

Abstract
Rationale
Vascular smooth muscle cell (VSMC) migration and proliferation are the hallmarks of restenosis pathogenesis after angioplasty. Cyclooxygenase (COX)-derived prostaglandin (PG)E2 is implicated in the vascular remodeling response to injury. However, its precise molecular role remains unknown.
Objective
This study investigates the impact of COX-2-derived PGE2 on neointima formation after injury.
Methods and Results
Vascular remodeling was induced by wire-injury in femoral arteries of mice. Both neointima formation and the restenosis ratio were diminished in COX-2 KO mice as compared to controls, whereas these parameters were enhanced in COX-1>COX-2 mice where COX-1 is governed by COX-2 regulatory elements. PG profile analysis revealed that the reduced PGE2 by COX-2 deficiency, but not PGI2, could be rescued by COX-1 replacement, indicating COX-2-derived PGE2 enhanced neointima formation. Through multiple approaches, the EP3 receptor was identified to mediate the VSMC migration response to various stimuli. Disruption of EP3 impaired VSMC polarity for directional migration by depressing small GTPase activity and retarded vascular neointimal hyperplasia while overexpression of EP3α and EP3β aggravated neointima formation. Inhibition or deletion of EP3α/β, a Gαs protein-coupled receptor, activated thecAMP/PKA pathway and depressed activation of RhoA in VSMCs. PGE2 could stimulate PI3K/Akt/GSK3β signaling in VSMCs through Gβγ subunits upon EP3α/β activation. Abolition of EP3 suppressed PI3K signaling and reduced GTPase activity in VSMCs, and altered cell polarity and directional migration.
Conclusions
COX-2-derived PGE2 facilitated the neointimal hyperplasia response to injury through EP3α/β-mediated cAMP/PKA and PI3K pathways, indicating EP3 inhibition maybe a promising therapeutic strategy for percutaneous transluminal coronary angioplasty.
Keywords: neointima formation, PGE2, EP3, VSMC migration, polarity
Percutaneous transluminal coronary angioplasty (PTCA) is commonly used for the treatment of coronary heart disease (CHD). However, restenosis after angioplasty and stent deployment, considered as a wound healing response to mechanical injury, continues to be problematic in coronary interventional treatment, although application of drug-eluting stents (DES) has dramatically increased success rate compared to regular bare-metal stents (BMS)1. Directional migration of vascular smooth muscle cells (VSMCs) from media to intima and their subsequent proliferation are the important processes for neointimal hyperplasia-lumen narrowing, followed by deposition of extracellular matrix, which together lead to self-remodeling of vessels (restenosis)2.
De-endothelialization promotes platelet activation and infiltration of inflammatory cells, which could initiate VSMC migration by releasing pro-inflammatory mediators such as MCP-1, IL-6, and prostaglandins (PGs)3. Elevated systemic inflammation markers such as MCP-1, C-reactive protein and C3 complement are associated with the increased risk of clinical angiographic restenosis4. Thus, investigators have proposed systemic anti-inflammatory approaches to prevent clinical restenosis5.
Cyclooxygenase (COX) is a key enzyme for PG biosynthesis. Both isoforms-COX-1 and COX-2 are expressed in the vasculature6 and can contribute substantially to the generation of vascular PG including PGE27. COX-2, an inducible isozyme and the dominant source of PGs in inflammation, is upregulated in vascular inflammation, such as atherosclerosis8, aortic aneurysm9 and balloon-injured arteries10, 11. Thus, elevated levels of PGE2 along with the induction of COX-2 in the vascular wall, is associated with the instability of plaque in the progression of atherosclerosis12. Pharmacological inhibition of COX-2, but not COX-1, reduces vascular neointimal hyperplasia in response to mechanical injury10, 11. Moreover, systematic inhibition of the production of PGE2 caused by genetic disruption of microsomal prostaglandin E2 synthase-1(mPGES-1) attenuates neointima formation after vascular injury13. These findings strongly suggest that COX-2-derived PGE2 might contribute to the pathogenesis of vascular restenosis.
A major source of the PGE2 formed in vivo is derived from mPGES-1, and other PGES isozymes do not compensate when mPGE-1 is deleted7. Along with notably direct suppression of PGE2 production, deletion of mPGES-1 reduces the neointimal hyperplasia response to vascular injury and results in cell specific differential utilization of the accumulated PGH2 substrates, such as predominant augmentation of prostacyclin (PGI2) in VSMCs and thromboxane A2 (TxA2) in macrophages14. Genetic deficiency of the PGI2 receptor (IP) enhances vascular proliferation response to wire injury; however, deletion of the TxA2 receptor (TP) depresses this response15. Targeted deletion of mPGE-1 in VSMCs and macrophages differentially modulates the response to vascular injury in mice16, indicating direct impact of mPGES-1-derived PGE2 on vascular remodeling, probably through its receptors known as EPs.
To test our hypothesis, we utilized COX-1>COX-2 mice, in which COX-1 is exchanged for COX-2 under the control of COX-2 regulatory elements17, as well as COX-2 knockout (KO) mice were utilized to address how COX-2-derived PGs are involved in the vascular remodeling in response to mechanical injury. We demonstrate here, PGE2, derived primarily from COX-2, accelerated vascular neointima formation in a vascular wire injury mouse model, by comparing the vascular responses and PG profiles in two strains of mice. By screening different pharmacological inhibitors, the EP3 receptor was identified to mediate the VSMC migration response to PGE2 stimulation. Disruption of EP3, in particular its α and β splice variants, impaired the polarity of VSMCs required for directional migration through inhibition of the activity of small GTPase activity, including RhoA, Rac1, and Cdc42. The activity of RhoA in VSMCs could be modulated by both EP3α and EP3β through the cAMP-dependent protein kinase (PKA) pathway. Blockage of EP3 suppressed PI3K signaling, reduced Rac1, RhoA, and Cdc42 activity in VSMCs, altered cell polarity and restrained directional migration.
Methods
The detailed Methods section is available in the Online data.
Results
Deletion of COX-2 Conveys Protection against Vascular Neointima Formation in Response to Injury
COX-1 and COX-2 were abundantly expressed in cultured VSMCs. Both mRNA and protein for COX-2, but not COX-1, were significantly upregulated by stimulation with lipopolysaccharide (LPS) (Figure 1A and 1B). Similarly, the level of expression of the mRNA of the dominant PGE2 synthase, mPGES-1, also increased significantly in response to LPS (Figure 1B). In the wire-injured artery model, COX-2 was strongly stained in the infiltrating inflammatory cells (CD68+ and CD11b+) and in the proliferating VSMCs (α-actin positive) in the neointima at different stages (Figure 1C and Supplemental Figure I), indicating that COX-2 activity contributes to the vascular response to the injury.
Figure 1. COX-2 deletion reduced vascular neointima formation in response to injury in mice.
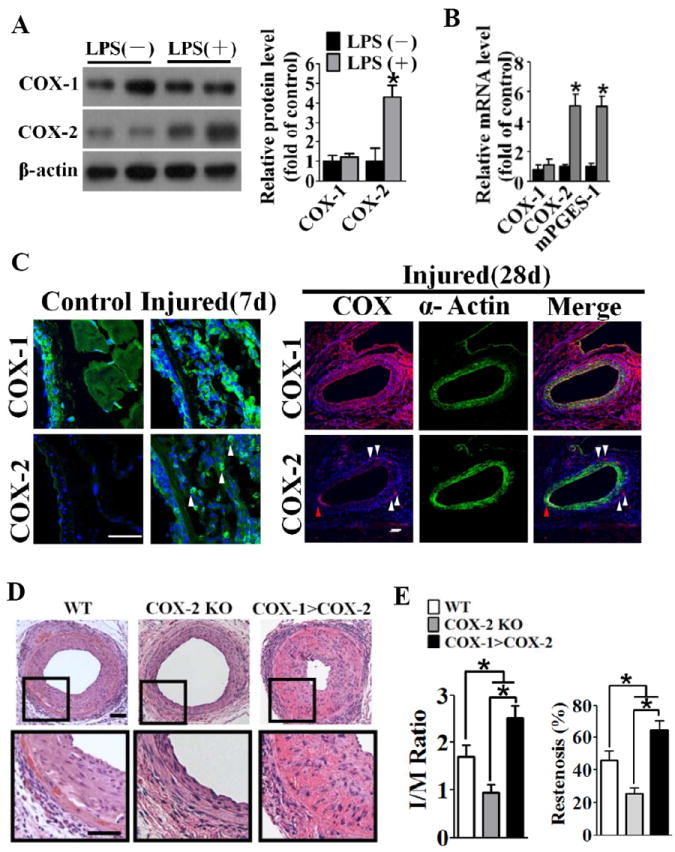
A, COX-1 and COX-2 expression in cultured VSMCs in the absence or presence of LPS. Left panel, representative western blots. Right panel, quantification of protein expression normalized to the levels of β-actin. *P < 0.05 vs. control without LPS treatment, n = 4. The results were repeated 3 times. B, RT-PCR analysis of mRNA expression of COX-1, COX-2, and mPGES-1 in cultured VSMCs in the absence or presence of LPS. *P < 0.05 vs. control without LPS treatment, n = 3. The results were repeated 3 times. C, Expression of COX-1 and COX-2 in the femoral artery in reponse to injury. White triangles indicate COX-2 staining in the perivasculature, red triangles indicate COX-1 staining in neointima at day 28 after injury. Scale bar, 50μm. D, Representative hematoxylin and eosin staining of crossections of wire-injured arteries from wild type (WT), COX-2 KO and COX-1>COX-2 mice. Scale bar, 50μm. E, Quantification of intima to media (I/M) ratio (left) and restenosis index (right) of post-injured arteries from WT, COX-2 KO and COX-1>COX-2 mice. *P < 0.05 vs. WT, n = 10-15.
To test our hypothesis, we performed wire injury in the femoral arteries of wild type (WT), COX-2 KO and COX-1>COX-2 and evaluated neointima formation at day 28 after injury. COX-2 deficiency, as anticipated, led to significant reduction of the intima-to-media (I/M) ratio (COX-2 KO, 0.95 ± 0.17 versus WT, 1.70 ± 0.25, P = 0.02) and the percentage of luminal narrowing (restenosis index, COX-2 KO, 25.59% ± 3.38%, versus WT, 46.00% ± 5.62%, P = 0.005) compared with WT with abundant neointima formation (Figure 1D and 1E). In contrast, the replacement of COX-1 in COX-1>COX-2 mice augmented the I/M ratio (COX-1>COX-2, 2.53 ± 0.25, vs. WT, 1.70 ± 0.25, P = 0.03) and the restenosis index (COX-1>COX-2, 64.5% ± 6.4%, vs. WT, 46.0% ± 5.6%, P = 0.04).
Immunohistochemical analysis revealed the presence of extensive inflammatory cell infiltration around the vasculature, as well as VSMC migration and proliferation in the intima in response to vascular injury (Supplemental Figure IA, IC, and IE). COX-2 deletion coordinately suppressed the inflammatory response as compared with WT controls, but no overt differences were found between COX-1>COX-2 and WT mice (Supplemental Figure IB and ID).
COX-2-derived PGE2 is Involved in the Development of Injury-Induced Neointima Formation
Given the different impacts of COX-2 deficiency and the replacement of COX-2 activity by COX-1 on vascular remodeling (Figure 1), we looked further into systemic PG metabolism and vascular PG generation of these strains. Vascular insult increased the excretion of all PG metabolites (Figure 2) consistent with induction of COX-2 in the vasculature of WT mice (Figure 1). Deletion of COX-2 led to the suppression of the levels of PGE2 metabolite (PGEM) and PGI2 metabolite (PGIM) in mice, since COX-2 is the dominant source of PGI26, and COX-1 and COX-2 contribute to the production of PGE27. However, the COX-1 insertion in COX-1>COX-2 mice rescued the decline in the levels of PGEM in COX-2 KO mice (10.3 ± 2.31 ng/mg creatinine in COX-2 KO vs. 24.1 ± 3.9 ng/mg creatinine in WT, P < 0.05; vs. 20.9 ± 3.7 ng/mg creatinine in COX-1>COX-2, P < 0.05) (Figure 2B), without significant influence on the suppression of urinary PGIM by deletion of COX-2 (Figure 2C). Similar PG measurements were observed after vascular injury, albeit with higher levels detected (19.5 ± 3.8 ng/mg creatinine in COX-2 KO vs. 38.1 ± 4.6 ng/mg creatinine in WTP < 0.05; vs. 35.1 ± 5.4 ng/mg creatinine in COX-1>COX-2, P < 0.05). Deletion of COX-2 had no impact on the excretion of urine PGD2 metabolite (PGDM) and Tx-M before and after injury. In cultured VSMCs, PGE2 and PGI2 were the dominant products of COX-2, and substitution of COX-1 for COX-2 can rescue notably PGE2 production, but PGI2 (measured as its stable hydrolysis product), which was depressed by COX-2 deletion (Supplemental Figure II). Thus, insertion of COX-1 in the COX-2 locus restored COX-2-derived biosynthesis of PGE2, not PGI2 in mice. Depletion of prostacyclin receptor (IP) in mice aggravated the vascular responses to injury15, as in COX-1>COX-2 mice with diminished PGI2 (Figure 2). Taken together, we surmise that the decrease of PGE2 in COX-2 KO mice provides protection against injury-induced vascular remodeling. This occurs despite reduced production of vasoprotective PGI2, by which COX-2 inhibition increases the risk of cardiovascular events in clinic8.
Figure 2. Urinary PG metabolite profile in WT, COX-2 KO and COX-1>COX-2 mice before and 3 days after wire-injury.
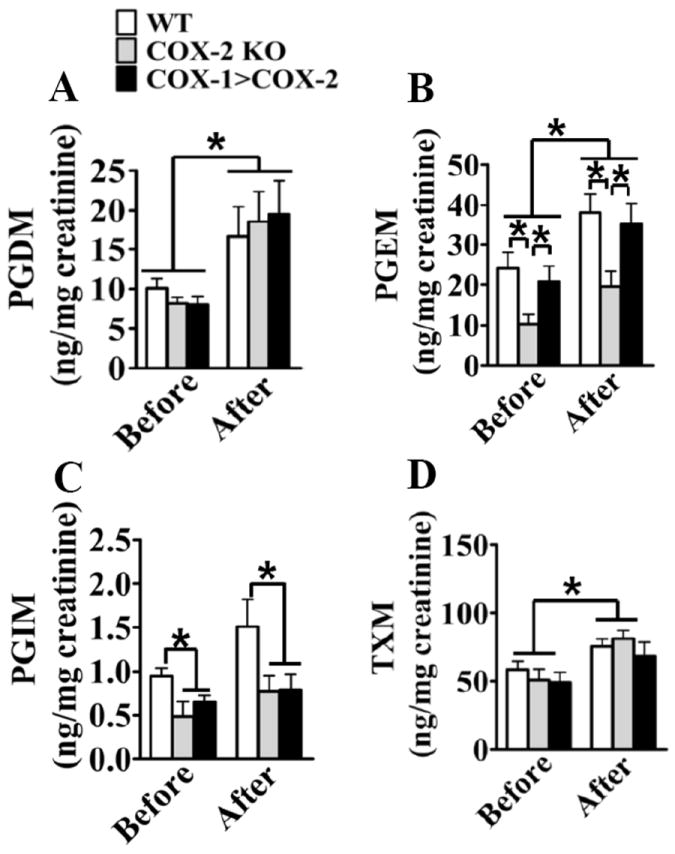
24-hour urine samples were collected from COX-2 KO, COX-1>COX-2 and WT male mice, and the levels of urinary PGD2 metabolite-PGD-M (A), PGE2 metabolite-PGE-M (B), PGI2 metabolite PGI-M (C), TxA2 metabolite-Tx-M (D) were measured. Data are presented as mean ± SEM, *P < 0.05 vs. WT controls, n = 10-15.
The EP3 Receptor Mediates Migration of VSMCs upon PGE2 Stimulation
In response to de-endothelialization induced by mechanical injury, VSMCs migrate directionally from arterial media into the intima and begin proliferating, resulting in neointima formation and restenosis2. We first monitored the migration of VSMCs from COX-2 KO mice by a traditional scratch wound healing assay. COX-2 deficiency significantly inhibited wound closure at all timepoints tested (Supplemental Figure III). In particular, at the first 12 h, cell proliferation is minimal and wound closure at this time primarily reflects cell spreading and migration18. Because COX-2-derived PGE2 is responsible for this process, and all 4 receptors for PGE2 (EP1, EP2, EP3, and EP4) are expressed abundantly in cultured VSMCs and arteries and are not significantly altered in response to LPS stimulation (Figure 3A). Using specific antagonists, we determined the identity of the receptor(s) involved in mediating the VSMC migration. The migration of VSMCs challenged with PGE2 was markedly reduced dose-dependently by pre-treatment with the EP3 receptor antagonist L-789,106 (Figure 3B and 3C), while no significant effects were detected using EP1, EP2 and EP4 receptor antagonists (Figure 3B). To rule out the impact of cell proliferation, we performed transwell migration assays of VSMCs within 3 h of treatment. Similarly, only the EP3 receptor antagonist L-789,106 strikingly attenuated VSMC migration (59% suppression compared with the vehicle control (Figure 3D), which again exhibited dose-dependent inhibition (Figure 3E). Therefore, PGE2, derived primarily from COX-2, promoted VSMC migration through the EP3 receptor.
Figure 3. Pharmacological inhibition of EP3 receptor attenuates VSMC migration in vitro.
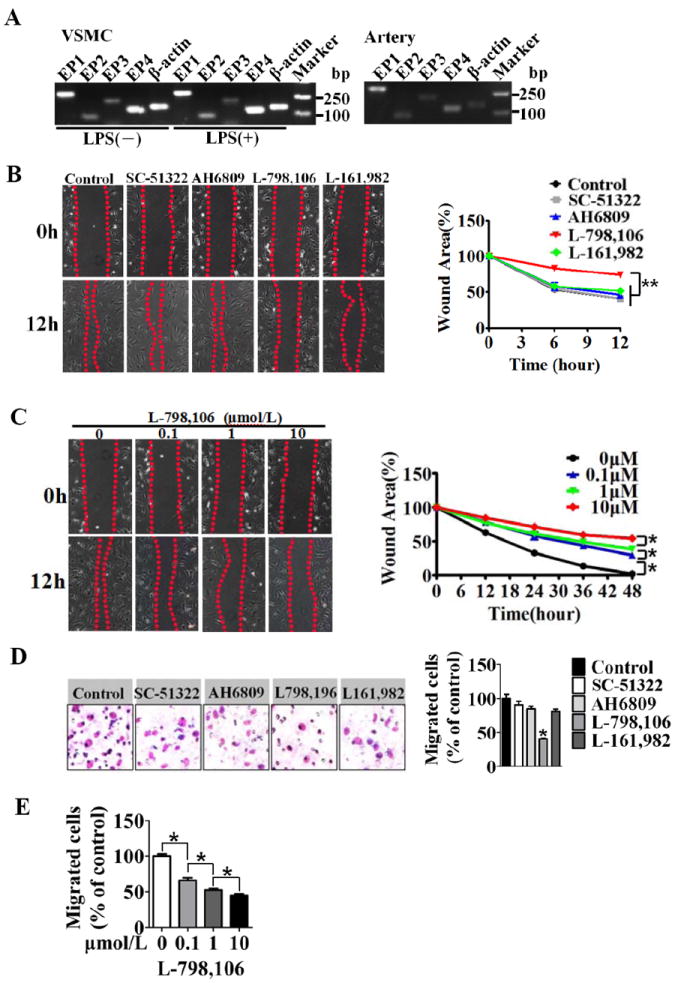
A, RT-PCR analysis of the expression of EP1, EP2, EP3 and EP4 receptors in cultured VSMCs (left) and arteries (right). B, The effect of PGE2 receptor antagonists on PGE2-stimulated VSMC migration in the wound healing assay. The EP1 antagonist, SC-51322; the EP2 antagonist, AH6809; the EP3 antagonist, L-798, 106; the EP4 antagonist, L-161, 982. Representative phase-contrast images were taken at 0, 6, and 12 h after scratching. The wound area was measured and quantitated as described in the Methods section. Data are normalized by the wound area at the 0 timepoint. ** P < 0.01 vs. control, n = 3. C, The effect of different concentrations of L-798, 106 on PGE2-stimulated VSMC migration in the wound healing assay. *P < 0.05 as indicated, n =4. D, The effect of PGE2 receptor antagonists on migration of VSMCs in a transwell assay. The number of migrating cells was normalized to the control group. *P < 0.05 vs. control, n = 3. E, The effect of EP3 antagonist L-798,106 on the migration of VSMCs. *P < 0.05 vs. control, n = 3.
Both EP3α and β Splice Variants are Involved in Regulation of VSMC Migration and Injury-Induced Neointimal Hyperplasia
Using EP3 small interfering RNA (siEP3), directed at a site within exon 2 to disrupt all three mouse EP3 splice variants, EP3 expression was reduced by 80% in primary VSMCs, resulting in a 48% decrease in VSMC migration through the transwell assay (Supplemental Figure IVA). Likewise, EP3 deletion caused similar impairment of VSMC migration as the EP3 inhibitor (Figure 4A). Real-time PCR data showed that all PG receptors were expressed in VSMCs and EP3 disruption did not significantly alter the expression of other PG receptors, with or without LPS treatment (Supplement Figure V). The 3 EP3 splice variants are expressed in VSMCs (Supplement Figure IVB). To identify the EP3 variant that mediates VSMC migration induced by PGE2, VSMCs from WT mice were subjected to transfection with siRNA specific for each variant. Real-time PCR demonstrated more than 70% reduction in the mRNA levels at 24 h post-transfection of the respective variants without significant compensation. Knockdown of EP3α and EP3β significantly impaired the capability of VSMC migration (EP3α inhibition, 78.00 ± 2.1, EP3β inhibition, 73.03 ± 2.4, vs. scrambled control, 100.0 ± 1.7, P < 0.05) (Supplemental Figure IVC). However, silencing of EP3γ did not significantly influence VSMC migration.
Figure 4. EP3α and EP3β are involved in the mediation of VSMC migration and vascular remodeling in response to injury.

A, EP3 deficiency significantly retarded VSMC migration. *P < 0.05 vs. WT, n = 3. B, Transwell migration analysis of the impact of re-expression of EP3α, EP3β, or EP3γ on the migration of the VSMCs isolated from EP3 KO mice. *P < 0.05 vs WT transfected with empty vectors, n = 3. C, Dose-dependent effect of transfected EP3α or EP3β expression vectors on VSMC migration. *P < 0.05 vs. EP3 KO transfected with empty vector or as indicated, n = 3. D, Representative hematoxylin and eosin staining of cross sections of wire-injured arteries from EP3 KO and WT mice 28 days after injury. Scale bar, 50μm. Quantification of intima to media (I/M) ratio (middle) and restenosis index (right) of injured arteries from EP3 KO and WT mice. *P < 0.05 vs. WT, n = 8-10. E, Representative hematoxylin and eosin staining of cross sections of wire-injured arteries from mice transduced with the lentivirus expressing each of the EP3 variants. F, Quantification of intima-to-media (I/M) ratio (left) and restenosis index (right). *P < 0.05, **P < 0.01 vs. control (WT infected by a GFP lentivirus), n = 8-10. Scale bar, 50μm.
We sought to determine whether re-expression/overexpression of EP3 variants could restore the ability of VSMC migration. Primary VSMCs isolated from EP3 KO mice were transfected with mouse EP3α, EP3β or EP3γ cDNAs which were tagged with the HA epitope at the N-terminus. Robust induction of the expression of each EP3 variant was observed at 24 h after transfection (Supplemental Figure IVD). Overexpression of EP3α or EP3β rescued the ability of VSMCs to migration, which was inhibited previously by EP3 deficiency (Figure 4B), and this renewed capability correlates positively with expression of EP3α or EP3β (Figure 4C), whereas EP3γ overexpression showed very little influence on migratory behavior (Figure 4B). These data demonstrate that PGE2-induced VSMC migration is mainly mediated by EP3α and EP3β. Consistent with these results, in this vascular injury model, EP3 abolition rendered a decrease in the I/M ratio (0.65 ± 0.16 vs. 1.34 ± 0.15, P = 0.005) and restenosis in EP3 KO mice (index, 23.4% ± 5.0% versus 42.2% ± 3.4%, P = 0.007) compared with control mice (Figure 4D). The levels of PG metabolites were not significantly influenced by EP3 disruption in mice, even when subjected to wire injury (Supplemental Figure VI).
To further confirm the involvement of EP3α and EP3β in vascular remodeling, we delivered lentiviral vectors, each constitutively expressing EP3α, EP3β, or EP3γ, to the femoral arteries after wire injury (Figure 4E). After 4 weeks, expression of the EP3α and EP3β transgenes significantly reversed the inhibition of neointimal hyperplasia in EP3 deficient mice (I/M ratio, EP3α overexpression, 1.68 ± 0.16, EP3β overexpression, 1.42 ± 0.08 vs. GFP control, 0.70 ± 0.07, P < 0.05), whereas vascular overexpression of EP3γ failed to significantly alter the vascular response to injury (Figure 4E and 4F). Thus, these data indicate the EP3α and EP3β receptors play important roles in the vascular remodeling in response to injury.
Activation of EP3 is Required for Polarity of Migrating VSMCs
The positioning of the microtubule organizing center (MTOC) relative to the nucleus toward the leading edge is a hallmark of migrating cell polarity19. Confluent cultures of VSMCs were wounded with a pipette tip and immunostained for the MTOC marker γ-tubulin and counterstained with DAPI to visualize cell nuclei at the wound edge (Figure 5A). The MTOCs that localized between the nucleus and the leading edge were determined as front-polarized20 (Figure 5B). Before wounding, MTOC was randomly distributed with 20.1% ± 1.0% of front-polarized cells. Six hours after wounding, the percentage of front-polarized VSMCs increased to 40.4% ± 8.5% (Supplemental Figure VII). Impairment of directional cell migration is accompanied by a loss of cell polarity toward the direction of migration20. Antagonism of the EP3 receptor activity by L-798,106 (10 μmol/L), markedly disrupted polarization of VSMCs and randomized the orientation of the MTOCs front-polarized cells of EP3 antagonist vs. control, 13.5 ± 3.4 vs 40.3 ± 8.5, P < 0.05, Figure 5B). Comparable results were observed in VSMCs from EP3 KO mice with nearly 16% front-polarized cells (EP3 KO vs. WT, 15.5 ± 5.1 vs. 42.3 ± 12.1, P < 0.05) (Figure 5A, C).
Figure 5. Pharmacological inhibition or genetic deletion of EP3 impairs the polarization of VSMCs for directional migration.

A, Representative confocal micrographs of primary post-confluent VSMCs isolated from WT and EP3 KO mice immunostained for γ-tubulin to localize the MTOCs (arrow) at 6 h after wounding in the presence or absence of the EP3 antagonist, L-798,106. Nuclei were stained with DAPI. The dashed box outlines the region enlarged to the right. Scale bar, 50μm. B, Schematic of the method used for quantifying the position of the MTOCs of wound-edge cells. C, Quantitation of front-polarized VSMCs from WT and EP3 KO mice, with or without L-798,106 treatment 6 h after wounding. *P < 0.05 vs. control for L-798,106 treatment or WT for EP3 KO, n = 3. Scale bar, 50μm. The results were repeated 3 times. D, Representative confocal micrographs of α-tubulin immunostaining of VSMCs from WT and EP3 KO mice, with or without L-798,106 treatment 6 h after wounding. Scale bar, 50μm. E, Representative confocal micrographs of F-actin (Left) immunostaining of VSMCs from WT and EP3 KO mice, with or without L-798,106 treatment 6 h after wounding. The white boxes outline the region enlarged to the lower panel. The number of cellular leading protrusions was quantitated as the number of leading protrusions divided by the number of cells at the leading edge (right). *P < 0.05 vs. control for L-798,106 treatment or WT for EP3 KO, n = 3. Scale bar, 50μm. The results were repeated 3 times.
Immunostaining for α-tubulin showed wound-facilitated formation of long and unbranched protrusions, which were oriented toward the direction of migration in intact VSMCs, while EP3 inhibition [L-798,106 or Gi protein blocker, pertussis toxin (PTX) ] or deletion of EP3, led to the formation of randomly oriented protrusions (Figure 5D). In migrating cells, protrusion of the leading edge requires the precise regulation of the lamellipodia and F-actin networks21. As indicated by F-actin staining, pharmacological inhibition of EP3 by L-798,106 or PTX sharply reduced the number of leading protrusions during VSMC migration (L-798,106 treatment, 1.29 ± 0.20; and PTX treatment, 1.0 ± 0.19; vs. control, 3.47 ± 0.38, P < 0.05, Figure 5E). Similar suppression of leading protrusions was replicated in VSMCs derived from EP3 KO mice compared with WT (1.11 ± 0.21 vs. 3.40 ± 0.36, P < 0.05, Figure 5E). These results suggest the migratory defect of VSMCs from EP3 KO is attributed to abnormal of cell polarity.
Activity of Small GTPases is Impaired in VSMCs from EP3 KO mice
Rho GTPases are pivotal regulators for cell polarization22 and directional migration23. We therefore performed standard pull-down assays using lysates prepared from primary VSMCs obtained from EP3 KO and WT mice, and examined GTP-bound RhoA, Rac1, and Cdc42 in response to PGE2 stimulation. RhoA activity was undetectable in untreated cultured VSMCs (data not shown), but was boosted robustly in the presence of PGE2, while EP3 disruption attenuated the induction of GTP-RhoA to 68% (Figure 6A). Overexpression of either EP3α or EP3β by transfection of EP3-deficient VSMCs resulted in up-regulated RhoA activity upon stimulation (Figure 6A). Again, Cdc42 and Rac1 activities that were low in VSMCs could be induced notably by PGE2. Deletion of EP3 resulted in a slight but significant increase in their activities, while re-expression of either EP3α or EP3β completely restored the suppression in EP3 KO VSMCs (Figure 6B). These observations indicate that either EP3α or EP3β was involved in regulating of RhoA, Cdc42 and Rac1 activation.
Figure 6. EP3 deletion reduces activation of RhoA, Rac1, and Cdc42 in VSMCs.
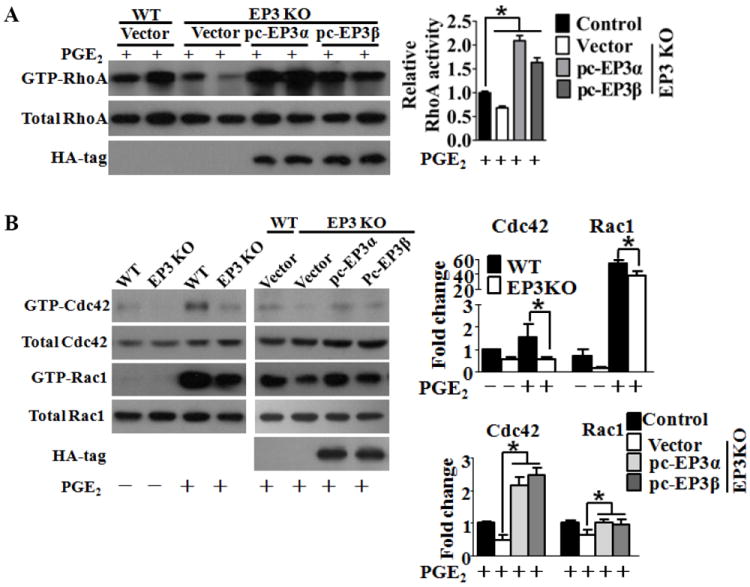
A, Western blot analysis of active (GTP-RhoA) and total RhoA in cultured VSMCs from WT and EP3 KO mice transfected with EP3α, EP3β or control vector (left). Densitometric quantification of GTP-RhoA levels normalized to the levels of total RhoA (right). *P < 0.05 vs. control, n = 6. B, Western blot analysis of Cdc42 (GTP-Cdc42), active Rac1 (GTP-Rac1), and total Cdc42 and Rac1 in cultured VSMCs from WT and EP3 KO mice transfected with EP3α, EP3β or control vectors as indicated (left). Densitometric quantification of Rac1 and Cdc42 activity normalized to that of total Rac1 and Cdc42 (right). *P < 0.05 vs. control. The data were collected from 3 independent experiments.
EP3α/β Variants are Coupled via Gαi Protein to Regulated RhoA Activity and VSMC Migration through a PKA Signaling Pathway
Evidence indicates EP3α and EP3β are coupled to Gαi24. To determine if these G protein interaction occurs in VSMCs, HA-tagged EP3α or EP3β was introduced into cultured VSMCs. Coimmunoprecipitation experiments displayed clear binding of EP3α or EP3β with Gαi using an HA-tag antibody (Figure 7A), suggesting that either EP3α or EP3β suppresses adenylate cyclase activity and cellular cAMP levels. As anticipated, the levels of cAMP were significantly depressed by overexpression of either EP3α or EP3β receptors upon stimulation with an EP3 agonist M&B28767 (Figure 7B). Accordingly, PKA activity was increased by 20% when EP3 was deleted in VSMCs, and reduced by 40% and 36% by overexpression of EP3α or EP3β, respectively (Figure 7C). Various lines of evidence suggest that PKA modulates activity of GTPases and influences cytoskeletal dynamics and cell migration25. To determine whether PKA is involved in the EP3-mediated RhoA activation, VSMCs were pretreated with a PKA inhibitor (H-89) before PGE2 stimulation. As shown in Figure 7D, overexpression of EP3α or EP3β markedly elevated RhoA activity. Blocking PKA activity by H-89 also enhanced RhoA activation, which was more apparent in EP3 KO cells. Surprisingly, we failed to detect a consistent impact of PKA on Cdc42 and Rac1 by using H-89 even in cells overexpressing EP3 (Supplement Figure VIII). Along with the increasing repositioning of MTOCs toward a scratch in the wound healing assay (EP3 KO, 15.5% ± 5.1% vs. H-89/EP3 KO, 32.6% ± 5.9%, P < 0.05, Figure 7E), antagonism of the induction of PKA activity by EP3 deletion partially rescued the ability of VSMCs to migration in the transwell assay (EP3 KO, 52.1% ± 1.7% vs. H-89/EP3 KO, 62.1% ± 1.1%, P < 0.05, Figure 7F), implying that additional EP3-mediated pathways may be involved. The inhibition of Rho signaling by the Rho-associated protein kinase (ROCK) inhibitor Y-27632 significantly delayed the migration of VSMCs, because ROCK1 and ROCK2 are the first effectors of Rho and participate in the formation of stress fibers and focal adhesions26.
Figure 7. EP3α/β are coupled via Gαi protein and to regulate RhoA activity and VSMC migration through the cAMP/PKA pathway.
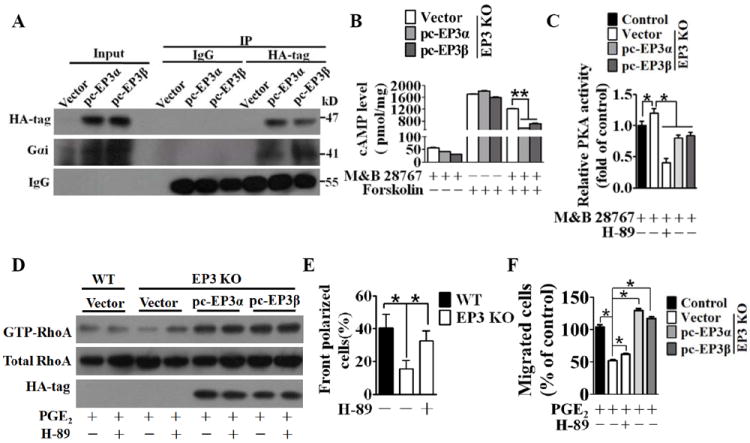
A, Western blot analysis of binding of EP3α/β and Gαi. B, Cellular cAMP levels were measured in VSMCs isolated from EP3 KO mice transfected with EP3α, EP3β, or control vector constructs treated with an EP3 agonist M&B 28767(10 μmol/L) and adenylate cyclase (AC) activator forskolin (10 μmol/L) as indicated. *P < 0.05 vs EP3KO transfected with control vectors, n = 3. The results were repeated 3 times. C, PKA activity was measured in VSMCs from EP3 KO or WT mice transfected with EP3α, EP3β, or control vector constructs treated with or without the PKA inhibitor H-89 (10 μmol/L). Data are normalized to the values of the control group. *P < 0.05, n = 3. The results were repeated 3 times. D, Western blot analysis of active RhoA in VSMCs from EP3 KO and WT mice transfected with EP3α, EP3β, or control vector constructs with or without, pretreatment with the PKA inhibitor H-89 (10 μmol/L). E, Effect of H-89 on reorientation of MTOC of EP3 KO and WT VSMCs. *P < 0.05, n = 3. The results were repeated 3 times. F, Effect of H-89 on the migration of VSMCs from EP3 KO and WT mice transfected with EP3α, EP3β, or control vectors constructs. The number of migrating cells was normalized to the control group. *P < 0.05 as indicated, n = 4.
The EP3α/β -Mediated PI3K-Akt-GSK3β Signaling Axis Modulates Migration and Polarization of VSMCs
Phosphorylation of GSK3β is implicated in the control of direction of cell protrusion by regulating the centrosome polarization27. To explore whether PGE2 activates the PI3K/Akt/GSK3β signaling pathway through the EP3 receptor, we first determined inhibitory phosphorylation of GSK3β at Ser9. We found that in response to PGE2 treatment, GSK3β underwent a rapid phosphorylation within 30 min reaching a peak at 6 h and fully recovered at 12 h (Supplement Figure IXA and IXB). The EP3 antagonist L-798,106 and Gαi blocker PTX suppressed PGE2-induced phosphorylation of GSK3β and Akt—PI3K signaling components (Supplement Figure IXB). The phosphorylation of GSK3β and Akt were also attenuated in EP3 KO mice in either the absence or presence of LPS, which activates the PI3K via Toll-like receptor 4 (TLR4) (Figure 8A). These results indicate that EP3 mediates the PI3K/Akt/GSK3β signaling by liberating Gβγ subunits upon PGE2 stimulation as previously described for other PGE2 receptors28.
Figure 8. EP3α/β-mediated PI3K-Akt-GSK3β signaling axis modulates migration and polarization of VSMCs.

A, Phosphorylation of Akt and GSK3β in VSMCs from EP3KO and WT mice in the presence or absence of LPS. B, The PI3K inhibitor Wortmannin (100 nmol/L) attenuated the phosphorylation of Akt and GSK3β induced by the overexpression of EP3α or EP3β in VSMCs isolated from EP3 KO mice. C, Wortmannin, SB216763(20 μmol/L) and HIMO (10 μmol/L) treatment reduced the upregulation of Rac1 activity in VSMCs overexpressing EP3α or EP3β isolated from EP3 KO mice. D, Wortmannin and LY294002 (20 μmol/L) treatment suppressed the upregulation of the Cdc42 activity in VSMCs overexpressing EP3α or EP3β isolated from EP3 KO mice, while HIMO and SB216763 treatment did not have effect. E, Wortmannin, HIMO and SB216763 treatment restrained MTOC reorientation (left) and transwell migration (right) by the EP3α or EP3β overexpression in VSMCs isolated from EP3 KO mice. MTOC reorientation: *P < 0.05, n = 3. The results were repeated 3 times. In transwell assay, the data were collected from 3 independent experiments, and the number of migrating cells was normalized to the control group.*P < 0.05 as indicated. F, Schematic diagram of EP3α/β-mediated polarization and directional migration of VSMCs through both cAMP/PKA and PI3K pathways upon PGE2 stimulation.
We next asked whether EP3α/β mediated the PI3K/Akt/GSK3β signaling, because these splice variants were involved in cell migration and polarization. Re-expression of either EP3α or EP3β in VSMCs from EP3 KO mice elevated the level of phosphorylation of Akt and GSK3β induced by PGE2. This effect was blunted by the PI3K selective inhibitor Wortmannin (Figure 8B). Moveover, re-expression of either EP3α or EP3β restored the phosphorylation of GSK3β in VSMCs isolated from EP3 KO mice, which was terminated by an Akt inhibitor HIMO or a GSKβ inhibitor SB216763 (Supplement Figure IXC). These findings indicate that the EP3α and EP3β receptors mediate the activation of PI3K/Akt/GSK3β signaling. GSK3β may be phosphorylated by PKA in neurons29. However, H-89 treatment did not significantly influence GSK3β phosphorylation even in VSMCs overexpressing EP3α/β with enhanced phosphorylation (Supplement Figure IXD), indicating that GSK3β phosphorylation occurs independently of PKA in VSMCs.
We next examined the effect of the EP3α/β-mediated activation of the PI3K/Akt/GSK3β pathway on MTOCs reorientation and on cell migration. Enforced ectopic overexpression of either EP3α or EP3β in EP3-deficient VSMCs restored cell polarity within 6 h by 3- or 2.8-fold, respectively, and augmented subsequent migration of VSMCs by 2- or 1.9-fold (Figure 8E), respectively. However, these restorations of cell polarity and migration responses were abolished by pretreatment with Wortmannin, HIMO, or SB216763, which specifically inhibit signaling through the PI3K pathway (Figure 8E). Thus, the PI3K/Akt/GSK3β signaling axis is involved in the regulation of VSMC migration and polarity through the EP3α/β receptors.
The activation of Cdc42 and Rac1, key regulators of cell migration30, was impaired in EP3-deficient VSMCs. Again, re-expressing of either EP3α or EP3β in EP3 deficient VSMCs markedly rescued the decreased activation of Rac1 and Cdc42 (Figure 8C and 8D). Pharmacological inhibition of PI3K (Wortmannin), Akt (HIMO) and GSK3β (SB216763) abrogated the activation of Rac1 induced by PGE2 in EP3α-expressing, EP3β-expressing and WT VSMCs (Figure 8B). Similarly, elevation of RhoA activity by transfection of VSMCs with either EP3α or EP3β was blocked by Wortmannin, HIMO and SB216763 (Supplemental Figure X). These results suggest that Rac1 and RhoA activity can be regulated by the EP3α/β-mediated PI3K/Akt/GSK3β signaling pathway (Figure 8F). However, elevation of Cdc42 activity by EP3α or EP3β transfection with either EP3α or EP3β cDNA was blunted by the selective PI3K inhibitors-Wortmanin and LY294002, but not affected significantly by HIMO or SB216763 (Figure 8B), indicating that the PGE2-induced Cdc42 activation depends on PI3K activity through the EP3α/β receptors (Figure 8F).
Discussion
Vascular remodeling is differentially regulated by PGs derived from COX-1 and COX-2. Pharmacological inhibition of COX-2 suppresses the vascular remodeling in response to balloon injury10, 11, whereas genetic knockdown (data not shown) or pharmacological inhibition of COX-1 does not effect this response. We found here that deletion of COX-2 or its downstream EP3 receptor attenuates vascular neointima formation after wire injury in mice; VSMC migration and cell polarization toward directional migration stimulated by PGE2 could be impaired by pharmacological inhibition or genetic modulation of EP3α or EP3β receptors through down-regulation of small GTPases. Activation of the EP3α or EP3β receptors directly fine-tune RhoA activity through inhibitory Gαi signaling, whereas liberated Gβγ subunits regulate Cdc42, Rac1, and RhoA activities in VSMCs through the PI3K/Akt/GSK3β pathway.
COX-2, expressed in vasculature, including VSMCs6, is markedly induced during vascular inflammation9-11, 31. Vascular COX-2 is the dominant source of PGI2 and PGE25. Thus, deletion of COX-2 significantly reduces systemic PGE2 and PGI2 production7, 32. As shown by the data presented in Figure 2 and Supplemental Figure I, COX-1 knocked-in to the COX-2 locus (COX-1>COX-2) could rescue PGE2, but not PGI2 production33, which aggravated the vascular response to injury, consistent with previous observations on the disruption of the IP receptor15. Analogous to pharmacological studies, COX-2 deficiency conferred protection against neointima formation along with decreased production of PGE2 and PGI2. Therefore, we reasoned that COX-2-derived PGE2, from both VSMCs and the infiltrated inflammatory cells, predominantly promoted vascular neointimal hyperplasia, because impaired PGE2 generation in mice attenuates the vascular response13.
In this study, COX-2 deletion impaired VSMC migration, while exogenous PGE2 accelerated VSMC migration. Pharmacological inhibition, gene silencing using siRNA, or genetic deletion of the EP3 receptor retarded PGE2-induced VSMC migration, indicating that COX-2-derived PGE2 mediated VSMC migration through EP3 α/β activation (Figure 2). Recently, EP3 was to shown to promote the migration of other cells such as airway smooth muscle cells34. Deficiency of EP3 attenuated the thickening of the neointima, while over-expression of EP3α or EP3β in vasculature augmented its formation in mice, which suggests that EP3-mediated VSMC migration contributes to the vascular remodeling in response to mechanical injury. Because endothelium loss triggers VSMC migration through fenestrae in the internal elastic lamellae into the intima and subsequent proliferation35, EP3 may be involved in mediating the early events in restenosis, although other PGE2 receptor subtypes, such as EP236, or perhaps EP437, might participate in later events including VSMC proliferation. However, different EP receptors play different roles in stability of atherosclerotic plaque38.
Small GTPases including Rho, Rac and Cdc42 are pivotal for MTOCs orientation and for cell migration39. Pharmacological inhibition or genetic deficiency of EP3 impaires MTOCs re-orientation and decreases the activities of Rac1, RhoA and Cdc42 upon PGE2 stimulation, which lead to retardation of VSMC migration and reduction of vascular neointima formation in response to injury in mice. These observations are also consistent with the notion that repositioning the centrosome stabilizes the chosen direction of movement through regulation of the microtubule system40. Cdc42 is believed to be a master regulator of cell polarity by directing MTOCs reorientation toward the direction of movement41, 42, despite accumulated evidence showing that Rho and Rac are involved in the coordinate regulation of cell polarization43-45. In the present study, pharmacological inhibition of PI3K signaling diminished Cdc42 activity and disrupted the reorientation of the MTOCs in cells overexpressing EP3α/β receptors. Surprisingly, blockage of Akt and GSK3β, which act downstream of PI3K, reduced Rac1 and RhoA activity in VSMCs without a detectable impact on Cdc42 and inhibited the reorientation of the MTOCs toward to directional migration, suggesting the involvement of Rac1 and RhoA in the microtubule rearrangement and cell polarity as described previously46.
EP3α and β splice variants couple with Gαi resulting in the inhibition of adenylyl cyclase, while activation of the EP3γ elevates cAMP probably through Gαs24. Stimulation of G protein-coupled receptors (GPCRs) activate PI3K/Akt/GSK3β signaling by free Gβγ or indirectly by Gα by competitive binding of axin and releasing the GSK3β substrate--β-catenin28. Here, we found that the EP3α or -β receptors, but not EP3γ, mediated VSMC migration and vascular neointimal hyperplasia in mice. Besides Gαi-mediated PKA pathway, signaling through PI3K/Akt/GSK3β pathway that mediated cell migration, was shown to occur upon activation of EP3α or –β receptors through the liberation of their coupled Gβγ subunits in VSMCs. PKA inhibits RhoA-dependent function by inducing Ser188 phosphorylation of RhoA and subsequent cytoplasmic sequestration of GTP-RhoA by Rho GDP dissociation inhibitor (RhoGDI)47. Coincidently, EP3 inhibition or deletion reduced, while overexpression of EP3α or -β elevated RhoA activity through a Gαi-mediated cAMP/PKA pathway. RhoA and its key downstream effector ROCK are involved in neointima formation after vascular injury48, probably through regulation of cell shape and migration by modulating stress-fiber formation49. We also observed here a partial but significant increase of VSMC migration through up-regulation of RhoA activity by inhibition of PKA, despite substantial suppression effect of ROCK inhibitor. Moreover, we failed to detect analogous inhibitory effects of H-89 on the activity of Rac1 and Cdc42 in VSMCs similar to that of RhoA (Supplement Figure IX); These observations strongly indicate additional non-cAMP/PKA pathways mediated by EP3α/β receptors are involved in the regulation of VSMC migration and activity of small GTPases.
The phosphorylation status of GSK3β, a key downstream substrate of PI3K and Akt, is essential for maintaining cell polarity27. PGE2 stimulation and wound scratch activated PI3K signaling and induced rapid inhibitory phosphorylation of GSK3β in VSMCs, which could be attenuated by genetic deficiency or pharmacological inhibition of EP3. Moreover, the reduced PI3K signaling in EP3 KO VSMCs could be rescued by the EP3α/β overexpression. Pharmacological inhibition of the EP3α/β-activated PI3K signaling reduced the capability of migration and impaired the repositioning of the MTOCs. These observations demonstrate that PI3K/Akt/GSK3β signaling mediated by the EP3α/β receptors is involved in the regulation of migration and polarization of VSMCs and the vascular remodeling response to injury.
In summary, we showed here that COX-2 derived PGE2 accelerates neointima formation in response to injury through the EP3α/β receptors in mice, and that the EP3α/β-mediated Gαi/cAMP/PKA and Gβγ/PI3K/Akt/GSK3β pathways play a role in regulation of the activation of small GTPases, and the subsequent VSMC polarization and migration upon stimulation with PGE2. Given that selective inhibition of COX-2 confers a cardiovascular hazard attributable to inhibition of COX-2-derived PGI2, inhibition of signaling through the EP3 receptor may be a viable target for prevention of in-stent restenosis.
Supplementary Material
Novelty and significance.
What Is Known?
Pharmacological Inhibition of Cyclooxygenase (COX)-2, but not COX-1, reduces vascular response to mechanical injury.
Genetic disruption of microsomal prostaglandin E2 synthase-1(mPGES-1) attenuates neointima formation after vascular injury.
What New Information Does This Article Contribute?
COX-2-derived PGE2 promotes vascular neointimal hyperplasia response to mechanical injury.
COX-2-derived PGE2 regulates polarization and directional migration of vascular smooth muscle cells (VSMCs) via both EP3α and EP3β receptors.
EP3α/β modulates small GTPase activity in VSMCs through both cAMP/PKA and PI3K pathways.
VSMC migration from media to intima and subsequent proliferation are the hallmarks of pathogenesis of restenosis after angioplasty. However, the mechanism that regulates the VSMC migration response to injury is not fully understood. In this study, we show that PGE2 derived from COX-2, not COX-1, promotes vascular neointima formation followed by mechanical injury. Both EP3α and β isoforms are identified to mediate PGE2-induced VSMC migration via multiple approaches. Deletion of EP3 receptor influences polarization and directional migration of VSMCs by depressing small GTPase activity and retards vascular neointimal hyperplasia while overexpression of EP3α and EP3β augments neointima formation. Activation of the EP3α or EP3β receptors directly fine-tunes RhoA activity in VSMCs through inhibitory Gαi signaling, and regulates other small GTPase activity through the liberated Gβγ subunits. Thus, COX-2-derived PGE2 facilitates neointimal hyperplasia response to injury through EP3a/β-mediated cAMP/PKA and PI3K pathways, indicating EP3 receptor may be a promising target for prevention of in-stent restenosis.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology of China (2012CB945100, 2011CB503906 and 2011ZX09307-302-01), and the National Natural Science Foundation of China (81030004), NSFC-CIHR joint grant (NSFC81161120538 and CIHR-CCI117951), The Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-R-09), Clinic Research Center at Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences (CRC2010007). Y.Y. was supported by the One Hundred Talents Program of the Chinese Academy of Sciences (2010OHTP10) and Pujiang Talents Program of Shanghai Municipality (11PJ1411100). C.D.F. is a Canada Research Chair holder and recipient of an Ontario Heart and Stroke Foundation Career Investigator award.
Non-standard Abbreviations and Acronyms
- PTCA
percutaneous transluminal coronary angioplasty
- CHD
coronary heart disease
- DES
drug-eluting stents
- BMS
bare-metal stents
- VSMCs
vascular smooth muscle cells
- PGs
prostaglandins
- COX
cyclooxygenase
- mPGES-1
microsomal prostaglandin E2 synthase-1
- PGI2
prostacyclin
- EP
PGE2 receptor
- IP
PGI2 receptor
- TP
TxA2 receptor
- PKA
cAMP-dependent protein kinase
- I/M
intima/media ratio
- MTOC
microtubule organizing center
- ROCK
Rho-associated protein kinase
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Inoue T, Node K. Molecular basis of restenosis and novel issues of drug-eluting stents. Circ J. 2009;2009:615–621. doi: 10.1253/circj.cj-09-0059. [DOI] [PubMed] [Google Scholar]
- 2.Costa MA, Simon DI. Molecular basis of restenosis and drug-eluting stents. Circulation. 2005;2005:2257–2273. doi: 10.1161/01.CIR.0000163587.36485.A7. [DOI] [PubMed] [Google Scholar]
- 3.Welt FG, Rogers C. Inflammation and restenosis in the stent era. Arterioscler Thromb Vasc Biol. 2002;2002:1769–1776. doi: 10.1161/01.atv.0000037100.44766.5b. [DOI] [PubMed] [Google Scholar]
- 4.Tanaskovic S, Isenovic ER, Radak D. Inflammation as a marker for the prediction of internal carotid artery restenosis following eversion endarterectomy--evidence from clinical studies. Angiology. 2011;2011:535–542. doi: 10.1177/0003319710398010. [DOI] [PubMed] [Google Scholar]
- 5.Horvath C, Welt FG, Nedelman M, Rao P, Rogers C. Targeting ccr2 or cd18 inhibits experimental in-stent restenosis in primates: Inhibitory potential depends on type of injury and leukocytes targeted. Circ Res. 2002;2002:488–494. doi: 10.1161/hh0402.105956. [DOI] [PubMed] [Google Scholar]
- 6.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Pure E, Funk CD, FitzGerald GA. Vascular cox-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;2012:132ra154. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin e synthase-1, and cardiovascular function. J Clin Invest. 2006;2006:1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grosser T, Yu Y, Fitzgerald GA. Emotion recollected in tranquility: Lessons learned from the cox-2 saga. Annu Rev Med. 2010;2010:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 9.King VL, Trivedi DB, Gitlin JM, Loftin CD. Selective cyclooxygenase-2 inhibition with celecoxib decreases angiotensin ii-induced abdominal aortic aneurysm formation in mice. Arterioscler Thromb Vasc Biol. 2006;2006:1137–1143. doi: 10.1161/01.ATV.0000216119.79008.ac. [DOI] [PubMed] [Google Scholar]
- 10.Yang HM, Kim HS, Park KW, You HJ, Jeon SI, Youn SW, Kim SH, Oh BH, Lee MM, Park YB, Walsh K. Celecoxib, a cyclooxygenase-2 inhibitor, reduces neointimal hyperplasia through inhibition of akt signaling. Circulation. 2004;2004:301–308. doi: 10.1161/01.CIR.0000135467.43430.16. [DOI] [PubMed] [Google Scholar]
- 11.Wang K, Tarakji K, Zhou Z, Zhang M, Forudi F, Zhou X, Koki AT, Smith ME, Keller BT, Topol EJ, Lincoff AM, Penn MS. Celecoxib, a selective cyclooxygenase-2 inhibitor, decreases monocyte chemoattractant protein-1 expression and neointimal hyperplasia in the rabbit atherosclerotic balloon injury model. J Cardiovasc Pharmacol. 2005;2005:61–67. doi: 10.1097/00005344-200501000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Wohlin M, Helmersson J, Sundstrom J, Arnlov J, Vessby B, Larsson A, Andren B, Lind L, Basu S. Both cyclooxygenase- and cytokine-mediated inflammation are associated with carotid intima-media thickness. Cytokine. 2007;2007:130–136. doi: 10.1016/j.cyto.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Wang M, Ihida-Stansbury K, Kothapalli D, Tamby MC, Yu Z, Chen L, Grant G, Cheng Y, Lawson JA, Assoian RK, Jones PL, Fitzgerald GA. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation. 2011;2011:631–639. doi: 10.1161/CIRCULATIONAHA.110.973685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin e synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci U S A. 2006;2006:14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane a2. Science. 2002;2002:539–541. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Yang G, Xu X, Grant G, Lawson JA, Bohlooly YM, Fitzgerald GA. Cell selective cardiovascular biology of microsomal prostaglandin e synthase-1. Circulation. 2013;2013:233–243. doi: 10.1161/CIRCULATIONAHA.112.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Y, Stubbe J, Ibrahim S, Song WL, Smyth EM, Funk CD, FitzGerald GA. Cyclooxygenase-2-dependent prostacyclin formation and blood pressure homeostasis: Targeted exchange of cyclooxygenase isoforms in mice. Circ Res. 2010;2010:337–345. doi: 10.1161/CIRCRESAHA.109.204529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mercure MZ, Ginnan R, Singer HA. Cam kinase ii delta2-dependent regulation of vascular smooth muscle cell polarization and migration. Am J Physiol Cell Physiol. 2008;2008:C1465–1475. doi: 10.1152/ajpcell.90638.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gundersen GG, Bulinski JC. Selective stabilization of microtubules oriented toward the direction of cell migration. Proc Natl Acad Sci U S A. 1988;1988:5946–5950. doi: 10.1073/pnas.85.16.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sabatini PJ, Zhang M, Silverman-Gavrila R, Bendeck MP, Langille BL. Homotypic and endothelial cell adhesions via n-cadherin determine polarity and regulate migration of vascular smooth muscle cells. Circ Res. 2008;2008:405–412. doi: 10.1161/CIRCRESAHA.108.175307. [DOI] [PubMed] [Google Scholar]
- 21.Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. Two distinct actin networks drive the protrusion of migrating cells. Science. 2004;2004:1782–1786. doi: 10.1126/science.1100533. [DOI] [PubMed] [Google Scholar]
- 22.Gundersen GG, Gomes ER, Wen Y. Cortical control of microtubule stability and polarization. Curr Opin Cell Biol. 2004;2004:106–112. doi: 10.1016/j.ceb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Nobes CD, Hall A. Rho, rac, and cdc42 gtpases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;1995:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 24.Dey I, Lejeune M, Chadee K. Prostaglandin e2 receptor distribution and function in the gastrointestinal tract. Br J Pharmacol. 2006;2006:611–623. doi: 10.1038/sj.bjp.0706923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howe AK. Regulation of actin-based cell migration by camp/pka. Biochim Biophys Acta. 2004;2004:159–174. doi: 10.1016/j.bbamcr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Riento K, Ridley AJ. Rocks: Multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;2003:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 27.Etienne-Manneville S, Hall A. Cdc42 regulates gsk-3beta and adenomatous polyposis coli to control cell polarity. Nature. 2003;2003:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- 28.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin e2 promotes colon cancer cell growth through a gs-axin-beta-catenin signaling axis. Science. 2005;2005:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 29.Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic amp promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol. 2000;2000:9356–9363. doi: 10.1128/mcb.20.24.9356-9363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Connor KL, Mercurio AM. Protein kinase a regulates rac and is required for the growth factor-stimulated migration of carcinoma cells. J Biol Chem. 2001;2001:47895–47900. doi: 10.1074/jbc.M107235200. [DOI] [PubMed] [Google Scholar]
- 31.Hui Y, Ricciotti E, Crichton I, Yu Z, Wang D, Stubbe J, Wang M, Pure E, FitzGerald GA. Targeted deletions of cyclooxygenase-2 and atherogenesis in mice. Circulation. 2010;2010:2654–2660. doi: 10.1161/CIRCULATIONAHA.109.910687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu Y, Fan J, Chen XS, Wang D, Klein-Szanto AJ, Campbell RL, FitzGerald GA, Funk CD. Genetic model of selective cox2 inhibition reveals novel heterodimer signaling. Nat Med. 2006;2006:699–704. doi: 10.1038/nm1412. [DOI] [PubMed] [Google Scholar]
- 33.Yu Y, Fan J, Hui Y, Rouzer CA, Marnett LJ, Klein-Szanto AJ, FitzGerald GA, Funk CD. Targeted cyclooxygenase gene (ptgs) exchange reveals discriminant isoform functionality. J Biol Chem. 2007;2007:1498–1506. doi: 10.1074/jbc.M609930200. [DOI] [PubMed] [Google Scholar]
- 34.Aso H, Ito S, Mori A, Suganuma N, Morioka M, Takahara N, Kondo M, Hasegawa Y. Differential regulation of airway smooth muscle cell migration by prostanoid ep receptor subtypes. Am J Respir Cell Mol Biol. 2013;2013:322–329. doi: 10.1165/rcmb.2012-0158OC. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz SM. Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest. 1997;1997:S87–89. [PubMed] [Google Scholar]
- 36.Zhu S, Xue R, Zhao P, Fan FL, Kong X, Zheng S, Han Q, Zhu Y, Wang N, Yang J, Guan Y. Targeted disruption of the prostaglandin e2 e-prostanoid 2 receptor exacerbates vascular neointimal formation in mice. Arterioscler Thromb Vasc Biol. 2011;2011:1739–1747. doi: 10.1161/ATVBAHA.111.226142. [DOI] [PubMed] [Google Scholar]
- 37.Gruzdev A, Nguyen M, Kovarova M, Koller BH. Pge2 through the ep4 receptor controls smooth muscle gene expression patterns in the ductus arteriosus critical for remodeling at birth. Prostaglandins Other Lipid Mediat. 2012;2012:109–119. doi: 10.1016/j.prostaglandins.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santovito D, Mezzetti A, Cipollone F. Cyclooxygenase and prostaglandin synthases: Roles in plaque stability and instability in humans. Curr Opin Lipidol. 2009;2009:402–408. doi: 10.1097/MOL.0b013e32832fa22c. [DOI] [PubMed] [Google Scholar]
- 39.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: Integrating signals from front to back. Science. 2003;2003:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 40.Panda D, Miller HP, Islam K, Wilson L. Stabilization of microtubule dynamics by estramustine by binding to a novel site in tubulin: A possible mechanistic basis for its antitumor action. Proc Natl Acad Sci U S A. 1997;1997:10560–10564. doi: 10.1073/pnas.94.20.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palazzo AF, Joseph HL, Chen YJ, Dujardin DL, Alberts AS, Pfister KK, Vallee RB, Gundersen GG. Cdc42, dynein, and dynactin regulate mtoc reorientation independent of rho-regulated microtubule stabilization. Curr Biol. 2001;2001:1536–1541. doi: 10.1016/s0960-9822(01)00475-4. [DOI] [PubMed] [Google Scholar]
- 42.Lee JS, Chang MI, Tseng Y, Wirtz D. Cdc42 mediates nucleus movement and mtoc polarization in swiss 3t3 fibroblasts under mechanical shear stress. Mol Biol Cell. 2005;2005:871–880. doi: 10.1091/mbc.E03-12-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.del Pozo MA, Vicente-Manzanares M, Tejedor R, Serrador JM, Sanchez-Madrid F. Rho gtpases control migration and polarization of adhesion molecules and cytoskeletal erm components in t lymphocytes. Eur J Immunol. 1999;1999:3609–3620. doi: 10.1002/(SICI)1521-4141(199911)29:11<3609::AID-IMMU3609>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 44.Wojciak-Stothard B, Ridley AJ. Shear stress-induced endothelial cell polarization is mediated by rho and rac but not cdc42 or pi 3-kinases. J Cell Biol. 2003;2003:429–439. doi: 10.1083/jcb.200210135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takesono A, Heasman SJ, Wojciak-Stothard B, Garg R, Ridley AJ. Microtubules regulate migratory polarity through rho/rock signaling in t cells. PloS One. 2010;2010:e8774. doi: 10.1371/journal.pone.0008774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi X, Liu M, Li D, Wang J, Aneja R, Zhou J. Cep70 contributes to angiogenesis by modulating microtubule rearrangement and stimulating cell polarization and migration. Cell Cycle. 2012;2012:1554–1563. doi: 10.4161/cc.19954. [DOI] [PubMed] [Google Scholar]
- 47.Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase a phosphorylation of rhoa mediates the morphological and functional effects of cyclic amp in cytotoxic lymphocytes. EMBO J. 1996;1996:510–519. [PMC free article] [PubMed] [Google Scholar]
- 48.Shibata R, Kai H, Seki Y, Kato S, Morimatsu M, Kaibuchi K, Imaizumi T. Role of rho-associated kinase in neointima formation after vascular injury. Circulation. 2001;2001:284–289. doi: 10.1161/01.cir.103.2.284. [DOI] [PubMed] [Google Scholar]
- 49.Seasholtz TM, Majumdar M, Kaplan DD, Brown JH. Rho and rho kinase mediate thrombin-stimulated vascular smooth muscle cell DNA synthesis and migration. Circ Res. 1999;1999:1186–1193. doi: 10.1161/01.res.84.10.1186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.