence of PLY (125 ng/ml) in H441 cells. (H) Influence of the PKC-α inhibitor Ro32-0432 (10 nM, 30 min) upon PKC-δ activation in H441 cells in the presence or absence of PLY (125 ng/ml). (I) Effect of TIP peptide pretreatment on PKC-δ and ERK1/2 activation in control and PLY-treated H441 cells.
The Lectin-like Domain of TNF Preserves ENaC Open Probability in the Presence of PLY by Stabilizing the ENaC-MARCKS-PIP2 Complex
Because the Po of ENaC is at least partially regulated by its complex formation with PIP2 and MARCKS (12–14), we assessed the influence of the TIP peptide on the ENaC–MARCKS association in the presence of PLY. As shown in Figures 5A and 5B, a 30-minute treatment of 2F3 cells with PLY (60 ng/ml) led to a significantly reduced association of ENaC-α with MARCKS, as compared to vehicle-treated cells. A 10-minute pretreatment with the TIP peptide (50 μg/ml) significantly preserved the ENaC–MARCKS association in PLY-treated cells. Together with our findings that the TIP peptide preserves ENaC-α protein expression in the presence of PLY, these data provide a rationale for the capacity of the peptide to increase ENaC activity in the presence of the pneumococcal toxin.
Figure 5.

(A) Representative immunoprecipitation Western blot showing that the association between apical epithelial Na+ channel (ENaC)and myristoylated alanine-rich C kinase substrate (MARCKS) is attenuated after treating Xenopus 2F3 cells with pneumolysin (PLY) (60 ng/ml) for 30 minutes, in comparison to vehicle-treated cells. The reduced association between ENaC and MARCKS was less pronounced in cells treated with TIP (50 μg/ml) 5 minutes before PLY treatment. WCL = whole-cell lysate. (B) Densitometry of immunoreactive bands corresponding to the ENaC-α subunit from three independent experiments. All values were normalized to the background signal.
TKI Mice Are More Susceptible to PLY-induced Impairment of ALC Than Wild-Type Mice
Up to 10% of pneumococcal pneumonia patients succumb days after their lungs are sterile following antibiotic treatment (48). A plausible explanation for this mortality is the massive release in the alveoli of the pneumococcal cholesterol-binding, pore-forming toxin PLY, an important mediator of permeability edema (48–50). In contrast to LPS, PLY does not induce a strong inflammatory response (51). We recently demonstrated that PLY impairs ENaC function in H441 cells (34).
To investigate whether the lectin-like domain of TNF can play a physiological role in ALC, we compared edema formation after intratracheal instillation of a low dose of PLY between wild-type (WT) and TKI C57BL6 mice, which produce a mutant TNF with a deficient Na+ uptake-activating capacity (23; see also Methods section). We selected a dose of PLY (1.5 μg/kg) that does not significantly impair alveolar epithelial and capillary barrier function, as shown in Figures 6A and 6C. Within 24 hours, this dose of PLY (1.5 μg/kg) caused a modest but significant increase in the generation of the proinflammatory cytokines IL-6 and TNF in the bronchoalveolar lavage fluid (Figure 6B). In the absence of barrier dysfunction (Figures 6A and 6C) and increased cell infiltration (data not shown), resident alveolar macrophages or AECs are the likely source of these mediators. The increase in TNF generation induced by PLY is comparable between WT and TKI mice, which allowed us to compare the actions of WT TNF, generated in the WT animals, and T104A-E106A-E109A mutant TNF in the TKI mice, on ALC, assessed as lung wet-to-dry ratio. The lung wet-to-dry ratio was significantly higher in TKI mice than in WT mice 24 hours after PLY instillation (Figure 6D). In lung homogenates from TKI mice, we detected a more important reduction in protein expression of ENaC-α, but neither the β nor γ ENaC subunits, than in WT animals upon PLY treatment (Figures 6E and 6F).
Figure 6.
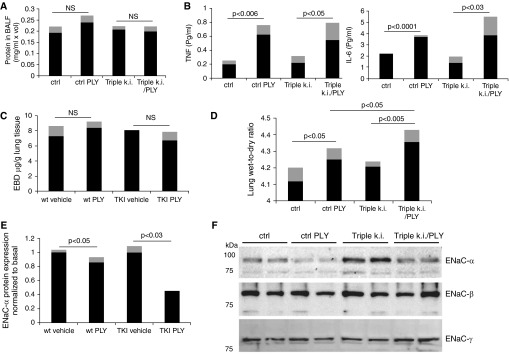
(A) Protein concentration in bronchoalveolar lavage fluid (BALF) in wild-type (WT) or triple-mutant TNF knock-in (TKI) mice (n = 6, mean [black bars] ± SD [gray bars]) treated for 24 hours with pneumolysin (PLY) (1.5 μg/kg). (B) TNF and IL-6 concentrations (in pg/ml; MCYTOMAG-70K assay; EMD Millipore, Billerica, MA) in BALF from WT or TKI mice treated or not with PLY (1.5 μg/kg, n = 4, mean [black bars] ± SD [gray bars]). (C) Capillary leak, measured as Evans blue dye incorporation (n = 6, mean [black bars] ± SD [gray bars]). (D) Lung wet-to-dry ratio in WT or TKI mice treated for 24 hours with PLY (1.5 μg/kg, n = 6, mean [black bars] ± SD [gray bars]). (E) Quantification of ENaC-α expression upon PLY instillation normalized to basal level in either WT or TKI mice (1.5 μg/kg PLY, n = 6, mean [black bars] ± SD [gray bars]). (F) Representative Western blot of apical epithelial Na+ channel (ENaC)-α, -β, and -γ subunit expression in lung homogenates from WT or TKI mice treated or not with PLY.
Discussion
In previous studies, our research group and other investigators have demonstrated that the lectin-like domain of TNF represents a potent stimulator of ALC in several mammalian species and involves activation of ENaC via an unknown mechanism (21, 24, 27–31, 38) . This study was designed to determine the mechanism by which the lectin-like domain of TNF can promote ENaC activity. We also investigated whether the lectin-like domain of TNF plays a physiological role in ALC during infection and inflammation. The results of this study demonstrate, for the first time to our knowledge, that ENaC activation by the TIP peptide involves multiple steps, starting with the interaction with glycosylated membrane components, possibly within the extracellular loops of ENaC (38) (Figure 1), followed by uptake within cholesterol-rich lipid rafts (Figure 3) and finally binding to the carboxy-terminal domain of the α subunit (Figure 2). A similar, multistep recognition and action mechanism was previously discovered in the interaction between TNF and African trypanosomes (52), where a conserved chitobiose-oligomannose (GlcNAc2-Man5–9) moiety of the variant-specific surface glycoprotein within the flagellar pocket of Trypanosoma brucei brucei serves as a binding ligand for TNF (53). TNF is subsequently internalized and transported to the lysosomes, where it induces lysosomal rupture and lysis of the parasite (53).
A limitation of our study is that we have not demonstrated that, like the TIP peptide, native TNF can be internalized. TNF can, however, increase ALC in mice lacking both TNF receptors (24), strongly suggesting TNF receptor–independent actions of the cytokine. Moreover, TNF has a membrane insertion capacity (54). Also, intra-alveolar catabolism is an important mechanism of protein removal, which is particularly important during recovery from pulmonary edema (55). In the presence of pore-forming toxins, such as PLY or the family member listeriolysin, pore formation was previously demonstrated to allow increased entry of large proteins in the affected cells (56). As such, it is possible that, in the presence of PLY, increased uptake of TIP peptide or TNF can take place in AECs. However, because the lectin-like domain of TNF can activate ENaC in conditions not involving pore formation, such as in untreated cells (Figure 1A) or in LPS-treated cells (data not shown), a role for a specific vesicular uptake of ENaC-TIP peptide or ENaC-TNF complexes in lipid rafts seems more likely than an unspecific pore-mediated entry as the main mechanism leading to intracellular ENaC activation. Our results demonstrate that the lectin-like domain of TNF activates ENaC, whereas TNF receptor 1 stimulation was previously shown to blunt the channel’s activity (19, 20). An increased generation of TNF occurs in AECs following stretch during ventilation (57, 58). It should be noted that in ventilator-induced lung injury, a dual role has been proposed for TNF receptors, with TNF receptor 1 hypothesized to mediate deleterious, TNF receptor 2–inducing protective activities (58).
The puzzling observation that oligosaccharides can blunt the binding of the TIP peptide to recombinant, and thus nonglycosylated, subunits can be explained by our previously published molecular docking study indicating that residues involved in the binding of human TNF to N,N′-diacetylchitobiose are different from, but spatially close to, those necessary for activation of Na+ uptake (40). Indeed, the mutant TIP peptide, which has a strongly reduced capacity to bind to ENaC-α and activate ENaC, still efficiently binds to N,N′-diacetylchitobiose (59). This suggests that direct binding of TNF to ENaC-α involves a region that partially overlaps, but is not identical to, the lectin-like domain of the cytokine. Our hypothesis is therefore that the association of TNF or of the TIP peptide with oligosaccharides sterically hinders its subsequent binding to ENaC-α. This hypothesis is supported by our observation that, although the TIP peptide fails to increase Na+ uptake in N-glycosidase F–treated H441 cells, it can still efficiently bind to ENaC-α in lysates from these cells (data not shown). Our data in this study strongly suggest that the primary target of the lectin-like domain of TNF, mimicked by the TIP peptide, is ENaC rather than Na+-K+-ATPase. These data do not contradict the findings described by Vadasz et al. (29), who proposed a stimulatory activity of the TIP peptide on Na+-K+-ATPase activity, because ENaC-mediated increases in the rate of Na+ entry across the apical membrane have been demonstrated to result in activation of the Na+-K+ pump (60).
We were able to demonstrate that the TIP peptide preserves ENaC-α protein expression and stabilizes the ENaC-PIP2-MARCKS complex, crucial for its open conformation, in the presence of the PLY. MARCKS is a substrate of both PKC-α and PKC-δ, the former of which is activated by PLY. Preservation of the ENaC-PIP2-MARCKS complex by TIP peptide prevents PIP2-mediated generation of diacylglycerol by PLC, as previously suggested by others (61). As such, the TIP peptide can also blunt subsequent PKC activation and PKC-mediated phosphorylation of MARCKS and ERK1/2.
Mutation of the lectin-like domain of TNF in TKI mice induced increased susceptibility to PLY-induced pulmonary edema, in correlation with a more important reduction in protein expression of ENaC-α. These results suggest that the lectin-like domain of TNF has a physiological role in ALC during lung inflammation. A limitation of our in vivo study is that we have studied a low PLY dose, which does not induce significant barrier dysfunction in the lungs. Yet, this dose allowed us to specifically investigate the effect of TIP on PLY-induced impairment of ALC rather than on barrier function. Moreover, we have previously shown that the TIP peptide possesses barrier-protective effects in permeability edema induced by instillation of higher doses of PLY (30).
Our approach to increasing lung liquid clearance by means of directly inducing ENaC activity in a cAMP-independent manner can potentially provide an interesting alternative treatment option for pulmonary edema. Limitations of this approach could come from conditions in which there is excessive damage of the alveolar epithelium, such that insufficient ENaC is expressed on their apical surface to be activated by the TIP peptide. Moreover, excess protein deposition in the alveolar compartment during the recovery phase from alveolar edema can potentially occur because the salt and water fraction of the edema fluid are cleared much faster than protein (55).
Taken together, our data suggest that the lectin-like domain of TNF activates ENaC by means of a unique mechanism involving direct binding to the ENaC-α subunit. This is, to our knowledge, the first example of a direct interaction between a cytokine and ENaC. The TIP peptide, mimicking the lectin-like domain of the cytokine, has been demonstrated to have promising protective effects in both preclinical and clinical settings of acute lung injury–associated edema, which should provide motivation for further evaluation of it as a potential novel therapeutic candidate for the treatment of pulmonary edema.
Acknowledgments
Acknowledgment
The authors thank Dr. Thomas Meergans (University of Konstanz, Germany) and Dr. Horst Bluethmann (F. Hoffmann-La Roche, Basel, Switzerland) for their help in generating the TKI mice and Dr. Rolf Hubmayr (Mayo Clinic, Rochester, MN) for reading the manuscript critically.
Footnotes
Supported by National Heart, Lung, and Blood Institute (NHLBI) Grant R01 HL094609 (R.L.), NHLBI Grant P01 HL101902 (A.V.), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant F32 DK093255 (A.A.), NHLBI Grant R37 HL51856 (M.A.M.), NIDDK Grant DK 037963/14 (D.C.E.), and SFB-TR 84 “Innate Immunity of the Lung” (DFG) (T.C., R.L. [Mercator Professor]).
Author Contributions: H.-F.B., D.K., S.S., H.-J.A., R.W., B.G., A.Z., M.P.-E., J.H., I.G.-G., Z.B., and A.L.: laboratory measurements and data analysis. A.V., A.W., B.F., J.F.P., W.S., and R.L.-G.: study design. T.C. and M.A.M.: study design and manuscript writing. D.C.E., I.C., A.A., and R.L.: study design, obtaining funding, laboratory measurements, data analysis, and manuscript writing.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201405-0833OC on July 16, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Vadász I, Raviv S, Sznajder JI. Alveolar epithelium and Na,K-ATPase in acute lung injury. Intensive Care Med. 2007;33:1243–1251. doi: 10.1007/s00134-007-0661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol. 2007;159:350–359. doi: 10.1016/j.resp.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthay MA. Resolution of pulmonary edema. Thirty years of progress. Am J Respir Crit Care Med. 2014;189:1301–1308. doi: 10.1164/rccm.201403-0535OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 6.Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol. 2009;71:403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- 7.Knight KK, Olson DR, Zhou R, Snyder PM. Liddle’s syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci USA. 2006;103:2805–2808. doi: 10.1073/pnas.0511184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kashlan OB, Kleyman TR. Epithelial Na(+) channel regulation by cytoplasmic and extracellular factors. Exp Cell Res. 2012;318:1011–1019. doi: 10.1016/j.yexcr.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in α-ENaC-deficient mice. Nat Genet. 1996;12:325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 10.Shimkets RA, Lifton RP, Canessa CM. The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J Biol Chem. 1997;272:25537–25541. doi: 10.1074/jbc.272.41.25537. [DOI] [PubMed] [Google Scholar]
- 11.Bobby R, Medini K, Neudecker P, Lee TV, Brimble MA, McDonald FJ, Lott JS, Dingley AJ. Structure and dynamics of human Nedd4-1 WW3 in complex with the αENaC PY motif. Biochim Biophys Acta. 2013;1834:1632–1641. doi: 10.1016/j.bbapap.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 12.Pochynyuk O, Tong Q, Staruschenko A, Ma HP, Stockand JD. Regulation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. Am J Physiol Renal Physiol. 2006;290:F949–F957. doi: 10.1152/ajprenal.00386.2005. [DOI] [PubMed] [Google Scholar]
- 13.Kooijman EE, Kuzenko SR, Gong D, Best MD, Folkesson HG. Phosphatidylinositol 4,5-bisphosphate stimulates alveolar epithelial fluid clearance in male and female adult rats. Am J Physiol Lung Cell Mol Physiol. 2011;301:L804–L811. doi: 10.1152/ajplung.00445.2010. [DOI] [PubMed] [Google Scholar]
- 14.Alli AA, Bao HF, Alli AA, Aldrugh Y, Song JZ, Ma HP, Yu L, Al-Khalili O, Eaton DC. Phosphatidylinositol phosphate-dependent regulation of Xenopus ENaC by MARCKS protein. Am J Physiol Renal Physiol. 2012;303:F800–F811. doi: 10.1152/ajprenal.00703.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goudenege S, Poussard S, Dulong S, Cottin P. Biologically active milli-calpain associated with caveolae is involved in a spatially compartmentalised signalling involving protein kinase C α and myristoylated alanine-rich C-kinase substrate (MARCKS) Int J Biochem Cell Biol. 2005;37:1900–1910. doi: 10.1016/j.biocel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Park JA, Crews AL, Lampe WR, Fang S, Park J, Adler KB. Protein kinase C delta regulates airway mucin secretion via phosphorylation of MARCKS protein. Am J Pathol. 2007;171:1822–1830. doi: 10.2353/ajpath.2007.070318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, Canessa CM, Pittet JF. Interleukin-1β decreases expression of the epithelial sodium channel α-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J Biol Chem. 2005;280:18579–18589. doi: 10.1074/jbc.M410561200. [DOI] [PubMed] [Google Scholar]
- 18.Peters DM, Vadász I, Wujak L, Wygrecka M, Olschewski A, Becker C, Herold S, Papp R, Mayer K, Rummel S, et al. TGF-β directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci USA. 2014;111:E374–E383. doi: 10.1073/pnas.1306798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamagata T, Yamagata Y, Nishimoto T, Hirano T, Nakanishi M, Minakata Y, Ichinose M, Dagenais A, Berthiaume Y. The regulation of amiloride-sensitive epithelial sodium channels by tumor necrosis factor-α in injured lungs and alveolar type II cells. Respir Physiol Neurobiol. 2009;166:16–23. doi: 10.1016/j.resp.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Bao HF, Zhang ZR, Liang YY, Ma JJ, Eaton DC, Ma HP. Ceramide mediates inhibition of the renal epithelial sodium channel by tumor necrosis factor-α through protein kinase C. Am J Physiol Renal Physiol. 2007;293:F1178–F1186. doi: 10.1152/ajprenal.00153.2007. [DOI] [PubMed] [Google Scholar]
- 21.Braun C, Hamacher J, Morel DR, Wendel A, Lucas R. Dichotomal role of TNF in experimental pulmonary edema reabsorption. J Immunol. 2005;175:3402–3408. doi: 10.4049/jimmunol.175.5.3402. [DOI] [PubMed] [Google Scholar]
- 22.Rezaiguia S, Garat C, Delclaux C, Meignan M, Fleury J, Legrand P, Matthay MA, Jayr C. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-α-dependent mechanism. J Clin Invest. 1997;99:325–335. doi: 10.1172/JCI119161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda N, Jayr C, Lazrak A, Wang Y, Lucas R, Matalon S, Matthay MA. Mechanisms of TNF-α stimulation of amiloride-sensitive sodium transport across alveolar epithelium. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1258–L1265. doi: 10.1152/ajplung.2001.280.6.L1258. [DOI] [PubMed] [Google Scholar]
- 24.Elia N, Tapponnier M, Matthay MA, Hamacher J, Pache JC, Brundler MA, Totsch M, De Baetselier P, Fransen L, Fukuda N, et al. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-α. Am J Respir Crit Care Med. 2003;168:1043–1050. doi: 10.1164/rccm.200206-618OC. [DOI] [PubMed] [Google Scholar]
- 25.Hession C, Decker JM, Sherblom AP, Kumar S, Yue CC, Mattaliano RJ, Tizard R, Kawashima E, Schmeissner U, Heletky S, et al. Uromodulin (Tamm-Horsfall glycoprotein): a renal ligand for lymphokines. Science. 1987;237:1479–1484. doi: 10.1126/science.3498215. [DOI] [PubMed] [Google Scholar]
- 26.Lucas R, Magez S, De Leys R, Fransen L, Scheerlinck JP, Rampelberg M, Sablon E, De Baetselier P. Mapping the lectin-like activity of tumor necrosis factor. Science. 1994;263:814–817. doi: 10.1126/science.8303299. [DOI] [PubMed] [Google Scholar]
- 27.Hamacher J, Stammberger U, Roux J, Kumar S, Yang G, Xiong C, Schmid RA, Fakin RM, Chakraborty T, Hossain HM, et al. The lectin-like domain of tumor necrosis factor improves lung function after rat lung transplantation—potential role for a reduction in reactive oxygen species generation. Crit Care Med. 2010;38:871–878. doi: 10.1097/CCM.0b013e3181cdf725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tzotzos S, Fischer B, Fischer H, Pietschmann H, Lucas R, Dupré G, Lemmens-Gruber R, Hazemi P, Prymaka V, Shabbir W. AP301, a synthetic peptide mimicking the lectin-like domain of TNF, enhances amiloride-sensitive Na(+) current in primary dog, pig and rat alveolar type II cells. Pulm Pharmacol Ther. 2013;26:356–363. doi: 10.1016/j.pupt.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vadász I, Schermuly RT, Ghofrani HA, Rummel S, Wehner S, Mühldorfer I, Schäfer KP, Seeger W, Morty RE, Grimminger F, et al. The lectin-like domain of tumor necrosis factor-α improves alveolar fluid balance in injured isolated rabbit lungs. Crit Care Med. 2008;36:1543–1550. doi: 10.1097/CCM.0b013e31816f485e. [DOI] [PubMed] [Google Scholar]
- 30.Lucas R, Yang G, Gorshkov BA, Zemskov EA, Sridhar S, Umapathy NS, Jezierska-Drutel A, Alieva IB, Leustik M, Hossain H, et al. Protein kinase C-α and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am J Respir Cell Mol Biol. 2012;47:445–453. doi: 10.1165/rcmb.2011-0332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartmann EK, Boehme S, Duenges B, Bentley A, Klein KU, Kwiecien R, Shi C, Szczyrba M, David M, Markstaller K. An inhaled tumor necrosis factor-α-derived TIP peptide improves the pulmonary function in experimental lung injury. Acta Anaesthesiol Scand. 2013;57:334–341. doi: 10.1111/aas.12034. [DOI] [PubMed] [Google Scholar]
- 32.Schwameis R, Eder S, Pietschmann H, Fischer B, Mascher H, Tzotzos S, Fischer H, Lucas R, Zeitlinger M, Hermann R. A FIM study to assess safety and exposure of inhaled single doses of AP301-A specific ENaC channel activator for the treatment of acute lung injury. J Clin Pharmacol. 2014;54:341–350. doi: 10.1002/jcph.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Migneault F, Boncoeur E, Morneau F, Pascariu M, Dagenais A, Berthiaume Y. Cycloheximide and lipopolysaccharide downregulate αENaC mRNA via different mechanisms in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;305:L747–L755. doi: 10.1152/ajplung.00023.2013. [DOI] [PubMed] [Google Scholar]
- 34.Lucas R, Sridhar S, Rick FG, Gorshkov B, Umapathy NS, Yang G, Oseghale A, Verin AD, Chakraborty T, Matthay MA, et al. Agonist of growth hormone-releasing hormone reduces pneumolysin-induced pulmonary permeability edema. Proc Natl Acad Sci USA. 2012;109:2084–2089. doi: 10.1073/pnas.1121075109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen XJ, Seth S, Yue G, Kamat P, Compans RW, Guidot D, Brown LA, Eaton DC, Jain L. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol. 2004;287:L366–L373. doi: 10.1152/ajplung.00011.2004. [DOI] [PubMed] [Google Scholar]
- 36.Lazrak A, Iles KE, Liu G, Noah DL, Noah JW, Matalon S. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J. 2009;23:3829–3842. doi: 10.1096/fj.09-135590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Czikora I, Alli A, Bao HF, Gorshkov B, Sridhar S, Lemmens-Gruber R, Shabbir W, Bagi Z, Chakraborty T, Matthay MA, et al. Novel mechanism of ENaC activation by the lectin-like domain of TNF. Am J Resp Crit Care Med. 2014;189(Meeting Abstracts):A3271. [Google Scholar]
- 38.Shabbir W, Scherbaum-Hazemi P, Tzotzos S, Fischer B, Fischer H, Pietschmann H, Lucas R, Lemmens-Gruber R. Mechanism of action of novel lung edema therapeutic AP301 by activation of the epithelial sodium channel. Mol Pharmacol. 2013;84:899–910. doi: 10.1124/mol.113.089409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lazrak A, Matalon S. cAMP-induced changes of apical membrane potentials of confluent H441 monolayers. Am J Physiol Lung Cell Mol Physiol. 2003;285:L443–L450. doi: 10.1152/ajplung.00412.2002. [DOI] [PubMed] [Google Scholar]
- 40.Dulebo A, Ettrich R, Lucas R, Kaftan D. A computational study of the oligosaccharide binding sites in the lectin-like domain of tumor necrosis factor and the TNF-derived TIP peptide. Curr Pharm Des. 2012;18:4236–4243. doi: 10.2174/138161212802430549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stockand JD, Staruschenko A, Pochynyuk O, Booth RE, Silverthorn DU. Insight toward epithelial Na+ channel mechanism revealed by the acid-sensing ion channel 1 structure. IUBMB Life. 2008;60:620–628. doi: 10.1002/iub.89. [DOI] [PubMed] [Google Scholar]
- 42.Copeland SJ, Berdiev BK, Ji HL, Lockhart J, Parker S, Fuller CM, Benos DJ. Regions in the carboxy terminus of α-bENaC involved in gating and functional effects of actin. Am J Physiol Cell Physiol. 2001;281:C231–C240. doi: 10.1152/ajpcell.2001.281.1.C231. [DOI] [PubMed] [Google Scholar]
- 43.Hill WG, An B, Johnson JP. Endogenously expressed epithelial sodium channel is present in lipid rafts in A6 cells. J Biol Chem. 2002;277:33541–33544. doi: 10.1074/jbc.C200309200. [DOI] [PubMed] [Google Scholar]
- 44.Balut C, Steels P, Radu M, Ameloot M, Driessche WV, Jans D. Membrane cholesterol extraction decreases Na+ transport in A6 renal epithelia. Am J Physiol Cell Physiol. 2006;290:C87–C94. doi: 10.1152/ajpcell.00184.2005. [DOI] [PubMed] [Google Scholar]
- 45.Shi H, Asher C, Chigaev A, Yung Y, Reuveny E, Seger R, Garty H. Interactions of β and γ ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem. 2002;277:13539–13547. doi: 10.1074/jbc.M111717200. [DOI] [PubMed] [Google Scholar]
- 46.Choi JY, Choi YS, Kim SJ, Son EJ, Choi HS, Yoon JH.Interleukin-1β suppresses epithelial sodium channel beta-subunit expression and ENaC-dependent fluid absorption in human middle ear epithelial cells Eur J Pharmacol 200756719–25 [DOI] [PubMed] [Google Scholar]
- 47.Awayda MS, Platzer JD, Reger RL, Bengrine A. Role of PKCα in feedback regulation of Na+ transport in an electrically tight epithelium. Am J Physiol Cell Physiol. 2002;283:C1122–C1132. doi: 10.1152/ajpcell.00142.2002. [DOI] [PubMed] [Google Scholar]
- 48.Anderson R, Steel HC, Cockeran R, von Gottberg A, de Gouveia L, Klugman KP, Mitchell TJ, Feldman C. Comparison of the effects of macrolides, amoxicillin, ceftriaxone, doxycycline, tobramycin and fluoroquinolones, on the production of pneumolysin by Streptococcus pneumoniae in vitro. J Antimicrob Chemother. 2007;60:1155–1158. doi: 10.1093/jac/dkm338. [DOI] [PubMed] [Google Scholar]
- 49.Rubins JB, Duane PG, Clawson D, Charboneau D, Young J, Niewoehner DE. Toxicity of pneumolysin to pulmonary alveolar epithelial cells. Infect Immun. 1993;61:1352–1358. doi: 10.1128/iai.61.4.1352-1358.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Witzenrath M, Gutbier B, Hocke AC, Schmeck B, Hippenstiel S, Berger K, Mitchell TJ, de los Toyos JR, Rosseau S, Suttorp N, et al. Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit Care Med. 2006;34:1947–1954. doi: 10.1097/01.CCM.0000220496.48295.A9. [DOI] [PubMed] [Google Scholar]
- 51.Maus UA, Srivastava M, Paton JC, Mack M, Everhart MB, Blackwell TS, Christman JW, Schlöndorff D, Seeger W, Lohmeyer J. Pneumolysin-induced lung injury is independent of leukocyte trafficking into the alveolar space. J Immunol. 2004;173:1307–1312. doi: 10.4049/jimmunol.173.2.1307. [DOI] [PubMed] [Google Scholar]
- 52.Magez S, Geuskens M, Beschin A, del Favero H, Verschueren H, Lucas R, Pays E, de Baetselier P. Specific uptake of tumor necrosis factor-α is involved in growth control of Trypanosoma brucei. J Cell Biol. 1997;137:715–727. doi: 10.1083/jcb.137.3.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Magez S, Radwanska M, Stijlemans B, Xong HV, Pays E, De Baetselier P. A conserved flagellar pocket exposed high mannose moiety is used by African trypanosomes as a host cytokine binding molecule. J Biol Chem. 2001;276:33458–33464. doi: 10.1074/jbc.M103412200. [DOI] [PubMed] [Google Scholar]
- 54.Baldwin RL, Stolowitz ML, Hood L, Wisnieski BJ. Structural changes of tumor necrosis factor α associated with membrane insertion and channel formation. Proc Natl Acad Sci USA. 1996;93:1021–1026. doi: 10.1073/pnas.93.3.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hastings RH, Folkesson HG, Matthay MA. Mechanisms of alveolar protein clearance in the intact lung. Am J Physiol Lung Cell Mol Physiol. 2004;286:L679–L689. doi: 10.1152/ajplung.00205.2003. [DOI] [PubMed] [Google Scholar]
- 56.Darji A, Chakraborty T, Wehland J, Weiss S. TAP-dependent major histocompatibility complex class I presentation of soluble proteins using listeriolysin. Eur J Immunol. 1997;27:1353–1359. doi: 10.1002/eji.1830270609. [DOI] [PubMed] [Google Scholar]
- 57.Vlahakis NE, Schroeder MA, Limper AH, Hubmayr RD. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol. 1999;277:L167–L173. doi: 10.1152/ajplung.1999.277.1.L167. [DOI] [PubMed] [Google Scholar]
- 58.Wilson MR, Goddard ME, O’Dea KP, Choudhury S, Takata M. Differential roles of p55 and p75 tumor necrosis factor receptors on stretch-induced pulmonary edema in mice. Am J Physiol Lung Cell Mol Physiol. 2007;293:L60–L68. doi: 10.1152/ajplung.00284.2006. [DOI] [PubMed] [Google Scholar]
- 59.Marquardt A, Bernevic B, Przybylski M. Identification, affinity characterisation and biological interactions of lectin-like peptide-carbohydrate complexes derived from human TNF-α using high-resolution mass spectrometry. J Pept Sci. 2007;13:803–810. doi: 10.1002/psc.902. [DOI] [PubMed] [Google Scholar]
- 60.Schultz SG, Dubinsky WP. Sodium absorption, volume control and potassium channels: in tribute to a great biologist. J Membr Biol. 2001;184:255–261. doi: 10.1007/s00232-001-0090-5. [DOI] [PubMed] [Google Scholar]
- 61.Glaser M, Wanaski S, Buser CA, Boguslavsky V, Rashidzada W, Morris A, Rebecchi M, Scarlata SF, Runnels LW, Prestwich GD, et al. Myristoylated alanine-rich C kinase substrate (MARCKS) produces reversible inhibition of phospholipase C by sequestering phosphatidylinositol 4,5-bisphosphate in lateral domains. J Biol Chem. 1996;271:26187–26193. doi: 10.1074/jbc.271.42.26187. [DOI] [PubMed] [Google Scholar]