

Abstract
Isopentenyl diphosphate dimethylallyl diphosphate isomerase (IDI) is a key enzyme in the isoprenoid biosynthetic pathway and is required for all organisms that synthesize isoprenoid metabolites from mevalonate. Type 1 IDI (IDI-1) is a metalloprotein and is found in eukaryotes, while the type-2 isoform (IDI-2) is a flavoenzyme found in bacteria and completely absent from human. IDI-2 from the pathogenic bacterium Streptococcus pneumoniae was recombinantly expressed in E. coli. Steady state kinetic studies of the enzyme indicated that FMNH2 (KM= 0.3 μM) bound before isopentenyl diphosphate (KM= 40 μM) in an ordered binding mechanism. An X-ray crystal structure at 1.4 Å resolution was obtained for the holo-enzyme, in the closed conformation with reduced flavin cofactor and two sulfate ions in the active site. These results helped to further approach the enzymatic mechanism of IDI-2 and, thus, open new possibilities for the rational design of antibacterial compounds against closely sequence and structure related pathogens such as E. faecalis or S. aureus.
Keywords: X-ray structure, steady-state kinetics, bisubstrate, flavoprotein
Introduction
Multi-drug resistant strains of pathogens have created a need for new antibiotic agents based on new drug targets.[1] The isoprenoid biosynthetic pathway, which produces essential metabolites in all organisms,[2] is an attractive target for development of antibacterial compounds. Isoprenoid molecules are constructed from two five-carbon building blocks, isopentenyl diphosphate (IPP) and its allylic isomer dimethylallyl diphosphate (DMAPP). These are synthesized by the mevalonate (MVA) pathway[3] in mammals, Archaea, fungi, plants, and some bacteria or by the methylerythritol pathway (MEP)[4] in algae, plant chloroplasts, and other bacteria. In the MVA pathway, IPP is the sole product from the ATP-dependant decarboxylation of mevalonate diphosphate and must be isomerized to DMAPP, while IPP and DMAPP are both produced during the reduction of hydroxydimethylallyl diphosphate in the MEP pathway. Thus, isopentenyl diphosphate: dimethylallyl diphosphate isomerase (IDI, EC 5.3.3.2) is an essential enzyme in organisms that rely on the MVA pathway.
IDI-1, the first isoform of the enzyme to be characterized, was identified over 40 years ago and has been extensively studied.[5–8] A second convergently evolved isoform (IDI-2), found in plant chloroplasts and many bacteria, was discovered in 2001 and requires reduced flavin mononucleotide and MgCl2 as cofactors.[9] In 2007, Rothman et al. proposed a mechanism for the isomerization where the reduced flavin cofactor acts as a general acid/base catalyst for protonation of IPP and deprotonation of the resulting carbocationic intermediate.[10] The cofactor can also provide π-cation stabilization for the intermediate (Scheme 1). This mechanism is similar to that proposed for IDI-1 where IPP is protonated by an active site glutamic acid.[7]
Scheme 1.
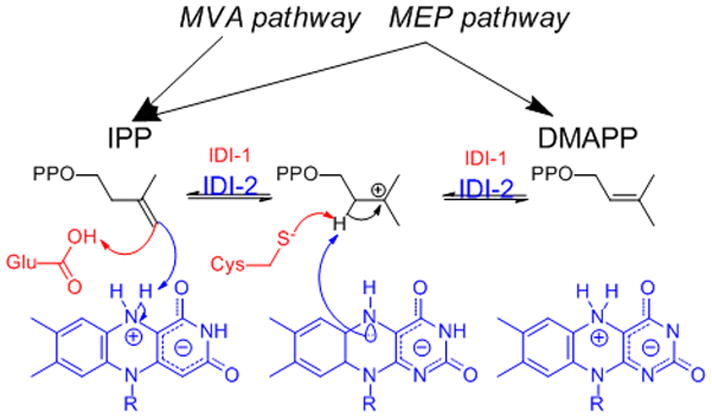
Currently proposed mechanisms for IDI-1 (red) and IDI-2 (blue). In the latter case the N5 nitrogen of FMN is likely candidate for the catalytic nucleophile.
The first crystal structure of IDI-2 was reported in 2003 for the enzyme from Bacillus subtilis (bs-IDI-2). Unfortunately, this structure was incomplete due to disorder of the N-terminal region, which included conserved residues involved in formation of the active site.[11] Subsequently, structures were reported for several IDI-2s at different levels of completeness and resolution (between 2 and 4 Å).[12, 13] Recently, the structures of an IDI-2 in complex with a variety of ligands were reported for the enzyme from the Archaeon Sulfolobusshibatae (ss-IDI-2).[14] Nevertheless, ss-IDI-2 shares less than 35% sequence identity with IDI-2s from pathogenic microorganisms such as Enterococcus faecalis (ef-IDI-2, 33%), Staphylococcus aureus (sa-IDI-2, 32%) or Streptococcus pneumoniae (sp-IDI-2, 30%).
We now describe characterization of the binding mechanism for IDI-2 from sp-IDI-2- and a crystal structure of the catalytic “closed” conformation of the enzyme at 1.4 Å resolution. The different structures and cofactor requirements of human IDI-1 and bacterial IDI-2, make IDI-2 an attractive target for development of new antiinfectious agents against pathogenic bacteria that synthesize isoprenoid compounds by the MVA pathway.
Results and Discussion
Overexpression, reconstitution and characterization of sp-IDI-2
Recombinant sp-IDI-2 was purified from 20 g of cell paste by Ni2+ affinity chromatography to give 5 mL of a 72 mg/mL solution of the protein. SDS-PAGE indicated that the purity of the protein was ~90%. By comparing the extinction coefficient for free FMN to that of FMN•sp-IDI-2, we determined the extinction coefficient of the complex to be 12,200 M−1s−1 at 454 nm. The mass of purified His-tagged sp-IDI-2 was 39874 Da (calculated 39868 Da) as determined by ESI/MS. The mass of the native protein determined by gel filtration chromatography of 173.6 kDa corresponds to a homotetramer, as previously reported for Thermus thermophilus (tt-), Pyrococcus furiosus (pf-)[15] and sa-IDI-2.[16]
During purification of sp-IDI-2, the FMN cofactor was released indicating a possible unfolding of the protein. A far UV CD spectrum of sp-IDI-2 was recorded in water, allowing reliable measurements down to 185 nm. A strong positive band centered at 195 nm appears together with a broad minimum between 208 nm and 222 nm, typical of the overlapping of various α-helix and β-sheet bands (Figure S1).[17] Thus, the CD spectrum of sp-IDI-2 indicates that the enzyme is folded and soluble under our experimental conditions.
Because the purified enzyme appears to have residual activity, it must have low levels of bound FMN that can interfere in the determination of steady state kinetic constants. To fully deflavinate the enzyme, we washed with 2 M KBr before elution from the Ni2+ column. A UV spectrum of the deflavinated enzyme did not have the characteristic peaks at 350 or 450 nm for FMN. Protein prepared in this manner had a residual activity less than 0.3%.
Steady state kinetic studies
Preliminary kinetic studies in buffer containing 8 μM FMN and varying concentrations of 14C-IPP indicated substrate inhibition at higher concentrations of IPP. Similarly, substrate inhibition was seen in buffer containing 150 μM IPP when the concentration of FMN was varied. Nonlinear regression analysis of the plot for varied IPP gave kinetic constants in good agreement with those reported for S. aureus IDI-2 [16] and for T. thermophillus IDI-2 (Table 1).[18] A related analysis for varied FMN gave a value of kcat similar to that reported for the T. thermophilus enzyme, but KmFMN = 0.34 ± 0.04 μM was much lower than KmFMN = 4.7 ± 0.6 μM reported for tt-IDI-2.[10]
Table 1.
Single Substrate Kinetic Parameters for IDI-2s
We then varied the concentrations of both IPP and FMN using deflavinated sp-IDI-2. A maximal concentration of 90 μM for IPP was below the onset of substrate inhibition. Double reciprocal plots gave intersecting lines, inconsistent with a ping-pong binding mechanism. A global fit to a sequential ordered mechanism where FMN binds first (Figure 1) gave kcat = 0.31 s−1, Kipp = 75 μM, and Kfmn = 1.6 μM (Table 2). Fits yielded negative parameter values when globally fit to an ordered mechanism where IPP binds first or a random mechanism (Table 2). The mechanism is consistent with the structure of the enzyme (see below), where FMN sits at the bottom of the active site with IPP stacked on top of the isoalloxazine nucleus. The enzyme is inhibited by higher concentrations of IPP and FMN, suggesting that the phosphate groups in FMN and IPP have an affinity for the phosphate-binding region of the other’s binding site.
Figure 1.
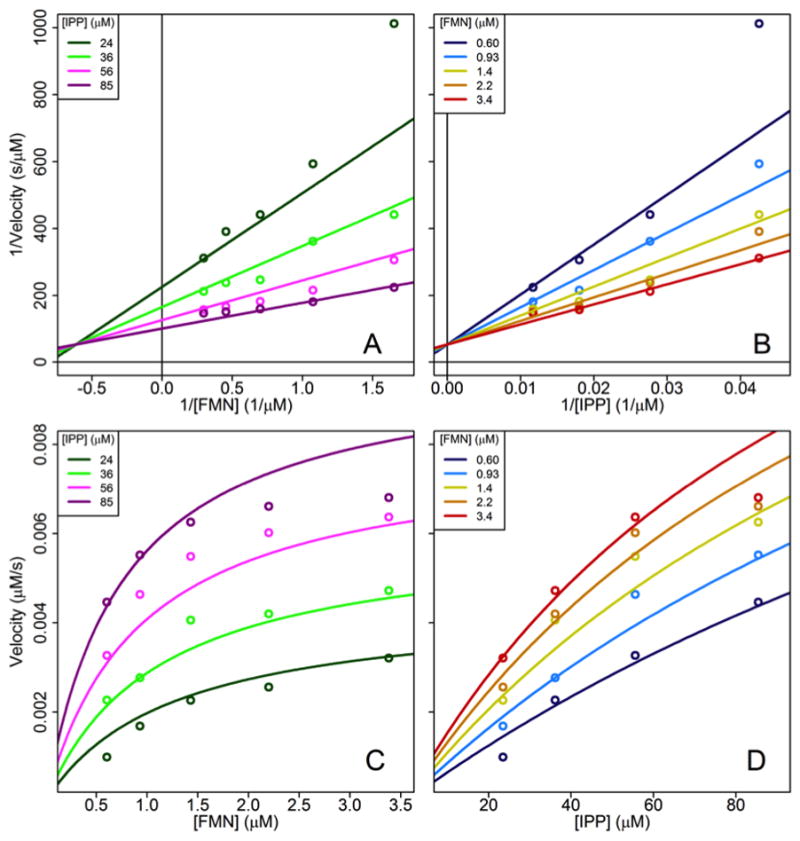
Bisubstrate kinetic plots for sp-IDI-2. Reciprocal plots (A and B) and standard plots (C and D) data points are the average from three experiments along with globally fit regression lines.
Table 2.
Bisubstrate Model Comparison for sp-IDI-2
| Mechanism | kcat (s−1) | Ki FMN (μM) | KIPP (μM) | KFMN (μM) | Residual sum of squares | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | S.E. | Mean | S.E. | Mean | S.E. | Mean | S.E. | ||
| Ordered Sequential FMN binding first | 0.311 | 0.062 | 1.63 | 0.37 | 75.4 | 23.8 | - | - | 0.004654 |
| Ordered Sequential IPP Binding first | 0.127 | 0.009 | −0.941 | 0.123 | −107 | 12 | - | - | 0.004279 |
| Random sequential | 0.203 | 0.050 | 3.12 | 1.51 | 34.8 | 19.1 | −0.672 | 0.265 | 0.004394 |
Assay conditions: 60 nM apo-S. pneumoniae type II IDI, 200 mM HEPES, pH 7.0, 2 mM MgCl2, 2 mM NADPH, 0.14 mg/mL BSA, 23.5 – 85.5 mM IPP and 0.930 – 3.38 mM FMN at 37 °C.
Crystal structure determination
Small crystals appeared within two days and reached their maximum size after seven days (Figure 2A, insert). A single crystal was harvested, cryoprotected and flash-cooled, prior to data collection. Data were collected to 1.4 Å resolution and processed in the space group I4 (a= b= 131.4 Å, c= 59.3 Å). The structure was determined by molecular replacement (Search models 1P0K and 3DH7) and subsequently refined (R=13.8 %, Rfree=15.8 %). Overall statistics for the data collection and refinement of the structure are presented in Table 3.
Figure 2.
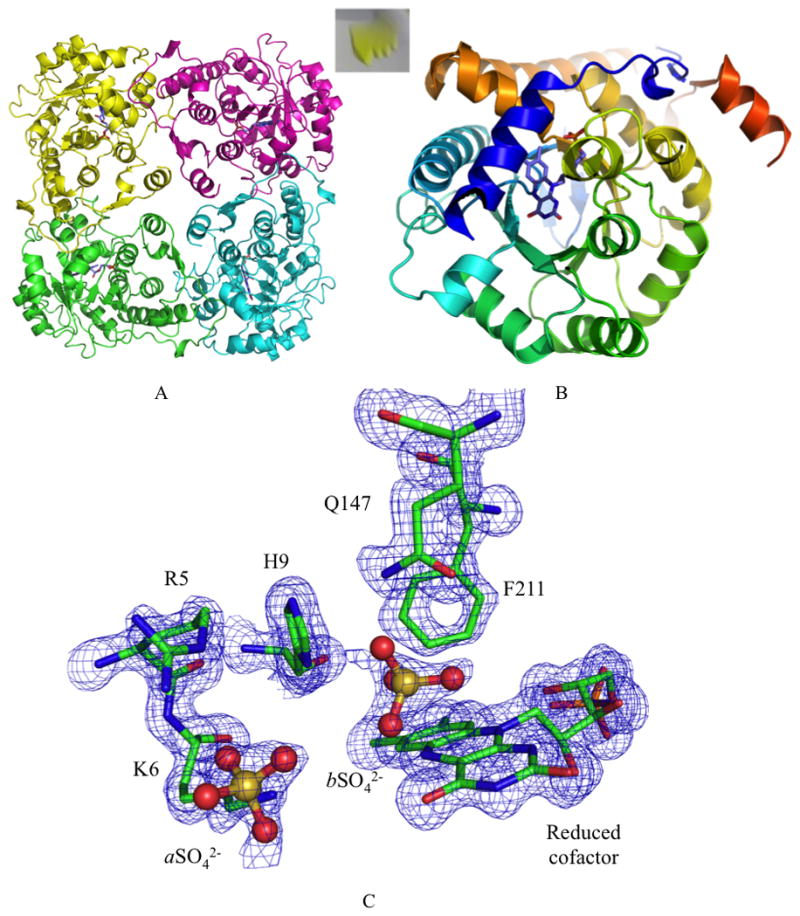
A) Tetrameric arrangement of the enzyme. FMN cofactor is represented in purple. Insert: Yellow crystals grown after 7 days. B) Cartoon representation of the typical α8β8 TIM-barrel structure of IDI-2 (N-terminus blue to C-terminus red). One end of the barrel is occupied by the reduced flavin cofactor the other is closed by two antiparallel β-sheets. The N-terminal α-helix caps the active site pocket. C) View of the binding pocket. The 2Fo-Fc electron density map (blue) is drawn at 1.0σ.
Table 3.
Data collection and refinement statistics of the sp-IDI-2.
| Crystal data | |
| Space group | I4 |
| Cell dimensions (Å) | a = b = 131.3 c = 59.4 |
| Subunit per asymmetric unit | 1 |
| Data set | |
| Resolution range (Å) | 33–1.4 (1.6–1.4) |
| Unique reflections | 98844 |
| Completeness (%) a | 99.5 (91.6) |
| Rmerge (%) a | 7.9 (79.8) |
| I/σ(I) a | 20.5 (2.1) |
| Redundancy | 5.1 (4.8) |
| Refinement | |
| Rcryst (%) | 13.8 |
| Rfree (%)b | 15.8 |
| Rmsd | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.03 |
| Mean B value (Å2) | 18.0 |
| Ramachadran plot c | |
| Favored (%) | 97.9 |
| Outliers (%) | 0.3 |
| PDB entry | |
| Accession code | 4N02 |
As previously described for other IDI-2 structures [11–14], sp-IDI-2 formed a planar tetrameric arrangement in the unit cell (Figure 2A) with, in our case, only one molecule in the asymmetric unit. The monomer folds as a classical α8β8 TIM-Barrel, with one end housing FMN and the enzyme active site, and the other end being capped by two antiparallel β-sheets (Figure 2B). After refinement of the apo-protein model, additional electron density was observed for FMN, which appeared to be in its reduced form. Indeed Despite crystal growth under anaerobic conditions, unbiased 3 σFo-Fc density revealed the presence of the reduced flavin cofactor in the active site (Figure S2) (17° between the aromatic planes of the isoalloxazine ring). Presumably the FMN was reduced by the synchrotron radiation emitted photoelectrons that can reduce initially oxidized moieties.[19, 20]
Comparison with related structures
We compared our structure with tt-IDI-2 in complex with inorganic pyrophosphate PPi (tt-IDI-2•PPi) [12] and ss-IDI-2 in complex with IPP (ss-IDI-2•IPP).[14] Despite a low sequence identity (33 and 30 % for tt- and ss-IDI-2), all three of these structures overlap tightly (RMSD on Cα for the whole protein of 1.5 and 1.7 Å). As observed previously, the cofactor is located in a pocket made of conserved residues and capped by the N-terminus of the protein.
The imidazole ring of H9 interacts with the diphosphate moiety of IPP in ss-IDI-2, with PPi in tt-IDI-2, and with the first sulfate ion (aSO4) located in the same region in sp-IDI-2. The sulfate also makes salt bridges with the guanidine group of arginine (R5) and amine moiety of lysine (K6) further strengthening the closed N-terminal position (Figure 2C).
We located residual positive Fo-Fc density on the si face of the isoalloxazine moiety (3 Å from the N5), which was modeled as a second sulfate ion from the crystallization buffer (Figure 2C). This ion is stabilized by interactions with the phenyl ring of phenylalanine (F211), the imidazole moiety of histidine (H9) and the amide group of glutamine (Q149). H9 and Q149 are highly conserved while Trp in some IDI-2s replaces F211.
By a closer inspection of the active sites we can assume that the two ions reported in our structure mimic the position of IPP into the active site. Indeed, second sulfate ion (bSO4) in the structure of sp-IDI-2 matches the position of a water molecule in tt-IDI-2•PPi and C1 of IPP in the ss-IDI-2•IPP complex. The sulfur atom of this ion is 4.2 and 4.4 Å from the N5 and C4a atoms of FMN, while C1-IPP in ss-IDI-2•IPP was reported to be 4.1 and 4.2 Å, respectively (Figure 3). Stabilization of these species at this location is also consistent with a protonation-deprotonation mechanism for isomerization of IPP to DMAPP, where the putative zwitter ionic form of the reduced cofactor stabilizes the bound sulfate (Scheme 1).
Figure 3.
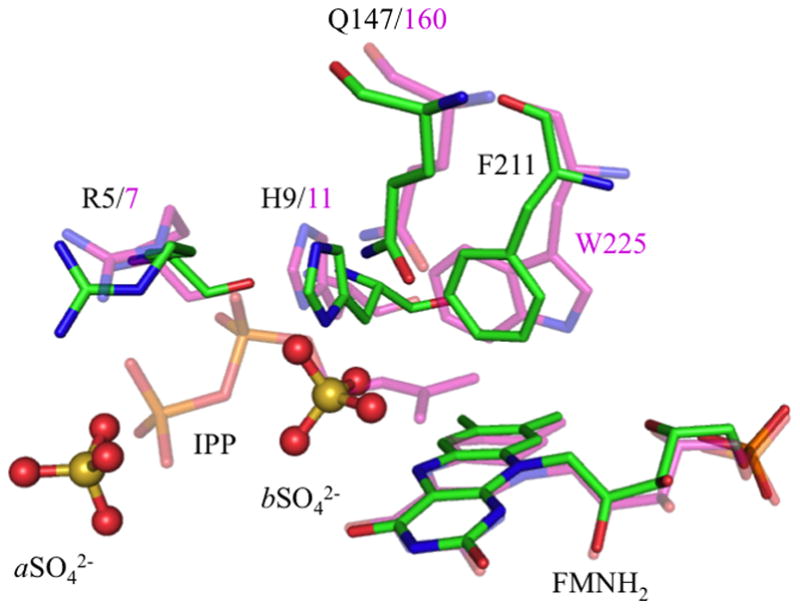
Superposition of ss-IDI-2 in complex with IPP and FMNH2 (purple) and sp-IDI-2 in complex with reduced cofactor (green) and sulfate ions (stick and balls). These ions are stabilized by ionic salt bridges with guanine from R5, and by interactions with amide moiety of Q149 and imidazole ring of H9. Interestingly, they lie at the same positions as the C2-atom and the β-phosphate within IPP observed in ss-IDI-2•IPP structure inside the binding pocket (sp numbering/ss numbering).
Implication for drug design
The proposed binding mechanism suggests that it might be possible to construct potent inhibitors of the enzyme that take advantage of interactions with both the IPP and reduced flavin cofactor regions of the active site. In this regard, the 1.4 Å crystal structure of the catalytic closed form of sp-IDI-2 should be a useful template for computationally guided drug design. Indeed, by structure alignment between sp and ss-IDI-2, we identified several changes in the amino acid sequence. In addition to the Trp mutated into Phe, an arginine in ss-IDI-2 is replaced by a serine (S92) in sp-IDI-2. This residue caps the active site of the enzyme and can thus be targeted to restrict the binding pocket availability for the substrate. Arg5 and Lys6 are also located at the surrounding of the active site and can be targeted by negatively charged moieties from potent inhibitors (Figure 4). We also located a Met (M151) surrounding the active site. These residues, and especially the sulphur moieties, are well known to interact with cisplatin or other Pt-derivative drugs. We identified, thus, three potential sites to design new inhibitors.
Figure 4.
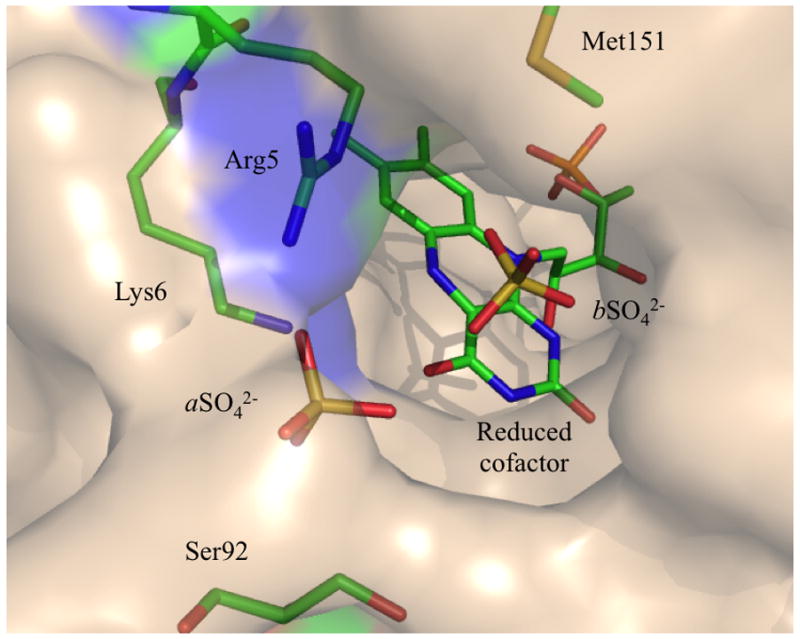
Surface representation of the active site pocket. Arg5, Lys6, Ser92 and Met151 can be targeted by competitive inhibitors to restrict access to the binding pocket. N-terminal segment is represented in blue and cap the active site in the “closed” conformation of the enzyme.
Conclusion
IDI-2 is an enzyme required for the synthesis of isoprenoid metabolites via the MVA pathway in S. pneumoniae. Although the protein binds the same substrates and catalyzes the same reaction as IDI-1 in humans, the two isoforms evolved convergently and have different protein folds, active site residues, and cofactor requirements. sp-IDI-2 has catalytic and structural features that make it an attractive antibiotic target against S. pneumoniae.
Reduced flavin mononucleotide (FMNH2) is a required cofactor for IDI-2. Typically, IDI-2 and the tightly bound oxidized cofactor (FMN) co-purify as an intense yellow complex. The active form of the enzyme is then obtained by reduction of FMN with NADPH or dithionite to give the FMNH2•IDI-2 complex. In contrast, most of the FMN is lost during purification of sp-IDI-2 and the cofactor must be added to the assay buffer along with the reductant. IPP and FMNH2 bind by a sequential ordered mechanism where the flavin (KM = 0.3 μM) binds before IPP (KM = 40 μM). Thus, the IPP and the FMNH2 loci in the active site are available for development of compounds that inhibit sp-IDI-2 by interacting with both regions of the active site.
As previously reported, when the active site is empty the N-terminal segment is disordered.[11, 12] Due to the presence of two sulfate ions, the N-terminal segment ofsp-IDI-2 is stabilized, and the enzyme is in the closed catalytic conformation in the crystal. Interactions of conserved R5 and K6 and one of the sulfate ions are key interactions for stabilizing the closed conformation. The other sulfate is located between the amide moiety in a highly conserved glutamine (Q147) and N5 of reduced flavin cofactor, which would be positively charged in the zwitter ionic form of the cofactor proposed for the catalytically competent enzyme-substrate complex.[10]
Following protonation of the double bond in IPP, the carbocationic intermediate is stabilized by the negatively charged isoalloxazine moiety in the flavin, dipolar interactions with the amide group in Q147, and quadrupolar π-cation interactions with the benzene ring in F211. Similar interactions were observed for tt-IDI-2 [12] and ss-IDI-2.[14] Quadrupolar π-cation stabilization of the carbocationic intermediate by an active-site tryptophan is also proposed for the structurally unrelated E. coli IDI-1.[7]
In conclusion, sp-IDI-2 binds IPP and FMNH2 by an ordered sequential mechanism, where the flavin binds first, in contrast to the behavior of other IDI-2s where the flavin is very tightly bound in the active site. This structure should also be useful for related studies with IDI-2s from other pathogenic bacteria such as sa-IDI-2 (40%) and ef-IDI-2 (41%) with high sequence identities to sp-IDI-2 relative to structures available from non-pathogenic bacteria with lower levels of sequence identity.
Experimental Section
Cloning of SP0384 from Streptococcus pneumoniae
The gene for IDI-2 was cloned from Streptococcus pneumoniae TIGR4 genomic DNA (ATCC), gene SP0384, with Klentaq LA polymerase (Sigma) using primers to introduce NcoI (N-terminus) and HindIII (C-terminus) restriction sites: 5′ – GGG CCA TGG CGA CAA ATC GTA AGG ACG A – 3′ and 5′ GGG AAG CTT CGC CTT TTT CAT CTG ATC CT – 3′. The PCR thermocycler set-up (25 cycles) was: 94 °C, 180 s initial denaturation; 94 °C, 10 s denaturation; 55 °C, 10 s denaturation; 72 °C, 60 s extension; 72 °C, 5 min final extension. The purified PCR product was ligated into the pGEM®-T vector (Promega) forming pSPIDI2a. E. coli DH5α™ (Invitrogen) cells were transformed with pSPIDI2a. The pSPIDI2a plasmid was purified from the transformant and digested with NcoI and HindIII (NEB). The SPIDI2a digested product gel purified and subcloned into the NcoI-HindIII sites of pBAD/Myc-His A expression vector (Invitrogen), to give pSPIDI2b. E. coli DH5α™ (Invitrogen) cells were transformed with and stored at −80 °C for long-term storage.
To produce enzyme with a cleavable His-tag, the spIDI2 gene was subcloned into the pQE30-Xa expression vector (Qiagen). The gene was amplified with Klentaq LA polymerase using primers to introduce an EcoRV (N-terminus) and HindIII (C-terminus) restriction sites: 5′ – GGC GAT ATC ACA ACA AAT CGT AAG GAC G – 3′ and 5′ – GGC AAG CTT TCA CGC CTT TTT CAT CTG ATC CT – 3′. Conditions for PCR (25 cycles) were: 94 °C, 180 s initial denaturation; 94 °C, 30 s denaturation; 47 °C, 30 s denaturation; 72 °C, 90 s extension; 72 °C, 5 min final extension. The purified PCR product was ligated into pGEM®-T Easy (Promega) to give SPIDI2c. E. coli DH5α™ (Invitrogen) cells were transformed with pSPIDI2c. The purified plasmid was digested with EcoRV and HindIII (NEB) and fragment SpxIDI2 was gel purified. The purified fragment was ligated into the StuI (blunt end)-HindIII sites of pQE-30 Xa to give pSPxIDI2. E. coli M15[pREP4] (Qiagen) cells were transformed with pSPxIDI2 and the resulting strain was stored at −80 °C.
Due to poor termination during expression, the stop codon was mutated from TGA to TAA T with Pfu DNA polymerase (Stratagene) using the primer: 5′ – P – AGC TCA GCT AAT TAA GCT TAT TAC GCC TTT TTC ATC TGA TCC – 3′. Conditions for PCR (25 cycles) were: 65 °C, 120 s/95 °C, 120 s initial denaturation; 95 °C, 45 s denaturation; 55 °C, 60 s denaturation; 65 °C, 8 min extension; 65 °C, 4 min final extension. The PCR mixture was digested with DpnI and the digested mixture transformed into E. coli XL1-blue competent cells (Invitrogen). Plasmid pSPIDI2taat was purified and transformed into E. coli M15 [pREP4]cells. The resulting strain was stored at −80 °C.
Overexpression and purification
E. coli M15 (p-sp-IDI-2) was grown at 37 °C to OD600 ~0.6. Isopropyl-β-D-thiogalactoside (IPTG, final concentration 0.4 mM) and flavin mononucleotide (FMN, final concentration 40 mg/L) were added and incubation was continued for 23 h at room temperature. Cells were pelleted and stored at −80 °C until needed. Frozen cells were thawed in binding buffer (20 mM sodium phosphate, pH 7.4, containing 500 mM NaCl and 20 mM imidazole) and disrupted with a Sonifier Cell Disruptor (8 cycles of 30 s, on ice).
sp-IDI-2 was purified by nickel-ion affinity chromatography on a HisTrap FF crude column (GE Heathcare) eluted with 50 mM Tris-HCl buffer, pH 7.4, containing 20–500 mM imidazole. Fractions of sp-IDI2 were pooled, concentrated with a 30 kD MWCO filter (Centriprep, Millipore) and dialyzed three times against 1 L of 10 mM Tris-HCl buffer, pH 8, containing 20% glycerol. This procedure gave sp-IDI-2 with a substoichiometric amount of bound FMN. An additional wash step with 2 M KBr in binding buffer just before elution with imidazole gave deflavinated apo-sp-IDI-2.[21] Protein concentrations were determined by the BCA assay (Pierce).
Gel filtration
The oligomeric state of sp-IDI-2 was estimated by gel filtration using a Superdex 200 (1 × 30 cm) column equilibrated with 50 mM potassium phosphate buffer, pH 8.0, containing 150 mM NaCl and 70 μL/L β-mercaptoethanol. Thyroglobulin (669 kDa), ferritin (440 kDa), BSA (67 kDa), ovalbumin (43 kDa) and ribonuclease A (14 kDa) were used as standards to calibrate the column.
Holoenyzme extinction coefficient and reconstitution of the apoenzyme
apo-sp-IDI-2 was incubated with 2 mM FMN, overnight at 4 °C. Reconstituted holoenzyme was loaded onto a Ni-IDA affinity chromatography column (PrepEase, Affymetrix) and washed with lysis-equilibrium-wash buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl) until the wash no longer appeared yellow (three washes). Protein was eluted with 50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 250 mM imidazole and analyzed by UV-vis spectroscopy (Agilent 8453 diode array spectrophotometer) and SDS-PAGE. Fractions containing IDI-2 were concentrated and combined.
The extinction coefficient for the flavoprotein was determined by the procedure of Macheroux.[22] Briefly, the absorption of a 75 μM solution of sp-IDI-2 in 10 mM Tris, pH 8.0 was measured at 445 nm. To this sample, 50% trichloroacetic acid (final concentration, ~8%) was added, upon which the protein precipitated. After centrifugation, the pH was adjusted to 8.0 using solid Na2CO3. The absorbance at 450 nm (εFMN = 12,200 M−1cm−1, [23]) of the supernatant was measured and used to determine the extinction coefficient of FMN bound sp-IDI-2.
Saturation and bisubstrate kinetic assays
Initial rates were measured by the acid-lability procedure as previously described.[24] Briefly, the reaction was initiated by adding enzyme diluted in 2 mg/mL BSA in 10 mM HEPES, pH 7.0 to assay buffer (saturation kinetic conditions in Table 1 and bisubstrate kinetic conditions in Table 2). After 10 min at 37 °C, the reaction was stopped by addition of 200 μL of methanol/concentrated HCl (4:1, v/v). The mixture was then incubated for 10 min at 37 °C and the radioactive products were extracted with 1 mL ligroin. A 500 μL portion of the ligroin extract was mixed with scintillation cocktail (UltimaGold, Perkin-Elmer). Disintegrations per minute (DPM) were counted by liquid scintillation spectrometry (TriCarb 2910TR, Perkin Elmer). Kinetic experiments were replicated three times; sample order for each replicate was randomly assigned.
Data analysis
Saturation kinetic data were fit to a single substrate with substrate inhibition equation and bisubstrate data were fit to the standard sequential ordered and random equations.[25] Analysis was performed using R version 3.0.0 [26] using the packages nlstools[27], calibrate [28]and dichromat.[29]
Circular dichroism measurements
Circular dichroism (CD) measurements in the far-UV region (185–260 nm) were performed at 20 °C, with a Jasco J-810 spectropolarimeter (Jasco, Japan), using a protein concentration of 0.14 mg/mL (3.5 μM) and a 0.1 cm cell path length. Spectra were acquired in water (pH ≈ 7) at a scan speed of 10 nm. min−1, with a 1 nm bandwidth and a 4 s DIT. The spectra were averaged after four accumulations and corrected by subtraction of the solvent spectrum obtained under the same conditions. Ellipticity measurements in mdeg were converted in mean residue ellipticity [30], by using a mean residue weight value of 111.7 Da, on the basis of the enzyme amino acid composition.
Calculation of the secondary structures from analysis of CD data was performed using CONTINLL [31, 32], CDSSTR [33, 34] and SELCON3 [35, 36] algorithms provided in the DichroWeb analysis server.[37, 38] Two protein reference databases (3 and 6) were used and the results from the individual analysis were averaged; the standard deviations between the calculated secondary structures are given in the text.
Crystallization experiments
For crystallization trials, the apoprotein was reconstituted by incubation in presence of 2 mM FMN overnight at 4°C. The reconstituted enzyme was loaded onto a Ni2+ affinity column and washed. After three washes, FMN no longer appeared. SDS-PAGE analysis of the washes and eluted fractions indicated that protein was only present in the elutions and not in the washes, as described above. The sitting-drop vapour-diffusion method was used to screen for crystal formation at 21 °C with a solution of 13 mg/mL of holoenzyme. Yellow crystals indicating the incorporation of oxidized flavin grew to their maximum size after 7 days equilibrated against a well containing 100 mM HEPES buffer, pH 7.5, 2 M (NH4)2SO4. Prior to data collection, crystals were cryoprotected using a reservoir solution containing 20% glycerol and were flash-cooled in liquid nitrogen.
Data collection and structure determination
Diffraction data were collected at 100 K, at the SSRL synchrotron (BL7-1). A complete dataset was recorded and processed with HKL2000.[39] Phasing was performed by molecular replacement using Phaser[40] (starting models: bs-IDI-2 and Thermus thermophillus IDI-2 (tt-IDI-2) (PDB IDs: 1P0K and 3DH7, respectively)) followed by rebuilding of the initial model and solvent incorporation using Arp/wArp. Subsequent all atom anisotropic refinement cycles were performed with Refmac[40] and Phenix.[41] Electron density maps were inspected with the graphic program Coot [42], and the quality of the model was analyzed with Molprobity.[43] PyMOL was used to generate high quality images of the enzyme.[44]
Supplementary Material
Acknowledgments
JdR thanks the FNRS for the position of postdoctoral Researcher. This work was supported by NIH grant GM 25521 (CDP). Authors wish to thank the proteomic core from the University of Utah for their help in mass spectrometry measurement. Portions of this research were carried out at the Stanford Synchrotron Radiation Light source, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH. This research used resources of the “Plateforme Technologique de CalculIntensif (PTCI)” (http://www.ptci.unamur.be) located at the University of Namur, Belgium, which is supported by the F.R.S.-FNRS. The PTCI is member of the “Consortium des Équipements de CalculIntensif (CÉCI)” (http://www.ceci-hpc.be).
References
- 1.Fischbach MA, Walsh CT. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sacchettini JC, Poulter CD. Science. 1997;277:1788–1789. doi: 10.1126/science.277.5333.1788. [DOI] [PubMed] [Google Scholar]
- 3.Bloch K. Science. 1965;150:19–28. doi: 10.1126/science.150.3692.19. [DOI] [PubMed] [Google Scholar]
- 4.Rohmer M, Knani M, Simonin P, Sutter B, Sahm H. Biochem J. 1993;295(Pt 2):517–524. doi: 10.1042/bj2950517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Durbecq V, Sainz G, Oudjama Y, Clantin B, Bompard-Gilles C, Tricot C, Caillet J, Stalon V, Droogmans L, Villeret V. Embo J. 2001;20:1530–1537. doi: 10.1093/emboj/20.7.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrigan CN, Poulter CD. J Am Chem Soc. 2003;125:9008–9009. doi: 10.1021/ja0350381. [DOI] [PubMed] [Google Scholar]
- 7.Wouters J, Oudjama Y, Barkley SJ, Tricot C, Stalon V, Droogmans L, Poulter CD. J Biol Chem. 2003;278:11903–11908. doi: 10.1074/jbc.M212823200. [DOI] [PubMed] [Google Scholar]
- 8.de Ruyck J, Durisotti V, Oudjama Y, Wouters J. J Biol Chem. 2006;281:17864–17869. doi: 10.1074/jbc.M601851200. [DOI] [PubMed] [Google Scholar]
- 9.Kaneda K, Kuzuyama T, Takagi M, Hayakawa Y, Seto H. Proc Natl Acad Sci USA. 2001;98:932–937. doi: 10.1073/pnas.020472198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rothman SC, Helm TR, Poulter CD. Biochemistry. 2007;46:5437–5445. doi: 10.1021/bi0616347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinbacher S, Kaiser J, Gerhardt S, Eisenreich W, Huber R, Bacher A, Rohdich F. J Mol Biol. 2003;329:973–982. doi: 10.1016/s0022-2836(03)00527-8. [DOI] [PubMed] [Google Scholar]
- 12.de Ruyck J, Pouyez J, Rothman SC, Poulter CD, Wouters J. Biochemistry. 2008;47:9051–9053. doi: 10.1021/bi801159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Unno H, Yamashita S, Ikeda Y, Sekiguchi S-y, Yoshida N, Yoshimura T, Kusunoki M, Nakayama T, Nishino T, Hemmi H. J Biol Chem. 2009;284:9160–9167. doi: 10.1074/jbc.M808438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagai T, Unno H, Janczak MW, Yoshimura T, Poulter CD, Hemmi H. Proc Natl Acad Sci USA. 2011;108:20461–20466. doi: 10.1073/pnas.1115749108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutoit R, de Ruyck J, Durisotti V, Legrain C, Jacobs E, Wouters J. Proteins. 2008;71:1699–1707. doi: 10.1002/prot.21863. [DOI] [PubMed] [Google Scholar]
- 16.Kittleman W, Thibodeaux CJ, Liu Y-n, Zhang H, Liu H-w. Biochemistry. 2007;46:8401–8413. doi: 10.1021/bi700286a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venyaminov SY, Yang JT. Circular Dichroism and the Conformational Analysis of Biomolecules. Plenum Press; New York: 1996. pp. 69–107. [Google Scholar]
- 18.de Ruyck J, Wouters J, Poulter CD. Curr Enz Inhib. 2011;7:79–95. doi: 10.2174/157340811796575317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Røhr ÅK, Hersleth H-p, Andersson KK. Angew Chem Int Ed. 2010;49:2324–2327. doi: 10.1002/anie.200907143. [DOI] [PubMed] [Google Scholar]
- 20.Garman E, Nave C. J Synchrotron Rad. 2009;16:129–132. doi: 10.1107/S0909049509005238. [DOI] [PubMed] [Google Scholar]
- 21.Hefti MH, Vervoort J, van Berkel JH. Eur J Biochem. 2003;270:4227–4242. doi: 10.1046/j.1432-1033.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- 22.Macheroux P. In: Methods in Molecular Biology. Chapman SK, Reid GA, editors. Vol. 131. Humana press; New Jersey: 1999. p. 5. [Google Scholar]
- 23.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for Biochemical Research. 2. Oxford University Press; Oxford: 1969. pp. 200–201. [Google Scholar]
- 24.Satterwhite DM. Methods Enzymol. 1985;110:92–99. doi: 10.1016/s0076-6879(85)10064-9. [DOI] [PubMed] [Google Scholar]
- 25.Segel IH. Enzyme Kinetics. Wiley; New York: 1975. pp. 274–329. [Google Scholar]
- 26.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: URL http://www.R-project.org/2013. [Google Scholar]
- 27.Baty F, Delignette-Muller ML. R package version 0.0-13. 2012. [Google Scholar]
- 28.Graffelman J. R package version 1.7.1. 2012. [Google Scholar]
- 29.Lumley T. R package version 2.0-0. 2013. [Google Scholar]
- 30.Schmidin FX. Protein Structure – A Practical Approach. Oxford University Press; Oxford: 1997. pp. 261–297. [Google Scholar]
- 31.Provencher SW, Glockner J. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 32.Van Stokkum IHM, Spoelder HJW, Bloemendal M, Van Grondelle R, Groen FCA. Anal Biochem. 1990;191:110–118. doi: 10.1016/0003-2697(90)90396-q. [DOI] [PubMed] [Google Scholar]
- 33.Manavalan P, Johnson WC., Jr Anal Biochem. 1987;167:76–85. doi: 10.1016/0003-2697(87)90135-7. [DOI] [PubMed] [Google Scholar]
- 34.Sreerama N, Woody RW. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 35.Sreerama N, Woody RW. Anal Biochem. 1993;209:32–44. doi: 10.1006/abio.1993.1079. [DOI] [PubMed] [Google Scholar]
- 36.Sreerama N, Venyaminov SY, Woody RW. Protein Sci. 1999;8:370–380. doi: 10.1110/ps.8.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitmore L, Wallace BA. DICHROWEB. 2004. [Google Scholar]
- 38.Whitmore L, Wallace BA. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Collaborative Computational Project, Number 4. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 41.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P, Cowtan K. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 43.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Acta Cryst. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The PyMOL Molecular Graphics System, Version 1.2r3pre. Schrödinger, LLC; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.