

Abstract
Chloroformates are important laboratory and industrial chemicals with almost one hundred listed in the catalogs of leading suppliers. They are, for example, of prime importance as protecting groups in peptide synthesis. In some instances, the more stable fluoroformate is preferred.
In recent years, the specific rates of solvolysis (k) for chloroformates and fluoroformates in solvents of widely ranging nucleophilicity and ionizing power have been studied. Analysis of these rates using the extended (two-term) Grunwald-Winstein equation has led to important information concerning reaction mechanism. Also assisting in this effort have been studies of kinetic solvent isotope effects (KSIE), of leaving group effects (especially kF/kCl ratios), and of entropies of activation from studies of specific rate variations with temperature. For solvolyses of chloroformate esters, two mechanisms (addition-elimination and ionization) are commonly encountered. For solvolyses of fluoroformates, mainly because of a strong C–F bond, the ionization pathway is rare and the addition-elimination pathway is in most situations the one encountered.
1. Introduction
Chloroformates are extremely important intermediates, such as in the preparation of herbicides, fungicides, and insecticides [1]. They have to be handled with reasonable care due to their ability to cause physiological damage. This led to their use as war gases during World War I [2–4]. The parent acid (HOCOCl) is unstable and decomposes spontaneously to HCl and CO2. Similarily, esters with a tertiary alkyl group, such as tert-butyl chloroformate (tC4H9OCOCl) and to a lesser extent benzyl and isopropyl chloroformates are unstable and decompose with loss of CO2 and formation of alkyl chloride and/or alkene plus HCl. This decomposition can lead to another hazard, in addition to toxicity, in that a pressure-induced explosion can occur. This danger can be minimized by periodically releasing the pressure and by storage at considerably reduced temperatures [5].
For many years, chloroformate esters have been used extensively in peptide and related syntheses [6] due to their ability to introduce good protecting groups, especially for hydroxyl groups. For a given situation, the choice from among many chloroformates that are available is determined largely by trial and error. They are also used as precursors in the syntheses of several potential prodrugs [7,8]. The major supply houses list many chloroformate esters. For example, the online Sigma-Aldrich catalog (2012–2014) yielded a listing of 83 chloroformate esters.
A review of the chemistry of chloroformate esters [9], concentrating on the preparation, physical properties, and reactions appeared in 1964. Consistent with the affiliation of the authors, it concluded with a survey of the many uses of chloroformates in polymer synthesis. The coverage was extensive with 750 references covering the period through 1962. A review in “The Chemistry of the Functional Groups” series volume entitled “The Chemistry of Acyl Halides” appeared [5] in 1972 and was written with a primary emphasis on reaction mechanism, concentrating on work appearing subsequent to the previously mentioned review [9].
Historically, chloroformate esters were extensively used for studies of substitution reactions proceeding at an acyl carbon due to the ground-state resonance (Eqn. 1) leading to initial state stabilization, which reduced the rates of the rapid reactions observed for conventional acyl chlorides with no stabilizing factor. This tends to place the rates of reaction of the chloroformate esters within a range convenient for determination by traditional titration techniques. Developing further the pioneering techniques of Hudson and Archer [10], rapid-response conductivity techniques can now be used to accurately determine the rates of solvolysis reactions of simple aliphatic and aromatic acyl chlorides [11–15].
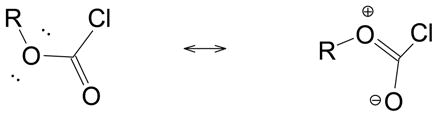 |
(1) |
In the present review, we concentrate on reaction mechanism studies, with emphasis on solvolysis reactions. Two major approaches have been applied in this area. When studies are also available for fluoroformates under otherwise identical conditions, there can be a consideration of Cl/F rate ratios. This was shown previously to be a very useful tool for assigning mechanism, with large ratios suggesting an important bond breaking at the transition state of the rate-determining step. Conversely, values close to unity suggest that little bond breaking has occurred at the transition state, such as when the addition step of an addition-elimination two-step mechanism is rate-determining. Many of the fluoroformates have been synthesized by treatment of the corresponding chloroformate with a fluoride salt in an inert solvent [5].
A second approach is in terms of the Grunwald-Winstein equations. The simple equation [16] was put forward in 1948 as a way of correlating the specific rates of solvolysis of ionization reactions, with the solvolysis of tert-butyl chloride in 80% ethanol being the standard substrate and solvent (Eqn. 2). In Eqn. 2, k and ko are the specific rates of solvolyses (first-order rate
| (2) |
coefficients) for solvolysis of a substrate RX in a given solvent and in 80% ethanol, m is the sensitivity to changes in solvent ionizing power Y, and c is a constant (residual) term. The equation was found to hold very well for solvolyses proceeding with negligible sensitivity towards accompanying changes in solvent nucleophilicity. It was found to apply less well when the solvolyses were SN2 in character [17] and it was suggested that a second term governed by the sensitivity (l) to changes in solvent nucleophilicity (N) should be added to the equation (Eqn. 3). The development and uses of the Grunwald-Winstein equations have been recently reviewed [18].
| (3) |
The original Y scale is now rarely used because it has been found [19,20] to include a nucleophilic component and to be dependent on the leaving group. A series of scales for a leaving group X have been developed based on the solvolyses of 1-adamantyl derivatives for the poorer leaving groups and of the slower reacting 2-adamantyl derivatives for the better leaving groups. This work has been reviewed and the review contains extensive tables of YX values [21].
The original solvent nuclophilicity scale [22] used in equation 3 was designated as NOTs and it was based on the SN2 solvolyses of p-toluenesulfonates. A major problem was that there was no independent way to arrive at the m value (mOTs) for insertion in the equation (l defined as unity) to incorporate with specific rate values, so as to arrive at the NOTs scale. A value of 0.3 for mOTs was estimated and used. A subsequent N scale (NT) was based on the SN2 solvolyses of S-methyldibenzothiophenium ion [23] and this system has the advantage that the leaving group is a neutral molecule and, to a good approximation [24], a consideration of solvent effects on its leaving-group ability can be neglected and log (k/ko) values can be taken as the values for the NT scale. The historical development and uses of solvent nucleophilicity scales have been reviewed and a table of available NT values is included [25]. The Eqn. 3, as it is applied to the chloroformates, can be rewritten in a more specific form (Eqn. 4).
| (4) |
In this review, we will show how the use of F/Cl ratios [26] and the l and m values of the extended Grunwald-Winstein equation (Eqn. 4), often considered as l/m ratios [27], can be useful indicators of solvolytic reaction mechanisms.
The chloroformates whose solvolysis will be considered include aryl chloroformates (the phenyl ester and ring-substituted derivatives), tertiary alkyl chloroformates (1-adamantyl and α,α,α-trichloro-tert-butyl esters), secondary alkyl chloroformates (2-adamantyl and isopropyl esters), primary alkyl chloroformates (methyl, ethyl, 2,2,2-trichloroethyl, n-propyl, isobutyl, neopentyl, and benzyl esters), and aliphatic chloroformates containing a double or triple carbon-carbon bond (allyl, vinyl, isopropenyl, 2-butyn-1-yl, and propargyl esters). Next, there will be a consideration of fluroformates with structures parallel to some of the above chloroformates and, finally, a brief consideration of methyl and ethyl chloroglyoxalates, with a second carbonyl group inserted between the alkoxy and carbonyl chloride groupings.
2. Aryl Chloroformates
The earliest mechanistic studies of the solvolyses of chloroformate esters were almost exclusively carried out with alkyl or cycloalkyl esters [5]. However, it was shown that a p-t-butyl substituent slightly reduced the rates of solvolysis of phenyl chloroformate in aqueous dioxane, consistent with nucleophilic attack in the rate-determining step [28].
Queen [29] carried out very detailed studies of the hydrolyses in 100% water of several chloroformate esters, including the phenyl ester, which reacted a little over an order of magnitude faster than the isopropyl ester and with smaller rate reductions on going to the similar rates for the n-propyl, ethyl, and methyl esters. The observations were considered to indicate a bimolecular mechanism except for the isopropyl ester, where a superimposed unimolecular mechanism was considered to be operative. Support for this viewpoint came from studies of the variation of specific rate with temperature, which indicated negative values for the entropy of activation, except for the isopropyl ester, where a positive value was observed. Such values are believed [30] to be indicative of bimolecular and unimolecular solvolyses, respectively.
A study of the methanolysis of phenyl chloroformate and derivatives [31] showed acceleration by electron-withdrawing substituents resulting in positive ϱ values [32]. The kinetic solvent isotope effects (KSIE) expressed as kMeOH/kMeOD were relatively large at 2.3 to 2.5. It was concluded that the reactions were of an associative nature, involving a concerted displacement due to the tetrahedral intermediate having a negligible lifetime (enforced concerted) [33].
Further study [34] of the p-nitrophenyl chloroformate extended the previous study [31] to water, and binary mixtures of water with acetone, acetonitrile, ethanol, and methanol. Grunwald-Winstein plots were carried out using the one-term equation (Eqn. 2). These showed only modest variation in specific rate as the solvent ionizing power was varied and shallow maxima in aqueous methanol and aqueous ethanol. In aqueous acetone, the curvature was only modest and an approximate m value of 0.12 was reported. These findings suggest a high dependence on solvent nucleophilicity, such that an analysis in terms of the two-term equation (Eqn. 4) would be more appropriate. The KSIE in methanol was corrected from 2.5 to 2.1 due to a redetermination of the specific rate in methanol-d.
A Bentley analysis [35] in terms of the four possible third-order rate coefficients in aqueous alcohols assuming general-base catalysis can involve catalysis by either water or alcohol to nucleophilic attack by water or alcohol, was found to give satisfactory agreement to experimental values for the p-nitrophenyl chloroformate solvolyses [34] and, also, the treatment could be applied to the solvolyses of the p-methoxy and p-nitro derivatives in methanol-water and ethanol-water [36]. Revisiting the plot against σ values, Hammett ϱ values in the range of 0.9 to 1.7 were found for the solvolyses in ethanol, methanol, 50% methanol, and water.
The finding of third-order behavior in the solvolytic studies is consistent with a previous finding of this type of behavior for methanolyses of p-nitrobenzoyl chloride in acetonitrile [37]. Kinetics were observed that were first-order in substrate and second-order in methanol. The mechanism proposed was formulated as in Scheme 1, with the use of a chloroformate as an example. A variation with attack by a dimer, rather than successive attack by two solvent molecules, would also be consistent with the observations.
Scheme 1.

There is also a recent study [38] of the solvolyses of phenyl chloroformate in aqueous dioxane and aqueous tetrahydrofuran, which concludes that the solvolyses are unimolecular in nature, as shown in Scheme 2. Although probably operating for the solvolyses of other chloroformate esters, the evidence for it operating for the solvolyses of phenyl chloroformate is weak and contrary to strong evidence for bimolecular reaction from other sources.
Scheme 2.
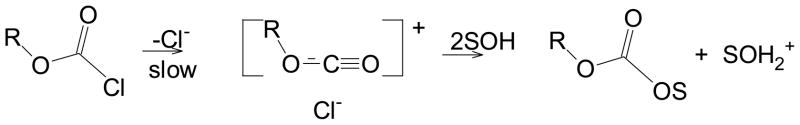
The strongest evidence for bimolecular reaction is probably from the application of the extended Grunwald-Winsetin equation (Eqn. 4). The parent compound, phenyl chloroformate was initially studied [39] in a good mix of 21 solvents of which nine had an appreciable fluoroalcohol, 2,2,2-trifluoroethanol (TFE) or 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), content. Inclusion of aqueous fluoroalcohols are very important to counteract the moderately strong multicollinearity observed when only “traditional” aqueous-organic solvents are used, such as aqueous ethanol and aqueous acetone. A good correlation was observed (Table 1) with a high l value of 1.65, appreciably higher than the l value of unity (by definition) when methyl p-toluenesulfonate or the S-methyldibenzothiophenium ion is solvolyzed.
Table 1.
Correlation of the specific rates of solvolysis of phenyl chloroformate and its p-methoxy and p-nitro derivatives using the extended form (Eqn. 4) of the Grunwald-Winstein equation.
| Substrate | n a | l b | m b | c b | l/m | R c | F d |
|---|---|---|---|---|---|---|---|
| MeOC6H4OCOCl | 44 e | 1.60 ± 0.05 | 0.57 ± 0.05 | 0.18 ± 0.06 | 2.81 ± 0.26 | 0.981 | 517 |
| 31 f | 1.46 ± 0.08 | 0.53 ± 0.03 | 0.19 ± 0.06 | 2.75 ± 0.22 | 0.964 | 182 | |
| C6H5OCOCl | 49 e | 1.66 ± 0.05 | 0.56 ± 0.03 | 0.15 ± 0.07 | 2.96 ± 0.18 | 0.980 | 568 |
| 21 g | 1.68 ± 0.10 | 0.57 ± 0.06 | 0.12 ± 0.03 | 2.95 ± 0.36 | 0.973 | 159 | |
| NO2C6H4OCOCl | 39 e | 1.68 ± 0.06 | 0.46 ± 0.04 | 0.08 ± 0.08 | 3.65 ± 0.34 | 0.976 | 363 |
| 38 h | 1.69 ± 0.07 | 0.46 ± 0.04 | 0.08 ± 0.08 | 3.67 ± 0.35 | 0.974 | 323 | |
| MeOC6H4OCOCl | 38 l | 1.58 ± 0.06 | 0.57 ± 0.04 | 0.17 ± 0.07 | 2.77 ± 0.22 | 0.974 | 320 |
| C6H5OCOCl | 38 i | 1.59 ± 0.07 | 0.54 ± 0.03 | 0.16 ± 0.08 | 2.94 ± 0.21 | 0.972 | 299 |
n is the number of solvents.
With associated standard error.
Multiple correlation coefficient.
F-test value.
Using all available data points.
Using the solvents in reference 40.
Using the solvents in reference 39.
Using all data points except the 90T-10E (not available for other two substrates in the table).
Using the same collection of solvents as in the 38 solvent entry for the p-nitro derivative.
The study by Koo, Lee, and coworkers [36] provided twenty additional data points and other data points [40] extended the ranges to 90% acetone and to 97% HFIP and included seven additional values for aqueous-TFE and TFE-ethanol solvolyses for the parent phenyl chloroformate substrate. Analyses using the extended Grunwald-Winstein equation for the full 49 solvents now available [40] led to essentially unchanged values for l and m and considerably improved goodness-of-fit values (Table 1). The unchanged l and m values indicate the data to be extremely robust. In contrast, use of the original one-term Grunwald-Winstein equation led to an m value (sensitivity to changes in solvent ionizing power) of −0.07 ± 0.11, c value of −0.46 ± 0.31, correlation coefficient (R) value of 0.093, and F-test value of 0.4 for an extremely poor fit; essentially no fit whatever. The two-term plot is shown in Figure 1.
Figure 1.

The plot of log (k/ko) vs. (1.66 NT + 0.56 YCl) for the solvolyses of phenyl chloroformate in pure and binary solvents at 25.0 ºC. Abstracted from ref. 40 with permission from the International Journal of Molecular Sciences.
The robust character of these correlations in terms of the extended equation (Eqn. 4) has led to the l and m values being extensively quoted as typical values for an addition-elimination (association-dissociation) mechanism (Scheme 1), with the addition process rate determining. With this mechanism the mY term will primarily relate not to the elongation of the carbon-halogen bond at the transition state but to the movement of π electrons of the carbonyl group onto the oxygen. The utility of the YCl scale suggests that it must also be a reasonably good scale for this process.
There are also situations where a third term can usefully be added to the extended Grunwald-Winstein equation to give Eqn. 5. In the additional term, h represents the sensitivity to changes in the aromatic ring parameter I [18,41]. This term is relevant when there are aryl groups on the α-
| (5) |
carbon (the one carrying the leaving group) or where a 1,2-aryl shift to the α-carbon is occurring (neighboring-group assistance). Reactions of aryl chloroformates do not fall in either of these categories and it was surprising to see that the hI term as a seemingly relevant contribution in a correlation of the specific rates of solvolysis of p-methoxyphenyl chloroformate in 31 solvents [40], where an h values of 0.85 ± 0.15 was obtained on application of Eqn. 5. However, it was pointed out that a potential problem with the analysis was that, of the 31 solvents, only two had a fluroalcohol component [39] and the remainder were binary mixtures of water with acetone, ethanol, and methanol. The possibility of multicollinearity is increased on going from a correlation against two scales to a correlation against three scales. In addition to an effective correlation of values of two of the scales against each other, multicollinearity can also occur if any of the scales correlates with any linear combination of the other two [42]. We have previously presented an example where neglect of a multicollinearity of this type led to a claim for the relevance of an hI term[43] for highly hindered tertiary alkyl derivative solvolyses, only for this contribution to the linear free energy relationship (LFER) to be no longer justified when multicollinearity of the type discussed above was found to operate over the range of solvents considered [44].
Similarly, after supplementing the listing of 31 specific rates with an additional twelve fluoroalcohol-containing solvents plus 90% ethanol [45], an analysis in terms of Eqn. 5 now gave l and m values similar to those obtained from Eqn. 4 and an h value of 0.29 ± 0.18, with a high 0.114 probability that the hI term is not statistically significant. The results from a two-term (Eqn. 4) treatment are included in Table 1 and the plot for the full 44 solvents is shown in Figure 2.
Figure 2.
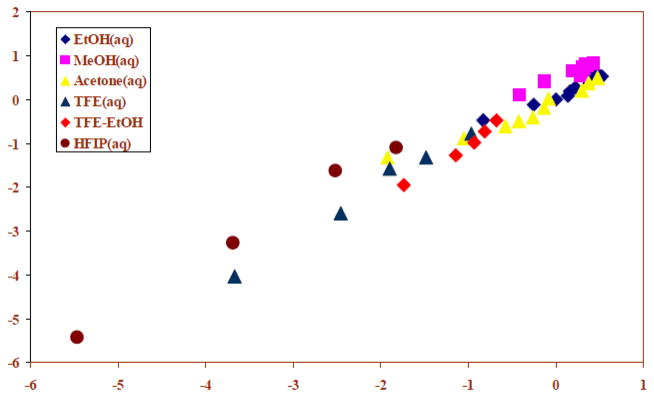
The plot of log (k/ko) vs. (1.60 NT + 0.57 YCl) for the solvolyses of p-methoxyphenyl chloroformate in pure and binary solvents at 25.0 ºC. Abstracted from ref. 45 with permission from the International Journal of Molecular Sciences.
The other extreme as regards substituents in the para-position is with the strongly electron-withdrawing nitro group. There are values available in the literature but, again, they were for aqueous acetone, ethanol, and methanol mixture [34,36]. Applying Eqn. 4 to these data, values were obtained of 1.85 ± 0.21 for l and 0.48 ± 0.05 for m, with a low multiple correlation coefficient (0.870) and a low F-test value (41). Subsequently, additional values for the specific rates of solvolysis were measured for five TFE-water, three HFIP-water, and two TFE-ethanol solvent compositions. With this new considerably improved mix of solvent type, the multiple correlation coefficient was raised to 0.976 and the F-test value to 363. The l and m values had lower standard errors and they were of values of 1.68 ± 0.06 and 0.46 ± 0.04, respectively [46]. The values are recorded in Table 1 and the correlation is shown in Figure 3.
Figure 3.

The plot of log (k/ko) vs. (1.68 NT + 0.46 YCl) for the solvolyses of p-nitrophenyl chloroformate in pure and binary solvents at 25.0 ºC. Abstracted from ref. 46 with permission from the International Journal of Molecular Sciences.
Also recorded in Table 1, so as to do a rigid comparison, are the correlation values obtained for phenyl chloroformate and the p-nitro and p-methoxy derivatives in the 38 solvents that are common to all three of the data sets. All indications are that all three substrates solvolyze over the full range of solvents studied by an addition-elimination pathway (Scheme 1), with the addition process rate-determining. Almost certainly for the solvolyses, as ϱ for the reactions with small amounts of alcohol in acetonitrile [37], general-base catalysis by a second nucleophilic center is involved [35]. This could involve a dimer attacking in the first step or, as formulated in Scheme 1, with an initial nucleophilic attack being followed by a second solvent molecule deprotonating the initially formed tetrahedral intermediate, such that the new intermediate on regeneration of the carbonyl group expels chloride ion in preference to alkoxide (or hydroxide). Without the deprotonation, one would expect an alcohol (or water) molecule to be ejected in preference to the chloride ion.
3. Tertiary Alkyl Chloroformates
The simplest tert-alkyl chloroformate, with a tert-butyl group, is unstable and decomposes with loss of carbon dioxide and with formation primarily of isobutylene and hydrogen chloride rather than tert-butyl chloride [47]. The decomposition can be retarded by replacement of hydrogen by chlorine and the 2,2,2-trichloro-1,1-dimethylethyl chloroformate (β,β,β-trichloro-tert-butyl chloroformate) is commercially available. Indeed, it is sufficiently often used in peptide and other syntheses that it has its specific abbreviation, TCBOC-chloride (based on naming as trichloro-tert-butoxycarbonyl chloride).
Its behavior under solvolytic conditions has been investigated in 33 well-chosen solvents including several with appreciable TFE- or HFIP-component [48]. Application of the extended Grunwald-Winstein equation led to the sensitivities towards solvent nucleophilicity and solvent ionizing power (l and m values), presented in Table 2, which are very similar to those observed for aryl chloroformates, and with an l/m ratio of 3.64, identical to that for p-nitrophenyl chloroformate (Table 1).
Table 2.
Correlation of the specific rates of solvolysis of alkyl chloroformates using the extended (Eqn. 4) form of the Grunwald-Winstein equationa,b.
| Substrate | nc | ld | md | cd | l/m | Re | Ff |
|---|---|---|---|---|---|---|---|
| 1-AdOCOCl | 11 | 0.08 ± 0.20 | 0.59 ± 0.05 | 0.06 ± 0.08 | 0.14 | 0.985 | 133 |
| 2-AdOCOCl | 19 | 0.03 ± 0.07 | 0.48 ± 0.04 | −0.10 ± 0.04 | 0.06 | 0.971 | 130 |
| i-PrOCOCl | 9 | 1.35 ± 0.22 | 0.40 ± 0.05 | 0.18 ± 0.07 | 3.38 | 0.960 | 35 |
| 16 | 0.28 ± 0.04 | 0.59 ± 0.04 | −0.32 ± 0.06g | 0.47 | 0.982 | 176 | |
| EtOCOCl | 28 | 1.56 ± 0.09 | 0.55 ± 0.03 | 0.19 ± 0.24 | 2.84 | 0.967 | 179 |
| 7 | 0.69 ± 0.13 | 0.82 ± 0.16 | −2.40 ± 0.27g | 0.84 | 0.946 | 17 | |
| n-PrOCOCl | 22 | 1.57 ± 0.12 | 0.56 ± 0.06 | 0.15 ± 0.06 | 2.79 | 0.947 | 83 |
| 6 | 0.40 ± 0.12 | 0.64 ± 0.13 | −2.45 ± 0.27 | 0.63 | 0.942 | 11 | |
| iso-BuOCOCl | 18h | 1.82 ± 0.15 | 0.53 ± 0.05 | 0.18 ± 0.07 | 3.43 | 0.957 | 82 |
| neo-PenOCOCl | 13 | 1.76 ± 0.14 | 0.48 ± 0.06 | 0.14 ± 0.08 | 3.67 | 0.977 | 226 |
| 8 | 0.36 ± 0.10 | 0.81 ± 0.14 | −2.79 ± 0.33g | 0.44 | 0.938 | 18 | |
| 1-AdCH2OCOCli | 18 | 1.84 ± 0.20 | 0.55 ± 0.05 | 0.23 ± 0.07 | 3.35 | 0.951 | j |
| 9 | 0.36 ± 0.09 | 0.86 ± 0.11 | −2.92 ± 0.23g | 0.42 | 0.966 | j | |
| MeOCOCl | 19 | 1.59 ± 0.09 | 0.58 ± 0.05 | 0.16 ± 0.07 | 2.74 | 0.977 | 171 |
| CCl3 C(CH3)2OCOClk | 33 | 1.42 ± 0.09 | 0.39 ± 0.05 | 0.16 ± 0.08 | 3.64 | 0.945 | j |
| CCl3CH2OCOCll | 30 | 1.28 ± 0.06 | 0.46 ± 0.03 | j | 2.78 | 0.973 | j |
Unless otherwise indicated by a footnote, abstracted from ref. 27.
When two entries are given for a substrate, the first refers to what is believed to be predominantly addition-elimination reaction and the second to the region with what is believed to be predominantly ionization reaction.
Number of solvents.
With associated standard error.
Multiple correlation coefficient.
F-test value.
Since the ko value (80% ethanol) is not available for the ionization reaction, log k was plotted and the intercept is (c + log ko), a close approximation to an estimate for log ko for ionization reaction.
Insufficient values in the ionization reaction region for a meaningful correlation.
From ref. 72.
Not reported.
From ref. 48.
From ref. 73.
Despite the tertiary structure for the alkyl group, the presence of three electron-withdrawing chlorine substituents leads to the addition-elimination pathway (Scheme 1) being followed. The solvent deuterium isotope effect for methanolysis of 2.14 is consistent with this assignment and with the operation of general-base catalysis from a second methanol molecule. Further support comes from the very negative (−28 to −36 cal mol−1 K−1) entropies of activation calculated from the temperature variation studies in five of the solvents.
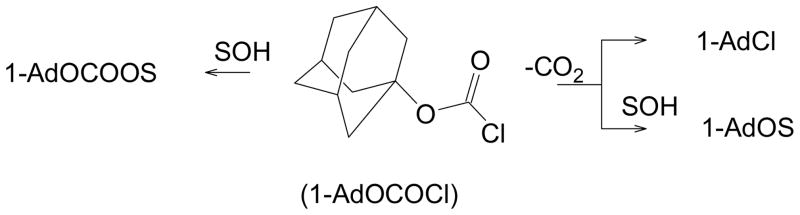
The bridgehead 1-adamantyl chloroformate is more stable that the tert-butyl ester and it can be conveniently prepared [49,50]. The increased stability is probably largely due to the alkene, which is the major product in the tert-butyl ester decomposition, being a disfavored high energy product when at a bridgehead (Bredt’s Rule). The fact that it is a solid as opposed to a liquid, which can serve as its own solvent, will also be disfavorable as regards decomposition. The decomposition to 1-adamantyl chloride with loss of carbon dioxide has been studied in nitrobenzene-benzene mixtures and an ionic process was proposed [50]. In acetonitrile, the initial products were approximately 60% 1-adamantyl chloride and 40% the acetonitrilium chloride, with the latter on addition of water, giving N-(1-adamantyl) acetamide or, if just left in solution, advancing to 1-adamantyl chloride as the thermodynamic product [50]. Alternatively, cycloaddition of added azide ion, gave 40–50% yields of 1-(1-adamantyl)-5-methyl tetrazole [51].
Under the more usual solvolytic conditions, both solvolysis and decomposition continue to be observed (Scheme 3). Product studies gave evidence for substitution with retention of the CO2 only in ethanol, where a small amount (<1%) of the 1-adamantyl ethyl carbonate was detected. The favored routes to products are the solvolysis and decomposition pathways to the right of the substrate.
Scheme 3.
In the kinetic analyses, the first-order rate coefficients (specific rates) were obtained for the overall process of producing products (by following the production of acid as a function of time). In conjunction with product studies, these could be subdivided into those for substitution, after loss of CO2, and decomposition. Data are available for 23 solvents [52,53]. The solvents were of relatively low ionizing power and, for many, the solvolyses of 1-adamantyl chloride were too slow to allow for convenient determination of YCl values. Accordingly, the original Y values, based on tert-butyl chloride solvolyses, were used in a treatment in terms of Eqn. 2, the one-term equation. A treatment in terms of the two-term equation was not attempted because of the limited number and poor mix of types as regards the solvents used in the study.
With all 23 solvents, only fair results were obtained for the correlations and the four aqueous acetone solvents were outliers, giving in themselves a good four point correlation with somewhat lower m value. Removal of these four data points, gave for 19 solvents an overall m value of 0.68, an m value of 0.77 for solvolysis, and an m value of 0.57 for decomposition. It was proposed that loss of CO2, either directly by a concerted pathway or by one involving the carboxylium ion (1-AdOCO+) as a reactive intermediate, leads to the 1-Ad+ Cl− ion pair. The difficulty in choosing between a concerted pathway and one involving a highly reactive intermediate in assigning mechanism is a common one in physical-organic chemistry [33,54]. The ion-pair then either collapses or solvent inserts, with separation of the ions (hence the higher m value for solvolysis), to give the solvent separated ion-pair. This process is usually followed by capture of the cation by the inserted solvent molecule (Scheme 4), even when it is the weakly nucleophilic acetonitrile.
Scheme 4.
It has been shown that to a rough approximation the original Y scale (here used) is related to YCl as in Eqn. 5 [19,21]. This allows conversion to approximate m values, based on the YCl scale, of 0.51 for the overall solvolysis-decomposition reaction, 0.57 for solvolysis, and 0.43 for decomposition.
| (5) |
Since the m value is by definition unity for the solvolyses of 1-adamantyl chloride (the standard solvolyses for the YCl scale), the value of about 0.5 for the solvolyses of the chloroformate is considerably lower. This may be a consequence of the fragmentation occurring during the solvolysis-decomposition process. The production of the stable CO2 molecule in the rate-determining process could result in an earlier transition-state, with less charge development than in the standard solvolysis of 1-AdCl. Another possible factor is that ion-pair return does not count as reaction in the solvolyses of the standard and this return can be reduced by use of a highly ionizing solvent. In contrast, after loss of CO2, the same ion-pair return does count as reaction, since 1-AdCl and not the original 1-AdOCOCl substrate is formed and the influence of the solvent ionizing power will be restricted to the forward, non-reversible reaction.
The entropies of activation in six solvents [53] were found to be of low value, with five of them being positive. This would not be expected for the addition-elimination pathway, or for any other bimolecular pathway, and it is much more consistent with the proposed ionization pathway [30].
4. Secondary Alkyl Chloroformates
Secondary alkyl derivatives as, for example, the chloride or tosylate have been found to exhibit borderline behavior and can react by unimolecular or bimolecular pathway or, frequently, by the two acting in tandem [55]. Using the extended Grunwald-Winstein equation (Eqn. 4), two secondary chloroformate esters have been studied: isopropyl chloroformate (the simplest in this category) and the 2-adamantyl chloroformate, with the chloroformate at a bridging position of the symmetrical cage structure.
Leimu [56] showed that, in methanolysis, the rate order for methyl, ethyl, and isopropyl chloroformate was Me > Et > i-Pr which was indicative of a dominant bimolecular mechanism. Crunden and Hudson [57] found the order in 65% aqueous acetone to be Me > Et < i-Pr, suggesting for the isopropyl ester a change over to a dominant unimolecular mechanism. Going to the much more ionizing solvent, formic acid, led to a rate order Me < Et < i-Pr, suggesting for ethyl and isopropyl, at least, the dominant pathway to have moved further towards ionization.
For solvolyses in the highly ionizing water as solvent, the kinetic solvent isotope effect (KSIE) kH2O/kD2O had a value at 25.0 ºC of 1.25 for isopropyl chloroformate and values in the range of 1.79 to 1.95 for phenyl, ethyl, and methyl chloroformates [29,58]. More recent studies [59] have shown for the parent phenyl chloroformate and for substrates with the introduction of p-methoxy, p-methyl, p-chloro, and p-nitro substituents very little variation of the kH2O/kD2O ratio with values in the range of 1.67 to 1.72. The values suggest considerably less bond formation to the isopropyl chloroformate at the transition state, consistent with the proposed ionization mechanism for this substrate, and an addition-elimination mechanism, as discussed earlier in this review, for the aryl choroformates.
A study of the isopropyl chloroformate solvolyses in terms of Eqn. 4 led to a less than satisfactory correlation and inspection of the data for the 24 solvolyses [60] showed appreciable deviation for 100% and 90% alcohol contents of both the ethanol-water and methanol-water solvents. Removal of these four data points, which presumably have an addition-elimination component superimposed on an ionization pathway, considerably improved the correlation (Table 2) with an l value of 0.28 and m value of 0.52 (l/m value of 0.54); values consistent with an ionization pathway with a rather weak nucleophilic solvation of the developing carbocation and with an earlier transition state due to fragmentation.
Subsequently, the study was revisited [61] and solvolyses in very high ionizing power and low nucleophilicity HFIP-H2O solvents were added. At 40 ºC, when two such solvents were added and the four previously removed were now also accompanied by the data points for 90, 80, 70% aqueous acetone and the 20TFE-80EtOH solvent, the remaining 16 solvents gave a correlation (Table 2) with l and m values almost identical to the earlier study and with an l/m ratio of 0.47, showing the data to be robust.
The 2-adamantyl chloroformate can be conveniently prepared from 2-adamantanol and triphosgene [62]. The solvolysis-decomposition can be conveniently followed, at 25.0 ºC, in the usual types of solvent and product studies were also carried out, showing yields of the decomposition product to range from only 4.8% in ethanol (with 88% formation of the direct substitution product, 2-adamantyl ethyl carbonate) to 59% in TFE (accompanied by 41% of the 2-adamantyl 2,2,2-trifluoroethyl ether). The mixed carbonate was not observed in TFE or in any of the aqueous TFE mixtures studied.
The k1-Ad/k2-Ad ratio of about 105 for tosylates and mesylates are reduced to values in the 400 to 1500 range for the solvolyses of the chloroformates as proceeding by the ionization pathway.
The specific rates of solvolysis have been analyzed in terms of Eqns. 2 and 4 [62]. Using the one-term equation moderately good correlation was obtained with an m value of 0.40 when all 23 solvents were included. With the two-term equation, the l value is negligible at 0.04 ± 0.10 (0.70 probability that the lNT term is statistically insignificant) and with the m value essentially unchanged from the value for the one-term treatment at 0.41 ± 0.05, with the l/m ratio essentially zero. The 100 and 90% ethanol and methanol points, as was the case in the solvolyses of isopropyl chloroformate, lie above the plot. Application of the one-term equation with removal of these points now gives an m value of 0.47 ± 0.03. Further details of these correlations are given in Table 2. Clearly, there is a bimolecular, probably addition-elimination, mechanism also operating in ethanol and methanol and in these alcohols with relatively small amounts of water added. There is far less than the number of solvents needed to apply Eqn. 4 to these apparently bimolecular solvolyses.
The KSIE for the solvolyses in methanol is a little lower at 1.87 ± 0.08 than usually observed for an addition-elimination pathway for methanolyses. This kMeOH/kMeOD ratio probably incorporates a component from the ionization reaction, with a lower ratio for this contributor to the overall ratio [63].
One can tentatively draw a conclusion from the observation that the 1-adamantyl and 2-adamantyl chloroformates in their solvolysis reactions by the ionization mechanism have essentially no sensitivity to changes in solvent nucleophilicity (l ~ 0) but the corresponding solvolyses of isopropyl chloroformate have an l value of about 0.28. The 1-adamantyl chloride, or other 1-AdX type compounds, undergoing a straightforward solvolysis has, by definition, zero dependence on solvent nucleophilicity. However, the experimental l value for the solvolysis of isopropyl tosylate has been reported at 0.53, coupled with an m value of 0.56, which adjusts to 0.63 when the m value needed for the standard methyl tosylate solvolyses, to set up the NOTs scale, is corrected from 0.3 to 0.55 [64]. The finding of an l value of 0.28 for solvolyses of the isopropyl ester and an l value essentially zero for solvolyses of the 1- and 2-adamantyl esters suggests a nucleophilic solvation at the alkyl group for the isopropyl ester which is prohibited by the cage structure for the 1-adamantyl ester and by interactions between the entering and leaving groups and γ-hydrogens of the cage for an SN2-like transition state [21,65]. It is difficult to see how this large difference in l values could arise if the nucleophilic interaction was at the carbonyl carbon.
One alternative pathway involving loss of chloroformate ion to give a carbenium ion is very unlikely to operate because 1-adamantyl trifluoroacetate (1-AdOCOCF3) is structurally similar to 1-AdOCOCl, but with a CF3 group replacing Cl. Since the CF3 and Cl have similar Charton σI values of 0.47 and 0.42, respectively [66], one would, if R+ formation was rate-determining, predict similar rates of reaction. Measurement has shown [67] that, in 80% ethanol at 50 ºC, the trifluoroacetate solvolyses 106 times slower than the chloroformate.
The most plausible mechanism for the ionization of the tertiary and secondary chloroformate esters appears to be the concerted one, with the fragmentation ejecting a stable CO2 molecule leading to a relatively early transition state, consistent with the low sensitivities towards changes in both solvent nucleophilicity and solvent ionizing power (Scheme 5).
Scheme 5.
With variable nucleophilic solvation of the developing R+ and variable electrophilic solvation of the developing Cl−
5. Primary Alkyl Chloroformates
In going from the tertiary alkyl chloroformates, where essentially all the reaction involves ionization with fragmentation, to a secondary alkyl chloroformate, it is found that appreciable amounts of ionization continue to dominate in the more ionizing solvents but, in ethanol and methanol and mixtures of those solvents with small amounts of water, addition-elimination becomes an important contributor towards the isopropyl chloroformate solvolysis. For the 2-adamantyl chloroformate, with a less stable secondary alkyl carbocation, in ethanol only 4.8% of the decomposition product is observed and 88% of the product is the mixed carbonate. For solvolysis in TFE, no mixed carbonate is observed and 59% of the product is the 2-adamantyl chloride and 41% the mixed ether [62].
On continuing to the primary alkyl chloroformates and then on to methyl chloroformate, we would expect these trends to continue. Also, due to the instability of primary and, especially methyl carbocations, there will be a tendency for any ionization reaction not to be accompanied by fragmentation but to proceed to the ROCO+ carboxylium ion.
We report in this section on the solvolyses of ethyl [68], n-propyl [63], n-octyl [69], iso-butyl [27,70], neopentyl [71], and 1-adamantylmethyl [72] chloroformates. Solvolyses of benzyl chloroformates, which could be considered as primary, are deferred until a later section. A study of 2,2,2-trichloroethyl chloroformate [73] is also delayed until a later section. After the consideration of the primary alkyl chloroformates, we follow with a consideration, in section 6, of the solvolyses of methyl chloroformate [74].
The chloroformate esters with a continuous chain of carbons (ethyl, n-propyl, and n-octyl) react at similar rates in their solvolyses. At 24.2 ºC, the relative rates are 1.00:1.08:1.03 in methanol, 1.00:0.97:1.06 in ethanol, 1.00:1.08:1.01 in 80% ethanol, and 1.00:1.08:0.98 in 80% acetone. The kinetic patterns are also very similar, with appreciably negative entropies of activation, for the hydrolysis and alcoholysis of ethyl chloroformate [75,76] and in several solvolyses of the n-propyl chloroformate [63], consistent with a bimolecular reaction pathway, which Orlov [77] considered as rate-determining addition in an addition-elimination sequence, consistent with current thought, but Kivinen considered as following a concerted SN2 mechanism [76]. For the n-propyl ester, the KSIE, kMeOH/kMeOD, was 2.1. Values in the range of 2.1 to 2.5 are generally considered to result from a bimolecular solvolysis assisted by general-base catalysis [78,79].
For the solvolyses of ethyl chloroformate, application of Eqn. 4 to 35 data points showed that there were two reaction channels. One was dominant in 28 of the solvents, including TFE-water compositions with at least 10% water and the other in 100% TFE and 97%TFE-3% water and all of the (97–50%) HFIP-water solvents. The two channels were considered [68] to represent the addition-elimination and ionization (to the carboxylium) pathways. The data obtained from the correlations are reported in Table 2. The l/m ratios of 2.84 and 0.84 are nicely consistent with the proposed pathways. The correlation data are also reported in Table 2 for the two channels operating for the n-propyl chloroformate solvolyses, which lead to very similar l/m ratios of 2.79 and 0.63 [63]. Only six specific rate values are available for the n-octyl ester, insufficient for a two-term correlation.
The sequential replacement of the two β-hydrogens of n-propyl chloroformate by methyl groups leads first to isobutyl chloroformate [27,70] and then to neopentyl chloroformate [71]. As with the variation of chain length in the “normal” series, the introduction of methyl substituents at the β-carbon has only a moderate influence on rate for substrates reacting by the addition-elimination mechanism, the relative rate ratio for the solvolyses of n-propyl [63], isobutyl [27], neopentyl [71] and 1-adamantylmethyl [72] chloroformates at 45.0 ºC are 1.00:1.11:1.14:0.84 in methanol, 1.00:1.12:1.44:1.02 in ethanol, and 1.00:1.05:0.95:0.88 in 80% ethanol. The KSIE for methanolysis, kMeOH/kMeOD is 2.20 for isobutyl chloroformate [70] and 2.19 for 1-adamantylmethyl chloroformate [72], consistent with the addition step of an addition-elimination mechanism being rate-determining and the operation of general-base catalysis from a second methanol molecule.
These branched chain primary alkyl chloroformates show similar behavior to the continuous chain ones in their extended Grunwald-Winstein plots, with a large set of solvents following the addition-elimination pathway and a smaller set, consisting of aqueous fluoroalcohols, following the ionization pathway. The plot for neopentyl chloroformate is shown in Figure 4.
Figure 4.
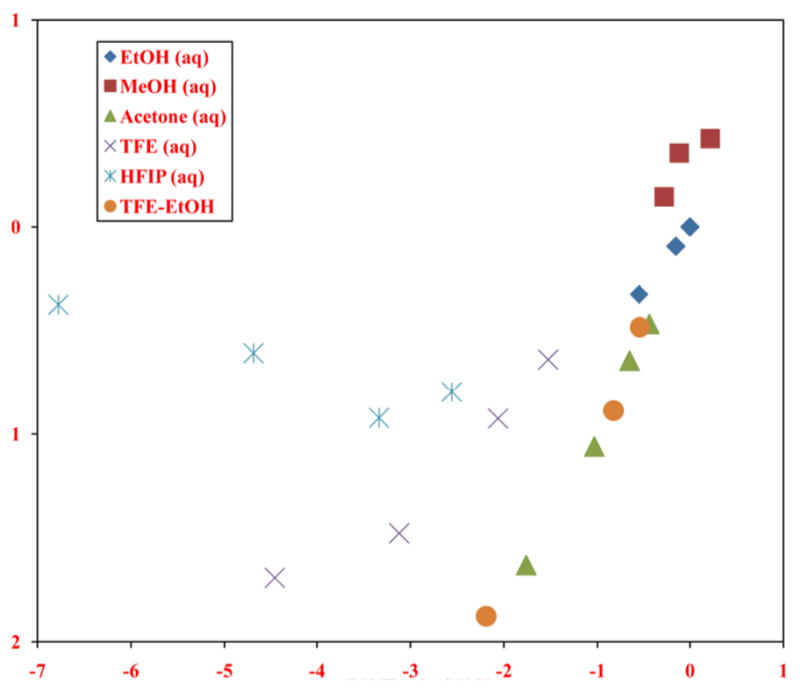
The plot of log(k/ko) values for solvolyses of neopentyl chloroformate at 45.0 °C against 1.76 NT + 0.48 YCl. Abstracted from ref. 71 with permission from the International Journal of Molecular Sciences.
Additional l/m ratio values have been reported [70] as 2.94 for isobutyl chloroformate in the addition-elimination (A–E) range and 1.19 in the ionization range, but some of the relatively large number of solvents assigned to the ionization range may have an A–E component. The 21 solvolyses studied for neopentyl chloroformate [71] can be divided into thirteen proceeding by the A–E mechanism with an l/m ratio of 3.67 and eight by the ionization mechanism with an l/m ratio of 0.44. With the neopentyl chloroformate in the more highly ionizing and weakly nucleophilic solvents, the ionization pathway may again feature a rate-determining fragmentation because of the energetically favorable 1,2-methyl shift, leading to the formation of a rearranged tertiary carbocation. The very large deviations from the plot illustrating the A–E mechanism (Fig. 4) for the data points for solvolyses in HFIP-H2O mixtures are consistent with this proposal.
In the solvolyses of 1-adamantylmethyl chloroformate [72], the same type of 1,2-shift is possible but in this case it would result in ring expansion to the 3-homoadamantyl cage structure [80]. The favorable aspects of going from the formation of a primary carbocation to the production of a tertiary carbocation would be countered by the introduction of ring strain. Product studies in ethanol and other aqueous ethanol compositions with up to 50% (by volume) water content gave no evidence for products based on the 3-homoadamantyl ring system and only very small amounts of 1-adamantylmethyl chloride (< 1%) and 1-adamantylmethyl ethyl ether (less than 4%) with the major products being 1-adamantylmethyl ethyl carbonate and 1-adamantylmethyl alcohol, formed by loss of CO2 from the 1-adamantylmethyl hydrogen carbonate [81]. As with other primary alkyl chloroformates, the solvolysis gave specific rates which correlated into two regions; the A–E mechanism operates in 18 solvents and, in nine solvents of low nucleophilicity and high ionizing power, the ionization pathway predominates. The correlation data are presented in Table 2 and the l/m values for the two branches, consistent with their assignment, are 3.35 and 0.42, respectively. The solvolyses of 9-fluorenylmethyl chloroformate also appear to have two branches in the extended Grunwald-Winstein plot and a KSIE, kMeOH/kMeOD, of 2.20 ± 0.03 [82].
6. Methyl Chloroformate
This can be considered as the parent compound for replacing one, two, or three hydrogens by alkyl groups to give the 3º, 2º, and 1º alkyl chloroformates discussed above. The trends observed in going from the 3º, to the 2º, and then to the 1º alkyl chloroformates of increasing amounts of reaction by the addition-elimination (A–E) with concurrent decrease in the amounts of ionization reaction continue when the study is extended to methyl chloroformate [74].
Only in the most extreme solvent studied in terms of high ionizing power and low nucleophilicity (90% HFIP) was there evidence, upward deviation from the plot in terms of Eqn. 4, for the operation of an ionization mechanism. The other 19 solvents fell nicely on the correlation, which yielded very typical values for solvolyses proceeding by the addition-elimination pathway. These are presented in Table 2. The l/m ratio has a value of 2.74, typical for solvolyses proceeding by the addition-elimination pathway.
7. Effect of Chlorine Substitution Within the Alkyl Group
Two compounds of this type have been studied, after replacement of a methyl group by a trichloromethyl group. One involves the replacement of one of the methyl groups of tert-butyl chloroformate to give 2,2,2-trichloro-1,1-dimethylethyl chloroformate (β,β,β-trichloro-tert-butyl chloroformate), Cl3CC(CH3)2OCOCl [48], and the other the replacement of the methyl group of ethyl chloroformate by the CCl3 group to give 2,2,2-trichloroethyl chloroformate, Cl3CCH2OCOCl [73].
For the trisubstituted tert-butyl ester, the Gruwald-Winstein treatment in terms of Eqn. 4 showed a good linear correlation with an l value of 1.42 and m value of 0.39 (l/m ratio of 3.64). These values strongly suggest that the introduction of the three chlorine atoms has changed the mechanism from what is for tertiary-alkyl chloroformates essentially ionization over the full range of solvents to addition-elimination over the full range. Further support comes from appreciably negative entropies of activation and a KSIE kMeOH/kMeOD of 2.14.
This dramatic change of mechanism is not unexpected because a trichloromethyl group is known to be powerfully electron-withdrawing. For example, Taft σ* value, for use for aliphatic systems is, +2.65, relative to methyl being zero by definition [83]. The influence of the one CCl3 outweighs the weaker electron-supplying influence of the two methyl groups.
Since the trichlorosubstituted tert-butyl compound proceeds by the A–E mechanism, then one would certainly expect the similarly substituted ethyl compound, without the two moderating methyl groups, to also proceed in this manner. That is indeed the case [48] and the l and m values are again similar to those previously reported for chloroformate esters undergoing solvolysis by the A–E mechanism (Table 2) and the l/m ratio has a value of 2.78, typical for such a mechanism.
At 25.0 ºC, the relative specific rates from CH3CH2OCOCl to CCl3CH2OCOCl to CCl3C(CH3)2OCOCl are 1.00:68.1:9.6 in methanol, 1.00:100:11.2 in ethanol, 1.00:97.3:5.7 in 80% ethanol, and 1.00:5.4:1.47 in 70% TFE. In the first three entries there is approximately two orders of magnitude increases followed by an approximately one order of magnitude decrease on adding the two methyl groups. In 70% TFE, the changes are in the same directions but the magnitudes are much smaller. The changes can be rationalized in terms of increased electrophilicity at the carbonyl carbon on introducing the three chlorine atoms, with this effect modified in presence of the two electron-supplying methyl groups and any steric hindrance effects would also lead to rate reductions.
8. Benzyl, Vinyl, Allyl, and Propargyl Chloroformates
Benzyl chloroformate(carbobenzoxy chloride; Cbz-chloride) is frequently the chloroformate of choice in peptide synthesis [84]. Subsequently, the Cbz group is readily removed by catalytic hydrogenolysis or by treatment with hydrogen bromide in acetic acid. The p-nitro derivative (PNZ-chloride) is also used and the PNZ group is easier to remove by hydrogenolysis and more difficult by use of acid. Allyl chloroformate (Alloc-chloride) is also used for the protection of amines.
The p-nitrobenzyl chloroformate gives as an acceptable correlation against NT and YCl values (Eqn. 4), with the sensitivity values typical for an addition-elimination mechanism [85]. The values from 19 data points are given in Table 3, the l/m ratio of 3.50 is consistent with a strong nucleophilic interaction by the solvent at the electrophilic carbonyl carbon. Even in 100% TFE, there was no evidence for an ionization component. Product studies showed no decomposition product and only the mixed carbonate from attack of alcohol at the carbonyl carbon and, with a water component to the solvent, the alcohol. The alcohol is believed to be formed via the hydrogen carbonate, which then loses CO2 [81].
Table 3.
Correlation of the specific rates of solvolysis of benzyl, alkenyl, and alkynyl chloroformates (ROCOCl) using the extended (Eqn. 4) form of the Grunwald-Winstein equationa.
| R in ROCOCl | nb | lc | mc | cc | l/m | Rd | Fe |
|---|---|---|---|---|---|---|---|
| C6H5CH2- | 15 | 1.95 ± 0.16 | 0.57 ± 0.05 | 0.16 ± 0.15 | 3.42 | 0.966 | 83 |
| 11 | 0.25 ± 0.05 | 0.66 ± 0.06 | −2.05 ± 0.11f | 0.38 | 0.976 | 80 | |
| p-NO2C6H4CH2- | 19 | 1.61 ± 0.09 | 0.46 ± 0.04 | 0.04 ± 0.22 | 3.50 | 0.975 | 157 |
| CH2=CH- | 12 | 1.67 ± 0.19 | 0.31 ± 0.07 | 0.10 ± 0.09 | 5.38 | 0.941 | 35 |
| 5 | 0.80 ± 0.03 | 0.59 ± 0.01 | −1.31 ± 0.03f | 1.36 | 0.999 | 578 | |
| CH2=C(CH3)- | 50g | 1.54 ± 0.06 | 0.54 ± 0.03 | 0.05 ± 0.06 | 2.85 | 0.968 | 347 |
| CH2=CH-CH2- | 28 | 1.43 ± 0.13 | 0.52 ± 0.03 | 0.10 ± 0.06 | 2.75 | 0.954 | 127 |
| 7 | 0.93 ± 0.12 | 0.66 ± 0.14 | −0.84 ± 0.30f | 1.41 | 0.974 | 36 | |
| H-C≡C-CH2- | 22 | 1.37 ± 0.10 | 0.47 ± 0.07 | 0.11 ± 0.11 | 2.91 | 0.970 | 152 |
| CH3-C≡C-CH2- | 14h | 1.50 ± 0.15 | 0.49 ± 0.08 | 0.15 ± 0.10 | 3.06 | 0.956 | 58 |
When two entries are given for a substrate, the first refers to what is believed to be predominantly addition-elimination reaction and the second to the region with what is believed to be predominantly ionization reaction.
Number of solvents.
With associated standard error.
Multiple correlation coefficient.
F-test value.
Since the ko value (80% ethanol) is not available for the ionization reaction, log k was plotted and the intercept is (c + log ko), a close approximation to an estimate for log ko for ionization reaction.
Omitting the (above the plot) data point for 97% HFIP.
Omitting data points in HFIP-H2O mixtures (above the plot).
The benzyl chloroformate was studied in 26 solvents. Since it is generally believed that, in studies of substitution reactions, one phenyl group is to a rough approximation equivalent to two alkyl groups [86], it would not be surprising to see mechanistic behavior similar to that discussed earlier in this review for isopropyl chloroformate. Indeed attempts to apply Eqn. 4 to all of the data showed that two branches are present, with the l and m values consistent with A–E and ionization. The fluoroalcohol water solvents and the TFE-ethanol solvents with at least 80% TFE (11 solvents) gave a linear plot with l and m values consistent with the ionization pathway (l/m value of 0.38) and the other 15 solvents gave a linear plot with typical values for addition-elimination (l/m value of 3.42). The detailed correlation values are given in Table 3.
Product studies for benzyl chloroformate solvolyses were also consistent with the duality of mechanism, with almost 50% of product being benzyl chloride in the most highly ionizing solvents. In the TFE-water mixtures appreciable amounts of benzyl 2,2,2-trifluoroethyl ether and benzyl alcohol were also observed. In all of the solvents only trace amounts of mixed carbonate were detected. In TFE-ethanol mixtures, with more than 20% ethanol the major product (70–85%) was the benzyl ethyl carbonate, with much smaller amounts of the two possible ether products. The detailed study of the products nicely match the claim for a dominant A–E pathway in the majority of the solvents and a dominant ionization mechanism in those with low nucleophilicity and high ionizing power. As regards the relative rates of the two arylmethyl chloroformates, in ethanol the p-nitro ester is more reactive than the parent benzyl ester by a factor of 3.6 and this factor is 3.3 in methanol, and 3.6 in 20% TFE-80% ethanol. These ratios are inverted in 80% TFE-20% ethanol, and the unsubstituted benzyl ester is faster by a factor of 1.9 and in 97% TFE-3% water this factor is 12.4. These ratios are consistent with the details of the proposed duality of mechanism.
It is somewhat unusual that, with a change in the mechanism as the solvent properties change, all 26 solvolyses used in the correlations can be assigned to one or the other two pathways. This is because the l values for the two pathways are very different and there is only a narrow range of solvent properties where both pathways are making a significant contribution to the overall reaction rate.
For the p-nitrobenzyl chloroformate in methanol the KSIE, kMeOH/kMeOD of 2.42 is a typical value for an A–E pathway assisted by general-base catalysis.
For vinyl and isopropenyl chloroformates any ionization reaction which might occur would not be expected to involve fragmentation, since the vinyl cation, like the phenyl cation, is of very high energy. Only with attached groups that will efficiently disperse the charge is formation of the cation likely. The one methyl group of the isopropenyl cation would not be sufficient. The vinyl group has a σ* (polar substituent constant for use with aliphatic systems) value of +0.65 [87], indicating it to be a fairly powerful electron-withdrawing group. As such, it should make the substrate favorable towards nucleophilic attack at the carbonyl carbon. Indeed, in ethanol at 25.0 ºC, the vinyl ester reacts [88] about three times faster than the phenyl ester and about 320 times as fast as the saturated equivalent, ethyl chloroformate. It appears for the vinyl chloroformate that there is an incursion of an ionization pathway for solvents rich in fluoroalcohol but not enough data could be obtained in the region where this pathway dominates for a meaningful application of the Grunwald-Winstein equation. For most of the solvolyses, the A–E mechanism is operating with consistent l and m values (Table 3) but with a somewhat larger than usual l/m ratio of 5.38, possibly resulting from the increased electrophilicity at the carbonyl carbon and the low steric hindrance to its approach.
Replacing the α-hydrogen of the vinyl chloroformate by a methyl group we arrive at isopropenyl chloroformate or, looking at it in another way, we are introducing a carbon-carbon double bond into isopropyl chloroformate. There have been three recent publications dealing with the specific rates of solvolysis of this substrate and their correlation [89–91].
Ryu carried out an extensive study, at 10.0 ºC, over a full range of mixtures of water with ethanol and methanol and with up to 90% acetone in acetone-water mixtures. There was less extensive coverage of TFE-water mixtures (80–40% TFE) and full coverage of TFE-ethanol mixtures. In KSIE studies, kMeOH/kMeOD had a value of 2.33, typical for general-base assisted A–E reaction (Koh [90] reported a value of 2.19 at 35.0 ºC) and a value for kH2O/kD2O of 2.08. Ryu reported an analysis, using the Bentley approach [35], in terms of third-order rate coefficients. He also carried out a correlation analysis in terms of Eqn. 4 for 40 solvents. A good linear plot was obtained with l and m values of 1.88 and 0.60, respectively, being incorporated (l/m ratio of 3.13). However, the regions where one would best observe any ionization component, HFIP-water mixtures and TFE-water mixtures rich in TFE, were not included in this study.
Koh [90], in a study at 35.0 ºC, included three HFIP-H2O solvents and 90% TFE to the solvents used by Ryu. The higher temperature moved the rates of these slow solvolyses into a region more amenable to study. A few studies at various temperatures, yielding very negative entropies of activation of −30 to −38 cal mol−1 K−1, were incorporated but, unfortunately, these studies did not include the new solvolyses not included in the Ryu study. The correlation at 35.0 ºC led to an l value of 1.42 and an m value of 0.36 (l/m ratio of 3.09). The lower l and m values will result, at least in part, from the neglect of the upward curvature that can be seen for the HFIP-H2O points in the presented plot. This curvature suggests the incursion of an ionization component.
Kevill, D’Souza and coworkers [91] extended the measurements of the new solvolyses studied by Koh and Kang [90] to other temperatures in the 25 to 65 ºC range and used Arrhenius plots to extrapolate to specific rates at 10.0 ºC, which could then be incorporated into the Ryu analyses. They also, extended the range of solvent type by measuring at higher temperatures and extrapolating to get specific rates at 10.0 ºC in 97% HFIP and 97% TFE. Analyses of data at 10.0 ºC were now repeated with, in addition to the 40 solvents of the Ryu study, a further 11 solvents, which had been chosen to extend the solvent composition ranges and to add the important HFIP-containing solvents. This enhanced analysis led to l and m values of 1.40 and 0.51, respectively, (l/m value of 2.75). The plot showed an exceptionally large deviation for 97% HFIP and removal of this point gave a considerably improved correlation with an l value of 1.54 and m value of 0.54 (l/m ratio of 2.85). Using these correlation values (more detail in Table 3) it can be estimated that in 97% HFIP, 97% of the overall reaction is with ionization and this value falls to 70% in 90% HFIP, 64% in 70% HFIP, and to only 5% in 50% HFIP. A value of 35% in 97% TFE was also estimated. Higher values for l, approaching the 1.80 of the Ryu study [89] would probably have been obtained if the data points for additional solvents were also omitted for the correlation.
There are two studies of the solvolyses of allyl chloroformate (CH2=CH-CH2OCOCl) available [88,92]. Allyl chloroformate has recently been prominent because of its use in surface coating technology to synthesize bacterial-resistant amphiphilic polymers [93].
The allyl cation is stabilized by resonance, which could give a driving force for the ionization reaction with fragmentation to the allyl cation. The reactivity of allyl derivatives in nucleophilic substitution reactions is frequently observed to be the same as for benzyl derivatives. This also applies to the chloroformate esters and, at 25.0 ºC, the allyl [88] to benzyl [85] rate ratios are, for example, 0.83 in 80% ethanol, 0.66 in methanol, 0.086 in 97% TFE, and 0.13 in 97% HFIP. Only small differences in methanol and aqueous ethanol and about an order of magnitude difference in fluoroalcohol-rich solvents.
Koh and Kang [92] placed all 30 data points obtained at 35.0 ºC on one plot, in terms of Eqn. 4, and obtained values for l of 0.93 and for m of 0.41. These values are quite similar to the values (1.00 and 0.54) for methyl p-toluenesulfonate solvolyses and, on this basis, they assumed an SN2 mechanism. However, examination of the plot presented in the paper shows a marked curvature, consistent with an ionization component in the fluoroalcohols. It follows that the values reported are probably composites from the combination of the specific rates for both pathways in the construction of the extended Grunwald-Winstein plot. They obtained a KSIE for kMeOH/kMeOD of 2.16, nicely consistent with addition-elimination assisted by general-base catalysis.
In a second study [88], with a total of 21 solvolyses at 25.0 ºC, it was found that, if the 97–70% TFE and HFIP solvents had their data points omitted, the remaining 12 solvents gave a good correlation (Table 3) with an l value of 1.78 and an m value of 0.43 (l/m = 4.14). It was proposed that the usual combination of addition-elimination and ionization mechanisms were operating and that there was no convincing evidence for any contribution from a conventional SN2 mechanism at the carbonyl carbon for the solvolyses of allyl chloroformate.
Two compounds with a triple bond introduced into the alkyl group of an alkyl chloroformate have been studied. Propargyl chloroformate (H-C ≡C-CH2OCOCl) [94] can be considered as being derived from n-propyl chloroformate and 2-butyn-1-yl chloroformate (CH3-C ≡C-CH2OCOCl) [95] can be considered as derived from n-butyl chloroformate. The two substrates can also be considered in terms of the second being derived from the first by replacement of the terminal hydrogen by a methyl group. Both substrates on ionization with fragmentation would lead to a carbocation stabilized by resonance. Also, the 2-butyn-1yl chloroformate would place part of the positive charge on a carbon now carrying a methyl group, which would give additional stabilization. In the absence of fragmentation the acetylenic grouping would be expected to be moderately electron-withdrawing. The H-C ≡C- group has a Charton σI value of 0.35 and the H-C ≡C-CH2- a value of 0.13 [96].
We can consider the relative rates of solvolyses, at 25.0 ºC, for the sequence of chloroformate esters (ROCOCl) with n-propyl, allyl, propargyl, and 2-butyn-1-yl as the R group. In ethanol, the rate ratios are: 1.00:5.05:15.9:8.00, in methanol 1.00:1.41:7.14:3.82, in 80% ethanol-20% water (v/v) 1.00:1.86:8.42:4.32, in 90% TFE-10% water (w/w) 1.00:3.39:3.66:1.8. In all instances the values successively rise on introducing the double and the triple bond and there is a fall off of almost exactly by a factor of two on introducing the γ-methyl group into the propargyl chloroformate. This fall off is consistent with a remote effect slightly modifying the electron-withdrawing assistance to an addition-elimination pathway and it is not consistent with an ionization reaction and, especially, not consistent with an ionization-fragmentation reaction, when it would be able to directly stabilize at the transition state a partial positive charge developing on the γ-carbon.
A good extended Grunwald-Winstein equation correlation (Table 3) was obtained over a full range of solvents with an l value of 1.37 and an m value of 0.47 (l/m of 2.91). Often useful in mechanistic studies is a direct linear free energy relationship (LFER) plot of the log (k/ko) values for a solvolysis under study with those for a substrate whose mechanism is believed to be well-understood. The solvolyses of phenyl chloroformate fall into this category. For a good linear correlation the requirement is that the l/m ratios be essentially identical, as they are for this correlation at 2.91 as opposed to 2.94 for the solvolyses of phenyl chloroformate. The slope of such a plot will be the ratio of either the l values or the m values for the two substrates. These values are 1.37/1.68 and 0.47/0.57, both giving a value of 0.82, in excellent agreement with the slope of the LFER correlation (Figure 5) of 0.86 ± 0.02.
Figure 5.
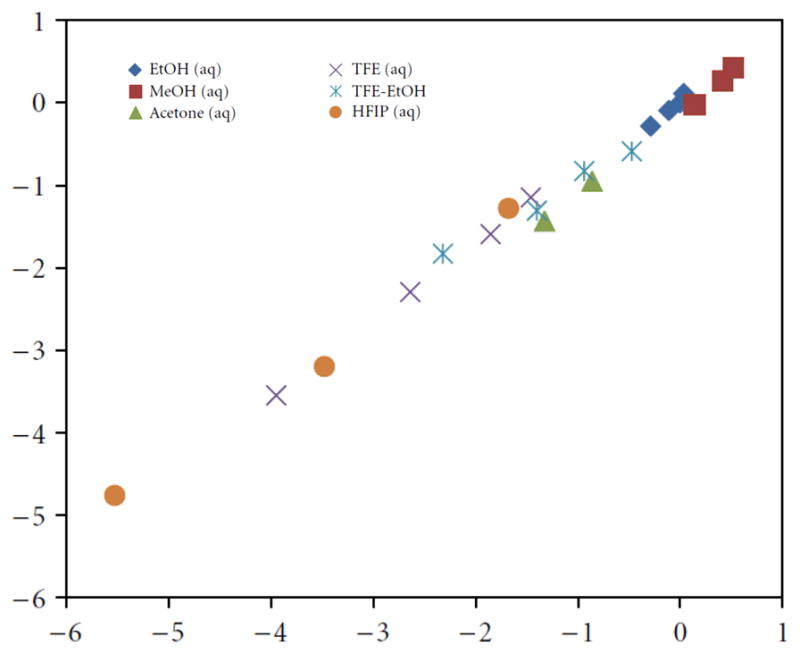
The plot of log (k/ko) for propargyl chloroformate against log (k/ko) for phenyl chloroformate in common pure and binary solvents at 25.03C. Abstracted from ref. 94 with permission from the International Scholarly Research Network (ISRN) Organic Chemistry.
Based on the correlation in terms of Eqn. 4 and the excellent LFER correlation against the phenyl chloroformate solvolyses, we can feel confident that propargyl chloroformate follows the same addition-elimination pathway, with the addition process rate-limiting.
As mentioned above, in four solvents the specific rates for 2-butyn-1yl chloroformate are about one-half of those of propargyl chloroformate. With only a rather remote electron-supplying methyl group as a variant, this is not surprising and, in mixtures of water with ethanol, methanol, acetone, and TFE together with TFE-ethanol mixtures, a good LFER was found between the two sets of data, with a multiple correlation coefficient of 0.995. When the above mentioned solvolyses of 2-butyn-1-yl chloroformate are analyzed in terms of an extended Grunwald-Winstein equation (Eqn. 4) a good correlation was obtained (Table 3), with l and m values of 1.50 and 0.49, respectively, and an l/m ratio of 3.06, as opposed to 2.91 for propargyl chloroformate [94].
9. Fluoroformates
Fluoroformates were briefly reviewed some forty years ago [5] in terms of preparation, properties, and reactivity. Very little had been done at that time in terms of rate measurements but one interesting observation was that, in solvolyses in 85% acetone-15% water at 0.0 ºC, the ethyl fluoroformate solvolyzed about thirty times faster than the chloroformate [97,98]. It had been realized earlier [26] that RCl/RF rate ratios can give important mechanistic information. For instance, in nucleophilic aromatic substitution, the faster reaction of the fluoride substrate [99] was consistent with rate-limiting addition of the nucleophile, without the breaking of the much stronger C–F bond, coupled with the greater electron-withdrawing influence of the fluorine giving increased electrophilicity at the attacked carbon.
In early preparations, fluoroformates were frequently prepared by the reactions of alcohols with COF2 or COFCl [100], paralleling the use of phosgene in chloroformate preparations. Alternatively, a metal fluoride, such as thallous fluoride, was used in an exchange reaction with the chloroformate ester [101,102]. A modification of the exchange reaction with the use of KF in the presence of 18-crown-6 [103] is now frequently used. In some cases, the chloroformate is unstable (tert-butyl, for example) or fluoride attacks at the α-carbon of the R group of ROCOCl (benzyl, for example) and Olofson and Senet devised a procedure involving an initial formation from the alcohol of a mixed carbonate by treatment with a 1-chloroalkyl chloroformate and then treatment of the mixed carbonate with KF and 18-crown-6 [104]. The latter two methods are those which have been used in preparing the fluoroformate esters discussed in this section.
In this section, we will discuss the solvolyses of two tertiary alkyl fluoroformates, the 1-adamantyl [105,106] and tert-butyl [107] esters, two secondary alkyl fluoroformates, 2-adamantyl [108] and isopropyl esters [109], five primary alkyl fluoroformates, the n-octyl [69], n-propyl [110], ethyl [111], isobutyl [112], and 1-adamantylmethyl [72], the “parent” methyl fluoroformate [113], and benzyl fluoroformate [114].
One complication in applying the extended Grunwald-Winstein correlation to fluoroformate esters, indeed to fluorides in general, is the lack of a scale of YF values. The 1-adamantyl fluoride, which would be the most desirable substrate for establishing a scale, solvolyzes only very slowly and it was shown that the solvolyses in 100% TFE were accelerated by three orders of magnitude by 7 x 10−3 M hydrogen fluoride [105]. The acceleration is sharply reduced but still appreciably on dilution with water. The very slow solvolyses and the acid-catalysis have so far thwarted attempts to establish a YF scale.
For solvolyses proceeding by the addition-elimination pathway (shown in Scheme 1 for replacement of chloride), in the rate-determining step attack of the nucleophile is driving the π electrons of the carbonyl group onto the oxygen and the YX scales are, strictly speaking, not applicable to this process. We find, however, that for carbonyl chlorides the YCl scale is adequate to also cover this situation. Actually, even if a YF scale was available, it might be best to continue using YCl values for parallel addition-elimination processes for fluoroformates.
The ionization process, shown in Scheme 2 for chlorides, is more problematic. Here the halide is being expelled in the slow step and YF values should ideally be applied. The incipient fluoride ion at the transition state will be heavily solvated since fluoride is a classical example of a species subject to hydrogen-bonding when appropriately situated hydrogens are available. Nonetheless, indications are that, in the few cases where this mechanism operates, use of the YCl does give reasonable correlations.
The kCl/kF ratios are extremely large for ionization processes, 105 for tert-butyl halides in 85% acetone, 106 for triphenylmethyl halides in that solvent [26], and 3 x 107 for p-(dimethylamino)benzoyl halides in water [115]. Accordingly, unless the specific rate of the addition-elimination process, underlying a dominant ionization process for a solvolysis of a given chloroformate ester, is at least 105 to 107 times lower than that for the operating ionization process (detailed magnitude depending on the system), on substituting fluorine for chlorine the observed mechanism will become by default addition-elimination.
Within the studied systems, we find that only for tertiary alkyl haloformates is this a consideration. For the solvolyses of tert-butyl fluoroformate [107] an extended Grunwald-Winstein correlation in 21 solvents gives reasonable correlation with an l/m value of 0.71. The four TFE-ethanol solvolyses lie below the plot, as is often observed [116], and with their omission, for 17 solvents, an l value of 0.41 and an m value of 0.65 (l/m ratio of 0.63) are observed (Table 4). The values are similar to those of tert-butyl chloride [20], suggesting an ionization process with nucleophilic solvation of a developing cation (as shown for 1-adamantyl chloroformate in Scheme 4). We cannot determine kCl/kF rate ratios because of the instability of the tert-butyl chloroformate [47].
Table 4.
Correlation of the specific rates of solvolysis of alkyl fluoroformates using the extended (Eqn. 4) form of the Grunwald-Winstein equationa,.
| Substrate | nb | lc | mc | cc | l/m | Rd |
|---|---|---|---|---|---|---|
| 1-AdOCOF | 10e | 2.78 ± 0.21 | 1.01 ± 0.06 | 0.09 ± 0.16 | 2.78 | 0.987 |
| 16f | ~ 0 | 0.70 ± 0.01 | −0.02 ± 0.05 | ~ 0 | 0.999 | |
| t-BuOCOF | 17 | 0.41 ± 0.05 | 0.65 ± 0.03 | 0.02 ± 0.04 | 0.63 | 0.989 |
| 2-AdOCOF | 17 | 1.92 ± 0.15 | 0.84 ± 0.06 | −0.02 ± 0.06 | 2.28 | 0.968 |
| i-PrOCOF | 20 | 1.59 ± 0.16 | 0.80 ± 0.06 | −0.12 ± 0.05 | 1.99 | 0.957 |
| n-OctOCOF | 19 | 1.67 ± 0.07 | 0.76 ± 0.03 | −0.08 ± 0.18 | 2.20 | 0.988 |
| n-PrOCOF | 16 | 1.72 ± 0.12 | 0.91 ± 0.08 | −0.05 ± 0.08 | 1.89 | 0.970 |
| EtOCOF | 17 | 1.34 ± 0.14 | 0.77 ± 0.07 | −0.06 ± 0.10 | 1.74 | 0.942 |
| MeOCOF | 14 | 1.33 ± 0.09 | 0.73 ± 0.06 | −0.08 ± 0.08 | 1.82 | 0.972 |
| C6H5CH2OCOF | 13 | 1.43 ± 0.13 | 0.70 ± 0.05 | −0.09 ± 0.17 | 2.04 | 0.974 |
| C6H5COFg | 41 | 1.58 ± 0.09 | 0.82 ± 0.05 | −0.09 ± 0.10 | 1.93 | 0.953 |
See text for references to the source of the specific rate values and the derived correlation values.
Number of solvents.
With associated standard error.
Multiple correlation coefficient.
By addition-elimination pathway.
By ionization pathway.
Correlation data for benzoyl fluoride from ref. 117.
For 1-adamantyl fluoroformate [105], the strain involved in introducing an sp2-hybridized carbon at a bridgehead of a cage structure is an additional feature discouraging the ionization pathway, with concurrent or subsequent decomposition involving loss of CO2. Only in highly ionizing and low nucleophilicity solvents was the solvolysis-decomposition mechanism paralleling that for the chloroformate (Scheme 4) observed. In the remainder of the studied solvents, the products, as determined by gas-chromatography, were primarily formed by attack at the acyl carbon. In ethanol and 80% ethanol about 90% of the products were formed in this way. An extended Grunwald-Winstein treatment was best analyzed in terms of these two pathways, with a division into ten and sixteen solvents for the A–E and ionization pathways, respectively. Details of the analyses are give in Table 4. Studies as a function of temperature variation were also consistent with the two pathways, with very negative entropies of activation (−32 to −42 cal mol−1 K−1) for the A–E solvolyses and considerably less negative (−8 to −15 cal mol−1 K−1) for solvolyses following the ionization pathway. For this substrate, the effect of varying pressure on the rates of solvolyses was also studied [106] and this study gave results which also suggested that the solvolysis follows two major reaction pathways.
For the studied primary and secondary alkyl chloroformates, with the exception of 2-adamantyl chloroformate, it was found that the addition-elimination pathway in the “traditional” ethanol-water, methanol-water, and acetone-water solvents transformed over to a dominant ionization mechanism in solvents rich in fluoroalcohol (HFIP or TFE) [Table 2]. For the 2-adamantyl chloroformate, only in 100% and 90% ethanol and methanol was there evidence for an appreciable addition-elimination component. In contrast, no evidence was found for a region with a dominant ionization pathway for any of the studied secondary alkyl fluoroformates [108,109], primary alkyl fluoroformates [69,72,110,112], methyl fluoroformate [113] and benzyl fluoroformate [114]. All the points in the extended Grunwald-Winstein equation correlations (Eqn. 4) lay, in all instances, nicely on one linear plot except the points for the TFE-ethanol solvolyses were frequently slightly below the plot [116], and the controlling l and m values, the sensitivity to changes in solvent nucleophilicity and solvent ionizing power, were consistent with a rate-limiting addition-elimination pathway (Scheme 1). These values are presented in Table 4.
The last entry in Table 4 is the values from a comprehensive study of benzoyl fluoride [117]. The correlation data are remarkably similar to those for the fluoroformate esters. Indeed, by coincidence, the l and m values are almost identical to the mean of the values from the solvolyses of the secondary, primary, methyl, and benzyl fluoroformates that are within the table.
The kCl/kF ratios of not very far removed from unity are also consistent with the assignment of a mechanism in which the carbon-halogen bond undergoes hybridization changes but is not being broken prior to the transition state for the overall process. A selection of kF/kCl values are given in Table 5. Note that, for convenience, the ratio is inverted from the way it is presented earlier in the review because for the addition-elimination pathway, the fluoro-compound, in most instances, has higher specific rate. The kF/kCl ratios reported for 80% acetone are in excellent agreement with the values determined earlier by Queen and Nour [118] for varying R in ROCOX at 30.1 ºC in 70% acetone: Me, 7.16; Et, 5.46; n-Pr, 4–95; i-Pr, 1.09.
Table 5.
The specific rate ratio (kF/kCl) for solvolyses of alkyl haloformates (ROCOX) in pure and binary solvents.
| Solvent (%)a | methylb | ethylc | n- propyld | i- butyle | n- octylf | i- propylg | 2- adamantylh | benzyli |
|---|---|---|---|---|---|---|---|---|
| 100 EtOH | 0.83 | 0.57 | 0.57 | 0.45 | 0.62 | 0.18 | 0.37 | 1.19 |
| 80 EtOH | 8.28 | 8.74 | 5.62 | 5.43 | 8.09 | 2.11 | 3.48 | 11.5 |
| 60 EtOH | - | 14.0 | - | 8.43 | 15.1 | 1.79 | 3.01 | 14.6j |
| 100 MeOH | 1.12 | 0.93 | 0.75 | 0.66 | 0.95 | 0.39 | 0.42 | 1.78 |
| 90 MeOH | 5.11 | 4.82 | - | 2.42 | - | 1.76 | 2.40 | 7.18 |
| 80 Me2CO | 3.71 | 3.90 | 4.24k | 2.60 | 2.86 | 0.53 | 0.65 | 5.89 |
| 70 TFEl | 27.2 | 19.3 | 7.72 | 7.72 | 10.2m | 0.067 | 0.011 | 6.36 |
Unless otherwise indicated on a volume/volume basis, at 25.0 ºC, with the other component water.
From ref. 113.
From ref. 111.
From ref. 110.
From ref. 112.
From ref. 69.
From ref. 109.
From ref. 108.
From ref. 114.
For 70% ethanol.
For 70% acetone.
On a weight-weight basis with the other component water.
For 80% TFE.
The entropies of activation presented earlier in this section for the two pathways followed for 1-adamantyl fluoroformate follow the same pattern as for the other studied fluoroformates. For the tert-butyl fluoroformate values are obtained of −3 to −7 cal mol−1 K−1 and much more negative values of −21 to −50 cal mol−1 K−1 for the fluoroformates proceeding by the addition-elimination pathway (with a majority of the values in the −35 to −45 cal mol−1 K−1 range).
The kinetic solvent isotope effect (KSIE) studies in methanol and methanol-d (kMeOH/kMeOD) are presented (Table 6) for six of the situations where both the chloroformate and fluoroformate are believed to react by the addition-elimination mechanism. It can be seen that, although all values are large and supportive of general-base catalysis by a second methanol molecule to the addition-elimination pathway, the values for the fluoroformates are consistently higher that for the chloroformate. This is in agreement with the proposal that bond formation is more advanced at the transition state for addition to a fluoroformate than for the corresponding addition to a chloroformate. For the methanolyses of tert-butyl fluoroformate, the considerably lower kMeOH/kMeOD value of 1.26 relative to other values for the fluoroformates presented in Table 6 (3.10 to 3.96) gives additional support to its assignment as a solvolysis proceeding with rate-determining ionization.
Table 6.
Kinetic Solvent isotope effect (KSIE) values, kMeOH/kMeOD, for methanolyses of several alkyl haloformates (ROCOX, X = Cl or F).
| R group : | Me | Et | n-Pr | i-Bu |
|---|---|---|---|---|
| (kMeOH/kMeOD)X=Cl: | 2.14 | 2.22 | 2.17 | 2.00 |
| (kMeOH/kMeOD)X=F: | 3.98 | 3.10 | 3.32 | 3.40 |
| Reference Number: | 113 | 111 | 110 | 112 |
| R group : | 1-AdCH2a | i-Pr | t-Bu | Benzyl |
|---|---|---|---|---|
| (kMeOH/kMeOD)X=Cl: | 2.19 | - | - | 2.42b |
| (kMeOH/kMeOD)X=F: | 3.50 | 2.53 | 1.26 | 3.19 |
| Reference Number: | 72 | 109 | 107 | 114 |
1-Ad represent 1-adamantyl.
Value for the p-nitro derivative.
This report on the fluoroformates does not include any study in terms of Grunwald-Winstein equations of an aryl fluoroformate. Since the aryl chloroformates (Table 1) all react by the addition-elimination pathway with no indication of ionization pathways, even in solvents rich in HFIP, it was clear that, with the enormous reduction in specific rate for the ionization pathway on going to the fluoroformate, the same addition-elimination pathway would also be the one followed for solvolyses of aryl fluoroformates. This was confirmed by a determination [118] of a very large kF/kCl ratio of 32.2 for solvolyses of p-methoxyphenyl haloformates in 60% dioxane. This value is larger than any of the values determined when R is an alkyl group (Table 5). In contrast, the lowest value reported in Table 5 (0.067) is for isopropyl haloformates in 70% TFE. Here the fluoroformate will react by the addition-elimination pathway but the chloroformate will be boosted by the addition of a large component from the ionization pathway (Table 2). This effect is even greater for 1-adamantyl haloformates, with very low values, approaching 10−5, in methanol and ethanol [105].
10. Chloroglyoxalates
Chloroglyoxalates (ROCOCOCl) contain relative to chloroformates (ROCOCl) an additional carbonyl group. The naming is based on replacing the hydrogen of an ester of glyoxalic acid with a chlorine. Alternative naming is based on consideration as an acid chloride derived from a half ester of oxalic acid. Hydrolysis leads to mono-substituted oxalic acid derivatives, which unlike the half esters of carbonic acids, as formed from chloroformate esters, are stable [5]. Unlike the methanolysis of 1-adamantyl chloroformate, which gives the mixed ether and 1-adamantyl chloride, both with loss of CO2 [52], the methanolysis of 1-adamantyl chloroglyoxalate proceeds smoothly to the mixed oxalate ester [119].
The additional carbonyl group negates the resonance stabilization involving the carbonyl of the carbonyl chloride group and reactivities are best considered relative to acyl chlorides. The presence of the powerfully electron-withdrawing alkoxycarbonyl group (ROCO-) (σ* value of +2.00 for methoxycarbonyl [87]) at the carbonyl carbon renders it of increased electrophilicity and very susceptible to nucleophilic attack by solvent. It is found [120] that solvolyses of methyl and ethyl chloroglyoxalates are about 106 times faster than the corresponding chloroformate solvolysis.
Even with a rapid-response conductivity technique, it was possible to follow the kinetics of solvolysis only at considerably reduced temperatures, which prevented studies with more than 10% water in aqueous-organic solvents. At −76 ºC, with seven measured and two extrapolated data points, values were obtained for l and m of 1.45 ± 0.16 and 0.30 ± 0.11, respectively, for methyl chloroglyoxalate solvolyses, with similar values of 1.57 ± 0.19 and 0.35 ± 0.14 for ethyl chloroglyoxalate solvolyses. These values are similar to those obtained for solvolyses of chloroformate esters, believed to solvolyze by the addition-elimination pathway (Table 2). Very negative entropies of activation, in the −16 to −31 cal mol−1 K−1 range gave further support to a bimolecular (or higher molecularity) pathway. The methyl ester, over a range of solvents and temperature, solvolyzed about 50% faster than the ethyl ester.
Acknowledgments
This publication was made possible by the Delaware INBRE (DE-INBRE) and Delaware EPSCoR (DE-EPSCoR) programs. Delaware INBRE is funded by a grant from the National Institutes of Health, National Institute of General Medical Sciences (NIH-NIGMS) IDeA program, grant number P20 GM103446. Delaware EPSCoR is funded by a National Science Foundation (NSF) EPSCoR grant (EPS-0814251). The DE-INBRE and DE-EPSCoR programs also acknowledge support from the State of Delaware (DE). The DE-INBRE and DE-EPSCoR grants were obtained through the leadership of the University of Delaware and the authors sincerely appreciate their efforts. Additional support from an NSF ARI-R2 grant 0960503 received by M.J.D. is also acknowledged.
Contributor Information
Malcolm J. D’Souza, Email: malcolm.dsouza@wesley.edu.
Dennis N. Kevill, Email: dkevill@niu.edu.
References
- 1.Herbicide Report. Chemistry and Analysis. Environmental Effects. Agricultural and Other Applied Uses. Report by Hazardous Materials Advisory Committee, United States Environmental Agency Science Advisory Board; Washington, DC, USA. May 1974. [Google Scholar]
- 2.Sartori M. Chemistry and Analysis. D. Van Nostrand Co., Inc; New York: 1939. The War Gases. [Google Scholar]
- 3.Wellings RE. School Sci and Math. 1942;42:331. [Google Scholar]
- 4.Trumpener U. J Mod Hist. 1975;47:460. doi: 10.1086/241340. [DOI] [PubMed] [Google Scholar]
- 5.Kevill DN. Chloroformate Esters and Related Compounds. In: Patai S, editor. Acyl Halides. John Wiley & Sons, Ltd; Chichester, UK: 1972. [Google Scholar]
- 6.Crich D, editor. Handbook of Reagents for Organic Synthesis: Reagents for Glycoside, Nucleotide, and Peptide Synthesis. Wiley; New York: 2005. [Google Scholar]
- 7.Parrish JP, Salvatore RN, Jung KW. Tetrahedron. 2000;56:8207. [Google Scholar]
- 8.Banerjee SS, Aher N, Patel R, Khandare J. J Drug Delivery. 2012:Article ID:103973. doi: 10.1155/2012/103973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matzner M, Kurkjy RP, Cotter RJ. Chem Rev. 1964;64:645. [Google Scholar]
- 10.Archer BL, Hudson RF. J Chem Soc. 1950:3259. [Google Scholar]
- 11.Ugi I, Beck F. Chem Ber. 1961;94:1839. [Google Scholar]
- 12.Johnson DE, Enke CG. Analyt Chem. 1970;42:329. [Google Scholar]
- 13.Daum PH, Nelson DF. Analyt Chem. 1973;45:463. [Google Scholar]
- 14.Kevill DN, Daum PH, Sapre R. J Chem Soc Perkin Trans. 1975;2:963. [Google Scholar]
- 15.Bentley TW, Carter GE, Harris HC. J Chem Soc Perkin Trans. 1985;2:983. [Google Scholar]
- 16.Grunwald E, Winstein S. J Am Chem Soc. 1948;70:846. doi: 10.1021/ja01190a523. [DOI] [PubMed] [Google Scholar]
- 17.Winstein S, Grunwald E, Jones HW. J Am Chem Soc. 1951;73:2700. [Google Scholar]
- 18.Kevill DN, D’Souza MJ. J Chem Res. 2008:61. [Google Scholar]
- 19.Bentley TW, Carter GE. J Am Chem Soc. 1982;104:5741. [Google Scholar]
- 20.Kevill DN, D’Souza MJ. J Chem Res Synop. 1993:174. [Google Scholar]
- 21.Bentley TW, Llewellyn G. Prog Phys Org Chem. 1990;17:121. [Google Scholar]
- 22.Schadt FL, Bentley TW, Schleyer PvR. J Am Chem Soc. 1976;98:7667. [Google Scholar]
- 23.Kevill DN, Anderson SW. J Org Chem. 1991;56:1845. [Google Scholar]
- 24.Kevill DN, Anderson SW. J Am Chem Soc. 1986;108:1579. [Google Scholar]
- 25.Kevill DN. In: Advances in Quantitative Structure-Property Relationships. Charton M, editor. Vol. 1. Jai Press; Greenwich, CT: 1995. pp. 81–115. [Google Scholar]
- 26.Swain CG, Scott CB. J Am Chem Soc. 1953;75:246. [Google Scholar]
- 27.D’Souza MJ, McAneny MJ, Kevill DN, Kyong JB, Choi SH. Beilstein J Org Chem. 2011;7:543. doi: 10.3762/bjoc.7.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urbanski J. Roczniki Chem. 1963;36:1441. [Google Scholar]
- 29.Queen A. Can J Chem. 1967;45:119. [Google Scholar]
- 30.Ingold CK. Structure and Mechanism in Organic Chemistry. 2. Cornell University Press; Ithaca, N.Y: 1969. pp. 471–473. [Google Scholar]
- 31.Yew KH, Koh HJ, Lee HW, Lee I. J Chem Soc, Perkin Trans. 1995;2:2263. [Google Scholar]
- 32.Exner O. In: Correlation Analysis in Chemistry. Chapman NB, Shorter J, editors. ch 10 Plenum; New York: 1978. [Google Scholar]
- 33.Jencks WP. Chem Soc Rev. 1981;10:345. [Google Scholar]
- 34.Koo IS, Yang K, Kang K, Lee I, Bentley TW. J Chem Soc, Perkin Trans. 1998;2:1179. [Google Scholar]
- 35.Bentley TW, Jones RO. J Chem Soc, Perkin Trans. 1993;2:2351. [Google Scholar]
- 36.Koo IS, Yang K, Kang K, Lee I. Bull Korean Chem Soc. 1998;19:968. [Google Scholar]
- 37.Kevill DN, Foss FD. J Am Chem Soc. 1969;91:5054. [Google Scholar]
- 38.Mousa MA, Diefallah EHM, Dessouki HA, Atwa ST. Thermochim Acta. 1989;141:1. [Google Scholar]
- 39.Kevill DN, D’Souza MJ. J Chem Soc, Perkin Trans. 1997;2:1721. [Google Scholar]
- 40.Kevill DN, Koyoshi F, D’Souza MJ. Int J Mol Sci. 2007;8:346. [Google Scholar]
- 41.Kevill DN, Ismail NHJ, D’Souza MJ. J Org Chem. 1994;59:6303. [Google Scholar]
- 42.Bentley TW, Garley MS. J Phys Org Chem. 2006;19:341. [Google Scholar]
- 43.Reis MC, Elvas-Leitão R, Martins F. Int J Mol Sci. 2008;9:1704. doi: 10.3390/ijms9091704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kevill DN, D’Souza MJ. Curr Org Chem. 2010;14:1037. doi: 10.2174/138527210791130505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Souza MJ, Reed D, Koyoshi F, Kevill DN. Int J Mol Sci. 2007;8:788. [Google Scholar]
- 46.D’Souza MJ, Shuman KE, Carter SE, Kevill DN. Int J Mol Sci. 2008;9:2231. doi: 10.3390/ijms9112231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choppin AR, Rodgers JW. J Am Chem Soc. 1948;70:2967. doi: 10.1021/ja01191a076. [DOI] [PubMed] [Google Scholar]
- 48.Koh HJ, Kang SJ, Kevill DN. Bull Korean Chem Soc. 2010;31:835. [Google Scholar]
- 49.Haas WL, Krumkalns EV, Gerzon K. J Am Chem Soc. 1966;88:1988. [Google Scholar]
- 50.Kevill DN, Weitl FL. J Am Chem Soc. 1968;90:6416. [Google Scholar]
- 51.Kevill DN, Weitl FL. J Org Chem. 1970;35:2526. [Google Scholar]
- 52.Kevill DN, Weitl FL. Tetrahedron Lett. 1971:701. [Google Scholar]
- 53.Kevill DN, Kyong JB, Weitl FL. J Org Chem. 1990;55:4304. [Google Scholar]
- 54.Jencks WP. Acc Chem Res. 1980;13:161. [Google Scholar]
- 55.Ref. 30, pp. 428–434.
- 56.Leimu R. Chem Ber. 1937;70:1040. [Google Scholar]
- 57.Crunden EW, Hudson RF. J Chem Soc. 1961:3748. [Google Scholar]
- 58.Kivinen A. Suomen Kemistilehti. 1965;38B:205. [Google Scholar]
- 59.Koo IS, Lee JI, Yang K, Kang K, Lee I. Bull Korean Chem Soc. 1999;20:573. [Google Scholar]
- 60.Kyong JB, Kim YG, Kim DK, Kevill DN. Bull Korean Chem Soc. 2000;21:662. [Google Scholar]
- 61.D’Souza MJ, Reed DN, Erdman KJ, Kyong JB, Kevill DN. Int J Mol Sci. 2009;10:862. doi: 10.3390/ijms10030862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kyong JB, Yoo JS, Kevill DN. J Org Chem. 2003;68:3425. doi: 10.1021/jo0207426. [DOI] [PubMed] [Google Scholar]
- 63.Kyong JB, Won H, Kevill DN. Int J Mol Sci. 2005;6:87. [Google Scholar]
- 64.Kevill DN, Rissmann TJ. J Org Chem. 1985;50:3062. [Google Scholar]
- 65.Fry JL, Lancelot CJ, Lam LKM, Harris JM, Bingham RC, Raber DJ, Hall RE, Schleyer PvR. J Am Chem Soc. 1970;92:2542. [Google Scholar]
- 66.Charton M. J Org Chem. 1964;29:1222. [Google Scholar]
- 67.Bentley TW, Roberts K. J Chem Soc, Perkin Trans. 1989;2:1055. [Google Scholar]
- 68.Kevill DN, D’Souza MJ. J Org Chem. 1998;63:2120. [Google Scholar]
- 69.Kevill DN, D’Souza MJ. J Chem Soc, Perkin Trans. 2002;2:240. [Google Scholar]
- 70.Lim GT, Lee YH, Ryu ZH. Bull Korean Chem Soc. 2013;34:615. [Google Scholar]
- 71.D’Souza MJ, Carter SE, Kevill DN. Int J Mol Sci. 2011;12:1161. doi: 10.3390/ijms12021161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park KH, Lee Y, Lee YW, Kyong JB, Kevill DN. Bull Korean Chem Soc. 2012;33:3657. [Google Scholar]
- 73.Koh HJ, Kang SJ. Bull Korean Chem Soc. 2012;33:1729. [Google Scholar]
- 74.Kevill DN, Kim JC, Kyong JB. J Chem Res Synop. 1999:150. [Google Scholar]
- 75.Hall HK., Jr J Am Chem Soc. 1955;77:5993. [Google Scholar]
- 76.Kivinen A. Acta Chem Scand. 1965;19:845. [Google Scholar]
- 77.Orlov SI, Chimishkyan AL, Grabarnik MS. J Org Chem USSR (Engl Transl) 1983;19:1981. [Google Scholar]
- 78.Ryu ZH, Shin SH, Lee JP, Lim GT, Bentley TW. J Chem Soc, Perkin Trans. 2002;2:1283. [Google Scholar]
- 79.Oh YH, Jang GG, Ryu ZH. Bull Korean Chem Soc. 2002;23:1089. [Google Scholar]
- 80.Fort RC., Jr . Bridgehead Carbonium Ions. In: Olah GA, Schleyer PvR, editors. Carbonium Ions. Vol. 4. Wiley; New York: 1973. Chap. 32. [Google Scholar]
- 81.Faurholt C, Gjaldback JC. Dansk Tids Farm. 1945;19:255. 1946. [Google Scholar]; Chem Abstr. 40:153. [Google Scholar]
- 82.Koh HJ, Kang SJ. Bull Korean Chem Soc. 2011;32:3799. [Google Scholar]
- 83.Shorter J. In: Advances in Linear Free Energy Relationships. Chapman NB, Shorter J, editors. Plenum Press; New York: 1956. p. 76. [Google Scholar]
- 84.Carrasco M, Jones RJ, Kamel S, Rapoport H, Truong T. Org Synth. 1992;70:29. [Google Scholar]
- 85.Kyong JB, Park BC, Kim CB, Kevill DN. J Org Chem. 2000;65:8051. doi: 10.1021/jo005630y. [DOI] [PubMed] [Google Scholar]
- 86.Ref. 30, p. 437.
- 87.Leffler JE, Grunwald E. Rates and Equilibria of Organic Reactions. Wiley; New York: 1963. pp. 219–225. [Google Scholar]
- 88.D’Souza MJ, Givens AF, Lorchak PA, Greenwood AE, Gottschall SL, Carter SE, Kevill DN. Int J Mol Sci. 2013;14:7286. doi: 10.3390/ijms14047286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ryu ZH, Lee HL, Oh Y. Bull Korean Chem Soc. 2005;26:1761. [Google Scholar]
- 90.Koh HJ, Kang SJ. Bull Korean Chem Soc. 2010;31:1793. [Google Scholar]
- 91.D’Souza MJ, Shuman KE, Omondi AO, Kevill DN. Eur J Chem. 2011;2:130. doi: 10.5155/eurjchem.2.2.130-135.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koh HJ, Kang SJ. Bull Korean Chem Soc. 2012;33:4117. [Google Scholar]
- 93.Hook AL, Chang CY, Yang J, Luckett J, Cockayne A, Atkinson S, Mei Y, Bayston R, Irvine DJ, Langer R, Anderson DG, Williams P, Davies MC, Alexander MR. Nature Biotechnol. 2012;30:868. doi: 10.1038/nbt.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.D’Souza MJ, Darrington AM, Kevill DN. ISRN Organic Chemistry. 2011;2011:Article ID 767141. doi: 10.5402/2011/767141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.D’Souza MJ, Knapp JA, Fernandez-Bueno GA, Kevill DN. Int J Mol Sci. 2012;13:665. doi: 10.3390/ijms13010665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Charton M. J Org Chem. 1964;29:1222. [Google Scholar]
- 97.Green M, Hudson RF. Proc Chem Soc. 1959:149. [Google Scholar]
- 98.Green M, Hudson RF. J Chem Soc. 1962:1055. [Google Scholar]
- 99.Miller J. Aromatic Nucleophilic Substitution. Elsevier; New York: 1968. p. 19. [Google Scholar]
- 100.Emeleus HJ, Wood JW. J Chem Soc. 1948:2183. [Google Scholar]
- 101.Goswami HC, Sarkar PB. J Indian Chem Soc. 1933;10:537. 1934. [Google Scholar]; Chem Abstr. 28:1332. [Google Scholar]
- 102.Ray PC. Nature. 1933;132:173. [Google Scholar]
- 103.Cuomo J, Olofson RA. J Org Chem. 1979;44:1016. [Google Scholar]
- 104.Dang VA, Olofson RA, Wolf PR, Piteau MD, Senet JPG. J Org Chem. 1990;55:1847. [Google Scholar]
- 105.Kevill DN, Kyong JB. J Org Chem. 1992;57:258. [Google Scholar]
- 106.Kyong JB, Kevill DN, Kim JC. J Korean Chem Soc. 1993;37:3. [Google Scholar]
- 107.Lee YW, Seong MH, Kyong JB, Kevill DN. Bull Korean Chem Soc. 2010;31:3366. [Google Scholar]
- 108.Kyong JB, Rhu CJ, Kim YG, Kevill DN. J Phys Org Chem. 2007;20:525. [Google Scholar]
- 109.Lee SH, Rhu CJ, Kyong JB, Kim DK, Kevill DN. Bull Korean Chem Soc. 2007;28:657. [Google Scholar]
- 110.Seong MH, Kyong JB, Kim DK, Kevill DN. Bull Korean Chem Soc. 2008;29:1747. [Google Scholar]
- 111.Seong MH, Kyong JB, Lee YH, Kevill DN. Int J Mol Sci. 2009;10:929. doi: 10.3390/ijms10030929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee Y, Park KH, Seong MH, Kyong JB, Kevill DN. Int J Mol Sci. 2011;12:7806. doi: 10.3390/ijms12117806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Seong MH, Choi SH, Lee YW, Kyong JB, Kim DK, Kevill DN. Bull Korean Chem Soc. 2009;30:2408. [Google Scholar]
- 114.Kyong JB, Ryu SH, Kevill DN. Int J Mol Sci. 2006;7:144. [Google Scholar]
- 115.Song BD, Jencks WP. J Am Chem Soc. 1989;111:8470. [Google Scholar]
- 116.Kevill DN, Miller B. J Org Chem. 2002;67:7399. doi: 10.1021/jo020467n. [DOI] [PubMed] [Google Scholar]
- 117.Kevill DN, D’Souza MJ. J Org Chem. 2004;69:7044. doi: 10.1021/jo0492259. [DOI] [PubMed] [Google Scholar]
- 118.Queen A, Nour TA. J Chem Soc, Perkin Trans. 1976;2:935. [Google Scholar]
- 119.Kevill DN, Weitl FL. J Chem Soc, Perkin Trans. 1972;1:2162. [Google Scholar]
- 120.Kevill DN, Park BC, Kyong JB. J Org Chem. 2005;70:9032. doi: 10.1021/jo050998m. [DOI] [PubMed] [Google Scholar]