

Abstract
Recent advances in genotyping technologies have facilitated genome-wide scans for natural selection. Identification of targets of natural selection will shed light on processes of human adaptation and evolution and could be important for identifying variation that influences both normal human phenotypic variation as well as disease susceptibility. Here we focus on studies of natural selection in modern humans who originated ~200,000 years go in Africa and migrated across the globe ~50,000 – 100,000 years ago. Movement into new environments, as well as changes in culture and technology including plant and animal domestication, resulted in local adaptation to diverse environments. We summarize statistical approaches for detecting targets of natural selection and for distinguishing the effects of demographic history from natural selection. On a genome-wide scale, immune-related genes appear to be major targets of positive selection. Genes associated with reproduction and fertility also appear to be fast evolving. Additional examples of recent human adaptation include genes associated with lactase persistence, eccrine glands, and response to hypoxia. Lastly, we emphasize the need to supplement scans of selection with functional studies to demonstrate the physiologic impact of candidate loci.
Keywords: adaptation, human genetics, population genetics, whole genome sequencing
1. Introduction
1.1. Recent advances in genomics
Recent advances in genotyping technologies and computational power have reduced the cost of obtaining high-quality genomic data. These advances have greatly increased our ability to understand how adaptation has shaped the genome of Homo sapiens. Genotyping technologies include whole genome sequencing (Drmanac et al 2010, Lam et al 2012, Margulies et al 2005, Shendure et al 2005), and microarrays that enable the genotyping of hundreds of thousands to millions of SNPs per sample (Jiang et al 2013). With the onset of affordable sequencing, we see the beginnings of population genomics (Black et al 2001, Goldstein & Weale 2001, Jorde et al 2001, Luikart et al 2003), whereby population genetic analyses can encompass entire genomes (1000 Genomes Project Consortium 2010, Lachance et al 2012). Importantly, genomic datasets allow selection on single genes to be distinguished from demographic processes with genome-wide effects. This review will not focus on the wide variety of genotyping technologies that are available. Instead, it will describe population genetic approaches that can be applied to genome-wide datasets to detect patterns of human adaptation (Figure 1) and shed light on our recent evolutionary history.
Figure 1. Genomic signatures of adaptation.

Positive selection can yield: A) an excess of nonsynonymous fixed differences, B) extended haplotype homozygosity (EHH), C) modified allele frequency distributions (note that the actual effects depend upon whether selective sweeps are still ongoing), and D) long branch length in evolutionary trees.
1.2. Human history
Humans diverged from chimpanzees 6 to 7 million years ago in Africa (Glazko & Nei 2003, Steiper & Young 2006), and notable adaptations in the past few million years include bipedalism, use of fire and tools, and an increase in brain size. Bipedalism was first observed in Australopithecus 4 million years ago, although the timing of this trait is a contentious topic (McHenry 2012). Potential benefits of bipedalism include carrying, new feeding adaptations, and adaptation to habitat changes. Another adaptation, the use of fire, dates back to as early as 800kya (Goren-Inbar et al 2004). Over the past few hundred thousand years our brain size has increased from a mean volume of 445 cm3 based on endocasts in A. afarensis to a mean volume of 1300 cm3 in modern humans, though this change has not been monotonic (Holloway et al 2009).
The earliest fossil and archeological evidence points to the emergence of anatomically modern humans in southern Ethiopia at least 150–190kya (McDougall et al 2005). Humans migrated out-of-Africa 50–100kya (Cavalli-Sforza & Feldman 2003, Klein 2008, Scally & Durbin 2012, Tishkoff et al 2009), and whole genome sequencing of an aboriginal Australian genome suggests that there have been multiple waves of migration out-of-Africa (Rasmussen et al 2011). This out-of-Africa migration had a profound effect on human genetic variation, and as humans expanded to explore the globe, variation was lost due to founder effects and serial population bottlenecks (DeGiorgio et al 2009).
Because of this demographic history, less genetic variation is found in non-African populations than African populations (McEvoy et al 2011, Tishkoff et al 2009). As humans expanded to cover the earth our ancestors encountered new environments and pathogens, potentially leading to many novel adaptations. However, conditions within Africa have also changed over the last hundred thousand years (Reader 1997) and evolution continues to shape the genomes of all human populations. Recent examples of human adaptation include lactose tolerance, vitamin D and skin pigmentation, immune function, and traits that are beneficial at high altitude (Bersaglieri et al 2004, Fumagalli et al 2011, Grossman et al 2013, Jablonski 2012, Tishkoff et al 2007, Yi et al 2010).
Multiple genome-enabled studies have recently examined whether there has been archaic admixture between ancient humans and other hominins, including Neanderthals. Whole genome sequences of Neanderthal and human genomes suggest that introgression occurred between Neanderthals and the ancestors of present-day Europeans and Asians, but not the ancestors of sub-Saharan Africans (Green et al 2010). Furthermore, interbreeding appears to have occurred between the ancestors of present-day humans living in Papua New Guinea and Denisovans (Reich et al 2010). Highly-divergent haplotypes have also been observed in African genomes, suggesting that archaic admixture with an unknown population may have occurred within Africa (Hammer et al 2011, Lachance et al 2012). Taken together, these findings present a complex picture of hominin evolution and population structure in which multiple populations have diverged over the last few hundred thousand of years only to exchange genes upon secondary contact (Lalueza-Fox & Gilbert 2011). These archaic introgression events can potentially involve alleles that were locally adapted.
Human environments and subsistence patterns have changed substantially during the past 10,000 years (Diamond 1997). Agriculture developed independently at multiple locations (including the fertile crescent, East Asia, and the Americas) 10,000 to 13,000 years ago (Vasey 1992). Multiple animal domestication events occurred around the same time, with evidence of pastoralism in North Africa and West Asia dating to around 10,000 years ago (Hanotte et al 2002, Loftus et al 1999). Due to these changes in subsistence patterns a mismatch may exist between our genomes and present-day environments. This results in what have been called diseases of modernity. One example of a disease of modernity is type 2 diabetes, and in the early 1960’s James Neel proposed the “thrifty genotype hypothesis” as an evolutionary explanation for why diabetes is so common (Neel 1962). This hypothesis suggests that alleles which enable individuals to deposit fat during periods of food abundance may have been adaptive to early hunter-gatherers (Neel 1962).
A secondary effect of these lifestyle changes has been the growth of cities and substantial increases in population densities. These changes have important implications for emerging infectious diseases (Dobson & Carper 1996, Jones et al 2008). Furthermore, as humans encounter new environments our genomes must adapt to novel challenges such as zoonotic diseases. Because environments and selection pressures differ from population to population, much of recent human adaptation may involve local adaptation.
2. Methods of detecting human adaptation
2.1. Comparative genomics
When genetic data are available from multiple species, a number of approaches exist for detecting natural selection. These approaches include calculating dN/dS ratios, using the McDonald-Kreitman test, and identifying conserved genomic regions. In addition to the genomes of outgroups such as chimpanzees (Auton et al 2012), resources for examining human genomic data include the encyclopedia of DNA elements (ENCODE) which classifies functional regions in the genome including regulatory elements (Dunham et al 2012), a comprehensive database of human genetic variation (dbSNP), a web-based tool for exploring signatures of selection in human genome diversity panel (HGDP) data (http://hgdp.uchicago.edu/cgi-bin/gbrowse/HGDP/), and an accessible genome browser (http://genome.ucsc.edu). One additional benefit of working with human data is that the human genome is relatively well annotated.
The dN/dS ratio is a widely used statistic that compares the number of non-synonymous substitutions (dN) to the number of synonymous substitutions (dS) (Yang & Bielawski 2000). Empirical dN/dS ratios from human data are < 1 for many genes, suggesting that many proteins are under purifying, rather than positive, selection (Zhang & Li 2005). However, because dN/dS ratios tend to be calculated across an entire gene it is possible for a single adaptive change to be dwarfed by the signatures of purifying selection acting on other sites. One solution is to allow dN/dS ratios to vary across sites and lineages, and this approach has been used to find evidence of positive selection in the BRCA1 tumor suppressor gene in primates (Yang & Nielsen 2002). dN/dS calculations can be also improved by using codon-based, as opposed to nucleotide-based, substitution models (Goldman & Yang 1994). A popular software package that incorporates codon-based substitution models and allows dN/dS ratios to vary across sites is PAML (Yang 2007). One limitation of dN/dS ratios is that they are restricted to analysis of protein-coding genes. Another limitation is that dN/dS ratios are relatively insensitive to the strength of selection when samples are drawn from a single population under selection (Kryazhimskiy & Plotkin 2008).
The McDonald-Kreitman (MK) test uses polymorphism data from a single species and fixed differences between multiple species to detect regions under selection (McDonald & Kreitman 1991). The McDonald-Kreitman test compares the ratio of nonsynonymous to synonymous variation within and between species (such as humans and chimpanzees). This acts as an internal control for differences in mutation rate amongst loci, and allows positive selection to be differentiated from lack of constraint. Although the McDonald-Kreitman test is traditionally used to examine protein-coding genes, it can be extended to other types of sequences, including regulatory DNA (Andolfatto 2005). The principle behind this test is that neutrally evolving genes are expected to show similar patterns within and between species.
Using McDonald-Kreitman tests, 9% of informative human genes show evidence of rapid amino acid substitution, and positively selected genes are enriched for functional categories that include apoptosis, gametogenesis, natural killer cell-mediated immunity, and sensory perception (Bustamante et al 2005). Additional studies have used McDonald-Kreitman tests to detect balancing selection in the malaria-resistance gene G6PD (Verrelli et al 2002), a gene involved in color perception OPN1LW (Verrelli & Tishkoff 2004), and selection related to Lassa fever in the genes IL21 and LARGE (Andersen et al 2012).
Relative levels of sequence conservation can also be used to identify targets of selection. Purifying selection results in conservation of functional genomic regions (Huang et al 2004) and adaptation results in a lack of conservation. If a genomic region is conserved in many related species but divergent in another it may imply that species-specific adaptation has occurred. However, an alternative explanation is that lack of sequence conservation has little to do with positive selection and instead involves pseudogenization and/or relaxation of constraint. Evolutionary conservation can be measured by GERP scores (Cooper et al 2005) and information-theory statistics (Capra & Singh 2007).
Once genomic regions with relaxed constraint in the human lineage are identified it is important to determine whether sequence changes lead to any functional differences. The bioinformatics tools SIFT and PolyPhen-2 can be used to assess putative functional consequences of mutations that result in amino-acid changes. SIFT classifies mutations as “tolerated” or “damaging” (Fu 1997), and PolyPhen-2 classifies mutations as “benign”, “possibly damaging”, or “probably damaging” (Beleza et al 2013). However, bioinformatics approaches to examine the function of adaptive loci are only the beginning, and the gold-standard of functional genetics remains in vivo experiments using mouse models and/or human cell lines.
2.2. Haplotype approaches
Due to linkage, the genetic signatures of natural selection extend beyond the loci that are actually selected. Specifically, genetic variation is reduced near targets of selection and linked SNPs are more likely to be homozygous. This reduction in variation due to positive selective sweeps has been called genetic hitchhiking (Smith & Haigh 1974). Haplotype statistics such as EHH and iHS use data from linked sites to identify past targets of natural selection based on the expectation that recent positive sweeps yield extended haplotypes that recombination has not had enough time to break down. These statistics are particularly well suited to detect selection that has occurred in the last 10,000 years (Sabeti et al 2002).
In 2002 Sabeti et al. introduced the extended haplotype homozygosity (EHH) statistic (Sabeti et al 2002). This statistic measures the probability that two randomly chosen chromosomes containing a core haplotype are homozygous (and identical by descent) for an extended interval extending out from the core region. Positive selection results in a high frequency haplotype that has higher EHH compared to other core haplotypes in a given genomic region. Statistical significance can then be found by simulating neutral haplotypes using a coalescent process. Using the EHH statistic, haplotypes at the G6PD and TNFSF5 genes were found to have signatures of positive selection (Sabeti et al 2002). Both of these genes confer resistance to the malarial parasite Plasmodium falciparum.
Building on the EHH statistic, Voight et al. developed the integrated haplotype score (iHS) (Voight et al 2006). This statistic compares the area under EHH vs. genetic distance curves for ancestral and derived alleles at a core locus. In neutral models low frequency alleles tend to be younger and associated with long haplotypes. Because of this, low frequency derived alleles that are associated with long haplotypes need not be indicative of adaptation. The iHS statistic controls for this effect by comparing extended haplotype curves of core SNPs that have the same allele frequency (Voight et al 2006). This standardized iHS score requires genome-wide data. The iHS statistic has greater statistical power to detect sweeps than Tajima’s D and Fay and Wu’s H (see section 2.3) for SNPs at low and intermediate frequency (Voight et al 2006). Using iHS statistics, a genome-wide scan of selection found evidence for a selective sweep near the muscle gene MYPN in Ju’/hoansi hunter-gatherers (Schlebusch et al 2012).
One limitation of haplotype statistics is that recombination hotspots can obscure signals of positive selection. A high quality recombination map (deCODE) has been generated from pedigree data of Icelandic individuals (Kong et al 2002), but it is unclear to what extent this fine-scale map also applies to other human populations (Hinch et al 2011). Furthermore, genes such as PRDM9 are known to influence recombination rates in humans (Kong et al 2010). A second limitation is that haplotype statistics have little power to detect soft sweeps, i.e. adaptation from standing genetic variation (Pennings & Hermisson 2006). When soft sweeps occur, adaptive alleles are found on multiple haplotype backgrounds. Because of this, a core genetic region need not have extended haplotype homozygosity. Population genetic theory suggests that soft sweeps may be widespread, especially for genes with large mutational target sizes, and present methods have low power to detect adaptation from standing variation (Pritchard et al 2010).
2.3. Allele frequency distributions
The allele frequency distribution, sometimes called the “site frequency spectrum”, summarizes the proportion of individuals that have SNPs at a particular frequency. Adaptation modifies allele frequency distributions, and these shifts in allele frequency distributions can be detected with population genetic data; loci under selection are likely to have allele frequency distributions that differ from neutral loci. Comparisons can be made between specific loci and either randomly sampled genomic regions or neutral expectations from theory.
A variety of summary statistics describe allele frequency distributions. These statistics include Tajima’s D, Fay and Wu’s H, and Li’s MFDM test. Tajima’s D compares the number of variants in a genomic region (using Watterson’s θ ) with the average number of pairwise differences (π) (Tajima 1989). Under neutral expectations both of these quantities are equal, yielding Tajima’s D = 0. When there is an excess of rare variants (such as when a genomic region has undergone a sweep in the recent past) Tajima’s D is negative. By contrast, when there is an excess of intermediate variants (such as when balancing selection is present) Tajima’s D is positive. Importantly, population size changes also change allele frequency distributions: expanding populations have an excess of rare variants and bottlenecked populations have a lack of rare variants. Because of this, demographic changes can cause neutral loci to have D ≠ 0. Thus, it is important to compare values of Tajima’s D to genome-wide distributions from the same population to identify putatively adaptive genomic regions. In addition, the presence of a rare haplotype in a single individual can lead to low values of Tajima’s D (Lachance et al 2012). One solution is to bootstrap individuals to identify genomic regions with low values of Tajima’s D (i.e. putatively adaptive regions) that are robust to data from a single individual.
Fay and Wu’s H uses genetic data from an outgroup such as Pan troglodytes to identify ancestral and derived alleles (Fay & Wu 2000, Zeng et al 2006). In neutral models the majority of derived alleles are expected to be rare. However, if a positive sweep has recently occurred then linked derived alleles are likely to have elevated frequencies due to genetic hitchhiking. Building upon this idea, Fay and Wu’s H is based on the observation of an excess of high frequency derived alleles over neutral expectations to detect signals of positive selection. However, population bottlenecks can also result in an excess of high frequency derived alleles (Jensen et al 2005). Another allele frequency statistic that can be used to detect adaptive regions of the genome is Li’s maximum frequency of derived mutations (MFDM) test (Li 2011). The MFDM test detects unbalanced gene trees and is relatively robust to population subdivision.
While summary statistics of allele frequency data can be informative, they involve a loss of information and an alternative approach is to use the entire allele frequency distribution. One method that utilizes the full allele frequency distribution is the Poisson Random Field (PRF) approach (Sawyer & Hartl 1992). PRF theory assumes an infinite-sites model with unlinked mutations that follow a Poisson process (Sethupathy & Hannenhalli 2008). In PRF models the distribution of sample allele frequencies is expressed as a function of mutation rate, effective population size, and scaled selection coefficients. Maximum likelihood can then be used to estimate parameter values, including the proportion of adaptive loci and the strength of selection (Nielsen et al 2005). The full set of allele frequencies can also be used to infer the distribution of fitness effects (Eyre-Walker & Keightley 2009). This distribution is used in turn to estimate the proportion of adaptive substitutions between species (Eyre-Walker & Keightley 2009). This approach is also able to account for the effects of demography on allele frequency distributions, including changes in population size.
One complication of using allele frequency data to detect signals of human adaptation is that large sample sizes are required; otherwise it is difficult to identify highly informative rare alleles. Also, high genotyping accuracy is required, as singletons can be due to genotyping error. Because of this, it is advisable to exclude singletons from allele frequency analyses if low-coverage whole genome sequencing is used.
2.4. Data from multiple populations
Allele frequency data from multiple human populations can be leveraged to detect adaptive loci. One benefit of this approach is that loci with small effects can be identified if enough time has passed for allele frequencies to differ substantially across populations (Stapley et al 2010). FST quantifies allele frequency differences between populations, and a classic method involves calculating the variance in FST across loci (Lewontin & Krakauer 1973). The variance in FST is expected to be larger for selected genes than neutral genes. The Lewontin-Krakauer test of variance in FST was initially met with criticism (Nei & Maruyama 1975, Robertson 1975). However, in recent years there has been resurgence in the use of FST-based approaches to identify locally adapted alleles (Narum & Hess 2011, Ronald & Akey 2005).
These approaches use genome-wide FST calculations to identify outlier SNPs. Genes that are enriched for high FST SNPs are then classified as putatively adaptive. So long as selection coefficients are at least five times the migration rate, FST approaches can detect adaptive loci (Beaumont & Balding 2004). One limitation of pairwise FST approaches is that they do not always reveal the population in which selection has occurred: large allele frequency differences can be due to adaptation in either population. Another method that uses allele frequency data from multiple populations is the XP-CLR test, or cross-population composite ratio test (Chen et al 2010). Here, allele frequency differences between populations are compared to null expectations from a neutral model.
An alternative approach is to use evolutionary trees and allele frequency data from greater than two populations. By using data from multiple populations, it is possible to identify genomic regions that have particularly long branch lengths in a single population. If only three populations are analyzed, a single unrooted population tree can be used. However, if data from greater than three populations are analyzed the population tree topology must already be known. Statistics that quantify relative branch lengths have been called locus-specific branch lengths (LSBL) (Shriver et al 2004) and population branch statistics (PBS) (Yi et al 2010). These statistics can then be calculated for each polymorphic site in genome-wide datasets to identify adaptive loci. Using LSBL statistics and SNP data from the International HapMap Project, signals of positive selection were found for SLC24A5 and other pigmentation genes (McEvoy et al 2006).
However, once genomic regions that are enriched for LSBL/PBS outliers have been found, assigning statistical significance remains a challenge. This is because the null expectations of these statistics are unknown. One option is to assume a particular model of demographic history and use coalescent simulators such as ms or SIMCOAL to generate neutral expectations of test statistics (Hudson 2002, Laval & Excoffier 2004). Empirical data can then be compared to neutral expectations to identify statistically significant outliers. An alternative option to determine statistical significance is to bootstrap data across individuals and/or loci.
2.5. Integration with environmental data
Correlations between allele frequencies and environmental variables can also be used to identify putatively adaptive loci. When selective pressures differ between geographic regions allele frequency differences between populations can be maintained in the face of gene flow. One issue is that allele frequencies of neighboring populations are not independent, and it is important to control for similarity based on isolation by distance (Coop et al 2010). A neutral null model is built using the covariance structure of alleles across populations. Bayes factors are then used to measure the support for the effect of environmental variables on individual SNPs compared to the neutral covariance matrix alone (Coop et al 2010).
This approach has been use to examine human adaptation to climate (Hancock et al 2011) and diet (Hancock et al 2010). Potential adaptations to climate include mutations in genes that are involved in response to UV radiation and thermoregulation (Hancock et al 2011). Potential dietary adaptations include a SNP in the MTRR gene that is associated with tuber-rich diets and a SNP in the pancreatic PLRP2 gene that is associated with use of cereals as a primary dietary component (Hancock et al 2010). Two complications that arise from this approach are that past environments may differ from present-day environments and geographic ranges of populations change over time. As the goal is to identify adaptations that occurred in the past, use of historical environmental data is preferable to use of present day environmental data. However, high quality historical data is not always available.
2.6. Phenotypes and QST-FST comparisons
Adaptive traits are more likely to differ between populations than neutral genetic markers, and QST-FST comparisons allow phenotypic differences to be compared to genetic differences. QST is the quantitative genetic analog of FST, and it measures the proportion of genetic variation in a trait that is due to differences among populations (Spitze 1993). QST = FST when trait differences between populations are due to genetic drift, QST > FST is indicative of directional selection that varies by population, and QST < FST is indicative of stabilizing selection acting in multiple populations. It is important that QST be compared to the full distribution of FST (Whitlock 2008), and, thus, genome-wide data is required.
One limitation of QST-FST comparisons is that calculation of QST requires familial data and minimal environmental effects (Leinonen et al 2013). When full-sib and half-sib data is not available it is possible to use purely phenotypic data (PST) in comparisons with FST. However, PST-FST comparisons lack the genetic rigor of QST-FST comparisons (Leinonen et al 2013). To date, most QST-FST comparisons involve studies of non-human adaptation (Leinonen et al 2013). One exception is the analysis of gene expression differences between Europeans and African-Americans (Price et al 2008). The application of QST-FST comparisons to future studies of human adaptation is likely to be fruitful.
2.7. Integrating multiple signatures of adaptation
Multiple signals of selection can be integrated by using a composite of multiple signals (CMS) approach (Grossman et al 2010). Results from different tests of selection (including FST, allele frequency, and haplotype approaches) are combined to yield CSMs scores. These CMS scores give the approximate posterior probability that a variant has been selected. Computer simulations indicate that the CMS approach is superior to single tests with respect to identifying causal variants and localizing signals of selection within fine genomic regions (Grossman et al 2010). Application of CMS to data from the 1000 Genomes Project led to the identification of numerous variants that are associated with infectious disease, including a nonsynonymous variant in the Toll-like receptor 5 (TLR5) gene (Grossman et al 2013).
2.8. Use of genomic data
Genomic, as opposed to genetic, data offers multiple advantages in detecting human adaptation. First, genome-wide datasets allow broad-scale patterns to be identified. This includes comparisons between coding and non-coding regions of the human genome (1000 Genomes Project Consortium 2010) and continental patterns of selection (Lohmueller et al 2008). Second, genome-wide datasets allow single genes to be placed in a broader context. Here, genome-wide data from putatively neutral sites can act as empirical baselines for demographic processes. For example, standardized versions of the iHS statistic require genome-wide data to distinguish regions that are outliers (Voight et al 2006). Third, genome-wide datasets also allow novel adaptive loci to be identified, including SNPs in non-coding DNA. By contrast, focused studies of candidate genes are less likely to result in the discovery of novel signatures of adaptation.
An additional benefit of using genomic data is that it is relatively free of ascertainment bias. SNP ascertainment bias refers to the systematic deviation of population genetic statistics from theoretical expectations. These deviations can arise from analyzing SNPs that have been ascertained in populations other than the one being studied (Clark et al 2005, Nielsen 2004). Use of unascertained data is particularly important for methods that use allele frequency and FST data. This is because ascertained SNPs are more likely to be older SNPs that have larger allele frequency differences between populations (Lachance & Tishkoff 2013).
One complication that arises from genome-wide datasets is the need to correct for multiple tests. Genotyping arrays and whole genome sequencing can yield data at more than a million SNPs, meaning that a p-value threshold of 0.05 will result in many false positives. A number of ways to correct for multiple tests of adaptation exist, including Bonferroni corrections and Benjamini-Hochberg false-discovery rates (Benjamini et al 2001, Johnson et al 2010, Purcell et al 2007). In human genetics, many genome-wide association studies (GWAS) correct for multiple tests by using a p-value threshold of 5 × 10−8 (Hindorff et al 2009). However, this threshold is arbitrary and it is best-suited for genotyping arrays that contain ~1 million SNPs. It is also important to note that genomic regions with the lowest p-values need not be regions that contain alleles with the largest effect size.
3. Examples of recent human adaptation
3.1. General patterns
On a genome-wide scale, natural selection in humans seems to be driven more by pathogen pressure than by adaptation to diet or climate. Using over 500,000 ascertained SNPs and genotype data from 55 distinct HGDP and HapMap populations, allele frequency distributions were analyzed and compared to a number of environmental variables (Fumagalli et al 2011). Although demography explained > 95% of the genetic variance among populations in this study, over 100 genes show a strong correlation between allele frequencies and pathogen pressure after correcting for demography. In addition, a recent study of polygenic adaptation found that pathways that are enriched for signals of adaptation tend to be involved in immune function (Daub et al 2013). These pathways include the IL-6 signaling pathway and cytokine-cytokine receptor interaction (Daub et al 2013).
Genes involved in reproduction and gametogenesis also appear to be evolving quickly in the human lineage. For example, dN/dS ratios for genes involved in spermatogenesis and the protamine gene complex show evidence of positive selection in humans (Torgerson et al 2002, Wyckoff et al 2000). Similarly, whole genome sequencing of 15 African hunter-gatherers revealed short (<10 kya) coalescence times for the sperm associated antigen 11 gene SPAG11A (Lachance et al 2012). Sexual selection facilitates the fast evolution of reproductive genes. Another reason why reproductive genes can evolve quickly is that many genes associated with reproductive traits have sex-specific and/or tissue-specific expression patterns, and are less likely to be under evolutionary constraint (Grassa & Kulathinal 2011).
An additional pattern observed in genome-wide scans of selection is that there is only a limited amount of overlap between signals of adaptation for populations on different continents (Pickrell et al 2009). It appears that local adaptation might be the norm (see Figure 2 for examples). However, because most scans of selection have greater power to detect recent selective seeps (i.e. < 10 kya) and humans expanded out of Africa 50–100 kya, the relative paucity of global signals of adaptation may be due to methodological limitations.
Figure 2.
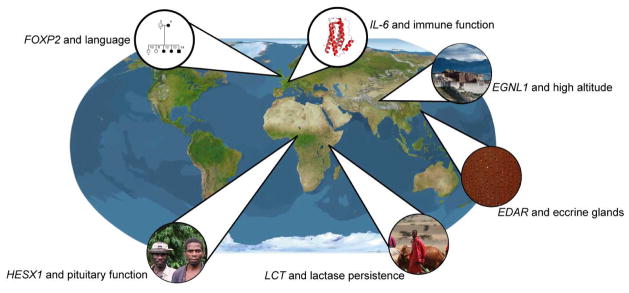
Genes associated with human adaptation
One contentious issue is the extent to which the human genome has been shaped by adaptive evolution. Using human polymorphism data and fixed differences between humans and chimpanzees, 304 out of 3374 (9%) informative loci appear to be positively selected (Bustamante et al 2005). Focusing on 50 genes with strong signals of positive selection, Nielsen et al. used a Poisson Random Field approach to conclude that 8% of mutations in these genes have been positively selected in the human lineage (Nielsen et al 2005). However, because this study focused on genes with particularly strong signals of selection, the 8% figure should be viewed as an upper bound. By contrast, evolutionary analysis of 182 housekeeping and 148 tissue-specific genes suggests that positive selection in the human genome may not be common (Zhang & Li 2005). Although many genomic regions might be linked to adaptive variants, the actual proportion of functionally adaptive sites can be very small.
3.2. Local adaptation among African hunter-gatherers
There have been multiple studies of selection in diverse African hunter-gatherers, such as central African Pygmies, Eastern African Hadza and Sandawe who speak languages that contain clicks classified as “Khoisan”, and southern African San populations who also speak languages classified as Khoisan. A recurring theme is that the genomes of each hunter-gatherer population have unique signatures of selection (Granka et al 2012, Lachance et al 2012). This observation is consistent with deep divergence times (>20 kya, dating back before the origin of agriculture) and unique diets, environment, and pathogen pressures for each hunter-gatherer population.
Using genome-wide SNP arrays and iHS statistics, a recent study of 220 individuals from southern Africa found strong evidence of selection near the major histocompatibility complex on chromosome 6 (Schlebusch et al 2012). This study also revealed signals of selection that overlap a gene involved in muscle growth (MYPN) in Ju’/hoansi hunter-gatherers, and allele frequencies at a gene associated with “fast-twitch” muscles (ACTN3) differ between Khoe-San populations and other African populations (Schlebusch et al 2012). Another recent study used a set of 500,000 SNPs to detect signals of adaptation in hunter-gatherer and other African populations (Granka et al 2012). Although signatures of local and shared adaptation were found, allele frequency differences between populations appear to be driven mainly by neutral demographic processes.
High-coverage whole genome sequences of African hunter-gatherers (Pygmies, Hadza, and Sandawe) have also been analyzed for signals of local adaptation (Lachance et al 2012). Evolutionary analysis of Pygmy genomes reveals signatures of selection in genes involved in olfactory transduction, metabolism, and spermatogenesis. Putatively adaptive regions of Pygmy genomes also overlap genes involved in pituitary function, including a homeobox gene that is statistically associated with short stature (HESX1) in African Pygmies (Lachance et al 2012). Hadza genomes contain selective signals that overlap genes involved in regulation of immune response (IL18R1/IL18RAP) and wound healing (VWF). Sandawe genomes contain signals of adaptation in the major histocompatibility complex and zona pellucida binding protein.
Interestingly, there appears to be minimal overlap between signals of selection between the Hadza and Sandawe, despite geographic proximity (~150km) and both populations having languages that contain clicks (Lachance et al 2012). One observation from these studies is that depending on genotyping technology and statistical approaches, different genes can be implicated. This illustrates the need for replication and follow-up studies that examine the functional biology of putatively adaptive regions of the genome.
3.3. EDARV370A and eccrine sweat glands
One particularly strong signal of selection in genome-wide scans of human adaptation involves the Ectodyspasin receptor gene EDAR in Asian populations (Grossman et al 2010, Sabeti et al 2007). The putatively adaptive haplotype of the EDAR gene includes a valine to alanine change (EDARV370A). Using approximate Bayesian computation (Beaumont et al 2002), the EDARV370A mutation appears to have originated approximately 30kya in central China (Kamberov et al 2013). In humans, mutations in the EDAR gene are associated with increased hair thickness and changes in tooth morphology (Fujimoto et al 2008, Park et al 2012). The EDARV370A mutation is also associated with mammary gland branching and fat size in transgenic mouse models and increased eccrine gland number in humans and mice (Kamberov et al 2013). Eccrine glands are the major sweat glands in humans, and they are essential for thermoregulation. Because EDAR is pleiotropic, it is unclear which trait(s) selection acted upon.
3.4. Lactase persistence
One of the best examples of local adaptation is the genetic basis of the lactase persistence (LP) trait (i.e. expression of the lactase enzyme in adults). In most humans the ability to digest the complex sugar lactose, present at high frequency in milk, decreases rapidly after weaning. However, anthropologists have long noted a correlation between the prevalence of the LP trait and the cultural practice of dairying and cattle domestication (Durham 1992). Thus, the LP trait is most common in northern Europe and decreases in frequency in central and southern Europe and the Middle East. It is also at low prevalence in East Asians, Native Americans, and most West Africans who practice agriculture. However, the LP trait is common in African pastoralist populations such as the Maasai.
The ability to digest milk as adults, due to persistence of lactase gene expression is a cis-regulated genetic trait. A mutation associated with LP (T-13010), located ~13 kb upstream of the lactase gene (LCT) in an intron of the neighboring MCM6 gene, was initially identified in Europeans (Enattah et al 2008). Several additional SNPs (C-14010, G-13915, G-13907) that are associated with the LP trait in Africa and the Middle East and that enhance transcription from the LCT promoter in vitro were identified (Ingram et al 2007, Ranciaro et al 2013, Tishkoff et al 2007). One LP-associated variant (C-14010) is most common in Tanzanian and Kenyan populations that herd cattle whereas the other two (G-13915, G-13907) are common in northern Sudan and Kenya. Genotyping of 123 SNPs across a 3 Mb region in these populations demonstrated that these African LP-associated mutations exist on haplotype backgrounds that are distinct from the European LP-associated mutation and from each other (Tishkoff et al 2007).
Chromosomes containing the LP-associated mutation most common in Kenya and Tanzania (C-14010) have extended haplotype homozygosity > 2 Mb, consistent with an ongoing selective sweep over the past 3,000–7,000 years (Tishkoff et al 2007). By contrast, chromosomes containing the European LP variant have extended haplotype homozygosity > 1 Mb, consistent with an origin of the selective sweep within the past 9,000 years (Bersaglieri et al 2004, Tishkoff et al 2007). These data indicate a striking example of convergent evolution and local adaptation due to strong selective pressure resulting from shared cultural traits (e.g. cattle domestication and adult milk consumption) in Europeans and Africans.
3.5. FOXP2 and language
Forkhead Box P2 is a highly conserved transcription factor that is involved in speech and language development and the FOXP2 gene appears to have undergone positive selection in recent human evolution. Speech and language problems often have a hereditary component, and pedigree data from the KE family in Britain have been particularly informative. Mutations in FOXP2 result in impaired pronunciation, grammar, and language comprehension (Vargha-Khadem et al 2005).
Comparative genomics analysis reveals that the human FOXP2 gene differs from that of other primates at two nonsynonmyous sites: T303N and N325S (Enard et al 2002). Accelerated evolution of FOXP2 in the human lineage opens up the possibility that this gene may have been important for the evolution of language (Zhang et al 2002). Furthermore, allele frequency distributions of FOXP2 targets show signatures of positive selection in Europeans (Ayub et al 2013). Intriguingly, the Neanderthal FOXP2 gene shares these two substitutions with H. sapiens. This suggests that both mutations may have been fixed >300kya (Krause et al 2007). However, near the human FOXP2 gene there is an excess of high-frequency derived alleles and minimal haplotype diversity. These patterns, coupled with potential gene flow between ancient humans and Neanderthals, suggest that selection may have occurred as recently as 42kya (Coop et al 2008).
3.6. Adaptation to high-altitude
Life at high altitude presents a number of physiological challenges and evidence of adaptation to high altitude has been found in populations from the Tibetan Plateau, the Andean Altiplano, and the Ethiopian Highlands. Reduced atmospheric pressure at high altitude contributes to hypoxia and potential complications of hypoxia include pulmonary hypertension (Scheinfeldt & Tishkoff 2010). In addition, pregnant women living at high altitude have increased risk of pre-eclampsia (Moore et al 2004).
Not surprisingly, genomic signatures of adaptation to high altitude have been found in a number of blood-related genes. Haplotype statistics of Tibetans reveal signatures of selection in EGLN1, a gene associated with hemoglobin levels (Simonson et al 2010), and allele frequency differences between Tibetans and Han Chinese implicate EPAS1, a gene that encodes a transcription factor involved in response to hypoxia (Yi et al 2010). However, genome-wide SNP data reveals that the hypoxia-inducible transcription factor (HIF) pathway is not over-represented for signatures of selection in Native Americans of Andean descent (Bigham et al 2009). Many SNPs with large allele frequency differences between high altitude Amhara and low altitude Oromo populations in Ethiopia are found in genes involved in pathogen defense rather than hypoxia (Alkorta-Aranburu et al 2012).
Combining genome-wide SNP screens for genes that are targets of natural selection and looking for correlations between variation at these genes and phenotypic data, another study found associations between the genes THRB and ARNT2 and hemoglobin levels in the Ethiopian Highlands (Scheinfeldt et al 2012). The physiological basis of adaptation to high altitude also varies across populations. For example, oxygen saturation profiles of Ethiopians differ from that of Andean and Tibetan populations (Beall 2006). Because the genetic architecture of adaptive traits can range from single genes of large effect to multiple genes of small effect, it is beneficial to use hierarchal scans of selection that analyze both individual genes and pathways.
4. Additional considerations
4.1. Uniquely human adaptation?
Humans have a relatively low effective population size compared to chimpanzees (Chen & Li 2001). From the nearly neutral theory of evolution, we know that the efficacy of selection depends on the magnitude of Ne × s, where Ne is the effective populations size and s is the selection coefficient. This means that a greater proportion of our genome is likely to have evolved neutrally compared to chimpanzees. Indeed, there is evidence from dN/dS ratios that a greater number of genes underwent positive selection in the chimpanzee lineage than the human lineage (Bakewell et al 2007).
Lower effective population sizes also influence the amount of slightly deleterious mutants that are segregating. If environmental conditions change, it is possible for the previously deleterious alleles to become adaptive. Effective population sizes also vary across human populations (Ne is smaller in non-African populations due to founder effects and serial bottlenecks). One implication of these Ne differences is that the relative number of nonsynonymous changes that result in potentially damaging amino acid changes in non-African genomes is greater than those observed in African genomes (Lachance et al 2012, Lohmueller et al 2008).
Comparative genomics can be used to identify genomic regions that are conserved among other mammals, but fast evolving in humans. Using this approach, 49 human accelerated regions (HARs) were found (Pollard et al 2006). The most dramatically changed element, HAR1, lies within two non-coding RNA genes that are expressed in the developing neocortex of the human brain, HAR1F and HAR1R (Pollard et al 2006). However, HAR1F and HAR1R are also expressed in ovaries and testes of adult humans, and it is possible that sexual selection has driven the accelerated evolution of this genomic region (Ponting & Lunter 2006).
In addition to genetic adaptation, culture has been important to our recent evolutionary history. However, humans are not unique among animals in having a complex culture. An intriguing question is whether human culture has been the leading or lagging factor relative to genetic evolution (Richerson et al 2010). However, answering this question requires accurate dating of adaptation events, something that is not always available. One aspect of culture with evolutionary implications is family size. A study of Quebecois range expansion in Saguenay Lac-Saint-Jean from 1686–1960 revealed that significantly greater numbers of offspring were likely for couples living at front of the advancing wave of colonization (Moreau et al 2011).
4.2. Dangers of adaptive storytelling
A major risk of scans of selection is that many outlier loci might not actually be adaptive. Gould and Lewontin famously described a “Panglossian Paradigm” whereby evolutionary biologists immediate jump to adaptive explanations (Gould & Lewontin 1979). An entertaining, if sobering, illustration of this can be found in a recent study that examines the limitations of functional annotation (Pavlidis et al 2012). Here, a neutrally evolving population of Drosophila melanogaster was simulated. Whole-genome scans of selection were conducted on this simulated dataset and outlier loci were analyzed using Gene Ontology enrichment software. The authors were then able to create a convincing narrative of a supposedly selected locus (Pavlidis et al 2012). Looking at empirical data, a meta-analysis of 21 genome-wide scans of selection found no less than 5,110 distinct regions that have been associated with signals of human adaptation (Akey 2009). One cautionary note arising from this meta-analysis is that only 14% of the 5,110 putatively adaptive regions were implicated in more than one study (Akey 2009). Replication of findings, preferably with functional data, is needed. However, even if studies can be replicated and functional effects of alleles are identified, for a variant to be adaptive it must be able to modify the fitness of an individual.
In some cases demographic history can yield patterns that mimic selection. One example of this is a phenomenon known as “allelic surfing.” Here, some mutations that arise during a range expansion are able to surf to high frequencies as they are propagated along a wave of advance (Edmonds et al 2004). This rapid increase in frequency makes it appear that the alleles in question are adaptive. When continental patterns of human genetic variation are analyzed, large allele frequency differences between populations can be explained by allele surfing and genetic drift, as opposed to natural selection (Hofer et al 2009).
GC-biased gene conversion can also yield patterns that are reminiscent of positive selection, including long branch lengths in evolutionary trees and elevated dN/dS ratios (Ratnakumar et al 2010). In this recombination-mediated process, AT/GC heterozygotes are more likely to produce gametes carrying G or C than A or T. It is possible to distinguish GC-biased gene conversion from positive selection by analyzing GC-content, taking into account local recombination rates, and examining the width of putative sweeps (Galtier & Duret 2007).
4.3. Relevance to human health and genome-wide association studies
The dangers of adaptive storytelling are particularly acute when it comes to describing human disease genes. It is rare to find a textbook that does not include details about HbS, sickle-cell anemia, and malaria resistance. While the HbS story is a great teaching tool it can falsely lead to the assumption that disease alleles must be adaptive. A more likely paradigm is that many disease variants are neutral or slightly deleterious, and that common disease alleles are able to reach intermediate frequencies because of genetic drift or genetic hitchhiking with other linked alleles. However, it is important to consider that environments change over time and some disease-causing alleles that are common now may have been beneficial in the past (Vasseur & Quintana-Murci 2013). Indeed, a meta-analysis of GWAS data revealed that that disease-associated alleles are enriched for signatures of natural selection (Lachance 2010). Additionally many genes that are targets of selection could have pleiotropic effects, e.g. APOL1 variants that play a role in resistance to trypanosome infection but also increase risk for kidney disease (Genovese et al 2010). Thus, identification of targets of natural selection in the genome could be important for identifying functionally important variation influencing both normal phenotypic variation and disease susceptibility.
5. Conclusion
Genome-wide data from SNP arrays and whole genome sequencing can be coupled to a wide array of statistical techniques to inform our understanding of human history. However, even when scans of selection yield plausible candidate loci, there is a need to supplement scans of selection with functional studies to demonstrate the physiologic impact of candidate loci.
SUMMARY POINTS.
Signatures of adaptation include relative levels of sequence conservation, reduced polymorphism, shifted allele frequency distributions, extended haplotype homozygosity, and large allele frequency differences between populations.
Genome-wide datasets allow selection acting on individual loci to be distinguished from demographic effects acting on entire genomes.
Different human populations have different signatures of selection
Immune genes appear to be under strong positive selection, and multiple genes involved in reproductive traits also appear to be adaptive.
One pitfall of selection scans is that they can yield many false positives and negatives. Ideally, genetic data can be integrated with historical data and follow-up studies to assay the functional consequences of variants. It is also important to demonstrate that putatively adaptive alleles can affect fitness traits.
Acknowledgments
This work was supported by an NIH grant (DP1 ES022577) to Sarah A. Tishkoff and an NIH NRSA postdoctoral fellowship (F32HG006648) to Joseph Lachance. The authors thank Laura Scheinfeldt and other members of the Tishkoff lab for helpful suggestions in the writing of this review.
Footnotes
Disclosure statement
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
References
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM. Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res. 2009;19:711–22. doi: 10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkorta-Aranburu G, Beall CM, Witonsky DB, Gebremedhin A, Pritchard JK, Di Rienzo A. The genetic architecture of adaptations to high altitude in Ethiopia. PLoS Genet. 2012;8:e1003110. doi: 10.1371/journal.pgen.1003110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen KG, Shylakhter I, Tabrizi S, Grossman SR, Happi CT, Sabeti PC. Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever. Philos Trans R Soc Lond B Biol Sci. 2012;367:868–77. doi: 10.1098/rstb.2011.0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P. Adaptive evolution of non-coding DNA in Drosophila. Nature. 2005;437:1149–52. doi: 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- Auton A, Fledel-Alon A, Pfeifer S, Venn O, Segurel L, et al. A fine-scale chimpanzee genetic map from population sequencing. Science. 2012;336:193–8. doi: 10.1126/science.1216872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayub Q, Yngvadottir B, Chen Y, Xue Y, Hu M, et al. FOXP2 Targets Show Evidence of Positive Selection in European Populations. Am J Hum Genet. 2013;92:696–706. doi: 10.1016/j.ajhg.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakewell MA, Shi P, Zhang J. More genes underwent positive selection in chimpanzee evolution than in human evolution. Proc Natl Acad Sci U S A. 2007;104:7489–94. doi: 10.1073/pnas.0701705104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall CM. Andean, Tibetan, and Ethiopian patterns of adaptation to high-altitude hypoxia. Integr Comp Biol. 2006;46:18–24. doi: 10.1093/icb/icj004. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Balding DJ. Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol. 2004;13:969–80. doi: 10.1111/j.1365-294x.2004.02125.x. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Zhang W, Balding DJ. Approximate Bayesian computation in population genetics. Genetics. 2002;162:2025–35. doi: 10.1093/genetics/162.4.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beleza S, Santos AM, McEvoy B, Alves I, Martinho C, et al. The timing of pigmentation lightening in Europeans. Mol Biol Evol. 2013;30:24–35. doi: 10.1093/molbev/mss207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–84. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, et al. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74:1111–20. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigham AW, Mao X, Mei R, Brutsaert T, Wilson MJ, et al. Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum Genomics. 2009;4:79–90. doi: 10.1186/1479-7364-4-2-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black WCt, Baer CF, Antolin MF, DuTeau NM. Population genomics: genome-wide sampling of insect populations. Annu Rev Entomol. 2001;46:441–69. doi: 10.1146/annurev.ento.46.1.441. [DOI] [PubMed] [Google Scholar]
- Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–7. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23:1875–82. doi: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Feldman MW. The application of molecular genetic approaches to the study of human evolution. Nat Genet. 2003;33(Suppl):266–75. doi: 10.1038/ng1113. [DOI] [PubMed] [Google Scholar]
- Chen FC, Li WH. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet. 2001;68:444–56. doi: 10.1086/318206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Patterson N, Reich D. Population differentiation as a test for selective sweeps. Genome Res. 2010;20:393–402. doi: 10.1101/gr.100545.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Bullaughey K, Luca F, Przeworski M. The timing of selection at the human FOXP2 gene. Mol Biol Evol. 2008;25:1257–9. doi: 10.1093/molbev/msn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Witonsky DB, Di Rienzo A, Pritchard JK. Using Environmental Correlations to Identify Loci Underlying Loci Adaptation. Genetics. 2010;185:1411–23. doi: 10.1534/genetics.110.114819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub JT, Hofer T, Cutivet E, Dupanloup I, Quintana-Murci L, et al. Evidence for Polygenic Adaptation to Pathogens in the Human Genome. Mol Biol Evol. 2013 doi: 10.1093/molbev/mst080. [DOI] [PubMed] [Google Scholar]
- DeGiorgio M, Jakobsson M, Rosenberg NA. Out of Africa: modern human origins special feature: explaining worldwide patterns of human genetic variation using a coalescent-based serial founder model of migration outward from Africa. Proc Natl Acad Sci U S A. 2009;106:16057–62. doi: 10.1073/pnas.0903341106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JM. Guns, germs, and steel : the fates of human societies. New York: W.W. Norton & Co; 1997. p. 480. 32 p. of plates. [Google Scholar]
- Dobson AP, Carper ER. Infectious Diseases and Human Population History. BioScience. 1996;46:115–26. [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham W. Coevolution, Genes, Culture, and Human Diversity. Stanford, CA: Stanford University Press; 1992. Cultural mediation: The evolution of adult lactose absorption; pp. 226–85. [Google Scholar]
- Edmonds CA, Lillie AS, Cavalli-Sforza LL. Mutations arising in the wave front of an expanding population. Proc Natl Acad Sci U S A. 2004;101:975–9. doi: 10.1073/pnas.0308064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard W, Przeworski M, Fisher SE, Lai CS, Wiebe V, et al. Molecular evolution of FOXP2, a gene involved in speech and language. Nature. 2002;418:869–72. doi: 10.1038/nature01025. [DOI] [PubMed] [Google Scholar]
- Enattah NS, Jensen TG, Nielsen M, Lewinski R, Kuokkanen M, et al. Independent introduction of two lactase-persistence alleles into human populations reflects different history of adaptation to milk culture. Am J Hum Genet. 2008;82:57–72. doi: 10.1016/j.ajhg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evol. 2009;26:2097–108. doi: 10.1093/molbev/msp119. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–13. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Ohashi J, Nishida N, Miyagawa T, Morishita Y, et al. A replication study confirmed the EDAR gene to be a major contributor to population differentiation regarding head hair thickness in Asia. Hum Genet. 2008;124:179–85. doi: 10.1007/s00439-008-0537-1. [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Sironi M, Pozzoli U, Ferrer-Admetlla A, Pattini L, Nielsen R. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 2011;7:e1002355. doi: 10.1371/journal.pgen.1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Duret L. Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet. 2007;23:273–7. doi: 10.1016/j.tig.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–5. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazko GV, Nei M. Estimation of divergence times for major lineages of primate species. Mol Biol Evol. 2003;20:424–34. doi: 10.1093/molbev/msg050. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–36. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Goldstein DB, Weale ME. Population genomics: linkage disequilibrium holds the key. Current biology. 2001;11:R576–9. doi: 10.1016/s0960-9822(01)00348-7. [DOI] [PubMed] [Google Scholar]
- Goren-Inbar N, Alperson N, Kislev ME, Simchoni O, Melamed Y, et al. Evidence of hominin control of fire at Gesher Benot Ya’aqov, Israel. Science. 2004;304:725–7. doi: 10.1126/science.1095443. [DOI] [PubMed] [Google Scholar]
- Gould SJ, Lewontin RC. The spandrels of San Marco and the Panglossian paradigm: a critique of the adaptationist programme. Proc R Soc Lond B Biol Sci. 1979;205:581–98. doi: 10.1098/rspb.1979.0086. [DOI] [PubMed] [Google Scholar]
- Granka JM, Henn BM, Gignoux CR, Kidd JM, Bustamante CD, Feldman MW. Limited evidence for classic selective sweeps in African populations. Genetics. 2012;192:1049–64. doi: 10.1534/genetics.112.144071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassa CJ, Kulathinal RJ. Elevated Evolutionary Rates among Functionally Diverged Reproductive Genes across Deep Vertebrate Lineages. Int J Evol Biol. 2011;2011:274975. doi: 10.4061/2011/274975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–22. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Andersen KG, Shlyakhter I, Tabrizi S, Winnicki S, et al. Identifying recent adaptations in large-scale genomic data. Cell. 2013;152:703–13. doi: 10.1016/j.cell.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Shlyakhter I, Karlsson EK, Byrne EH, Morales S, et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010;327:883–6. doi: 10.1126/science.1183863. [DOI] [PubMed] [Google Scholar]
- Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. Genetic evidence for archaic admixture in Africa. Proc Natl Acad Sci U S A. 2011;108:15123–8. doi: 10.1073/pnas.1109300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, Witonsky DB, Alkorta-Aranburu G, Beall CM, Gebremedhin A, et al. Adaptations to climate-mediated selective pressures in humans. PLoS Genet. 2011;7:e1001375. doi: 10.1371/journal.pgen.1001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, Witonsky DB, Ehler E, Alkorta-Aranburu G, Beall C, et al. Colloquium paper: human adaptations to diet, subsistence, and ecoregion are due to subtle shifts in allele frequency. Proc Natl Acad Sci U S A. 2010;107(Suppl 2):8924–30. doi: 10.1073/pnas.0914625107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanotte O, Bradley DG, Ochieng JW, Verjee Y, Hill EW, Rege JE. African pastoralism: genetic imprints of origins and migrations. Science. 2002;296:336–9. doi: 10.1126/science.1069878. [DOI] [PubMed] [Google Scholar]
- Hinch AG, Tandon A, Patterson N, Song Y, Rohland N, et al. The landscape of recombination in African Americans. Nature. 2011;476:170–5. doi: 10.1038/nature10336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer T, Ray N, Wegmann D, Excoffier L. Large allele frequency differences between human continental groups are more likely to have occurred by drift during range expansions than by selection. Ann Hum Genet. 2009;73:95–108. doi: 10.1111/j.1469-1809.2008.00489.x. [DOI] [PubMed] [Google Scholar]
- Holloway RL, Sherwood CS, Hof PR, Rilling JK. Evolution of the brain in humans - paleoneurology. In: Binder MD, Hirokawa N, Windhorst U, editors. Encyclopedia of Neuroscience. New York: Springer; 2009. pp. 1326–34. [Google Scholar]
- Huang H, Winter EE, Wang H, Weinstock KG, Xing H, et al. Evolutionary conservation and selection of human disease gene orthologs in the rat and mouse genomes. Genome Biol. 2004;5:R47. doi: 10.1186/gb-2004-5-7-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–8. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Ingram CJ, Elamin MF, Mulcare CA, Weale ME, Tarekegn A, et al. A novel polymorphism associated with lactose tolerance in Africa: multiple causes for lactase persistence? Hum Genet. 2007;120:779–88. doi: 10.1007/s00439-006-0291-1. [DOI] [PubMed] [Google Scholar]
- Jablonski NG. Human skin pigmentation as an example of adaptive evolution. Proc Am Philos Soc. 2012;156:45–57. [PubMed] [Google Scholar]
- Jensen JD, Kim Y, DuMont VB, Aquadro CF, Bustamante CD. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics. 2005;170:1401–10. doi: 10.1534/genetics.104.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Willner D, Danoy P, Xu H, Brown MA. Comparison of the performance of two commercial genome-wide association study genotyping platforms in Han Chinese samples. G3 (Bethesda) 2013;3:23–9. doi: 10.1534/g3.112.004069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RC, Nelson GW, Troyer JL, Lautenberger JA, Kessing BD, et al. Accounting for multiple comparisons in a genome-wide association study (GWAS) BMC Genomics. 2010;11:724. doi: 10.1186/1471-2164-11-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, et al. Global trends in emerging infectious diseases. Nature. 2008;451:990–3. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde LB, Watkins WS, Bamshad MJ. Population genomics: a bridge from evolutionary history to genetic medicine. Hum Mol Genet. 2001;10:2199–207. doi: 10.1093/hmg/10.20.2199. [DOI] [PubMed] [Google Scholar]
- Kamberov YG, Wang S, Tan J, Gerbault P, Wark A, et al. Modeling recent human evolution in mice by expression of a selected EDAR variant. Cell. 2013;152:691–702. doi: 10.1016/j.cell.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RG. Out of Africa and the Evolution of Human Behavior. Evolutionary Anthropology. 2008;17:267–81. [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–7. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature. 2010;467:1099–103. doi: 10.1038/nature09525. [DOI] [PubMed] [Google Scholar]
- Krause J, Lalueza-Fox C, Orlando L, Enard W, Green RE, et al. The derived FOXP2 variant of modern humans was shared with Neandertals. Curr Biol. 2007;17:1908–12. doi: 10.1016/j.cub.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance J. Disease-associated alleles in genome-wide association studies are enriched for derived low frequency alleles relative to HapMap and neutral expectations. BMC Med Genomics. 2010;3:57. doi: 10.1186/1755-8794-3-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance J, Tishkoff SA. SNP ascertainment bias in population genetic analyses: Why it is important and how to correct it. BioEssays. 2013 doi: 10.1002/bies.201300014. under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance J, Vernot B, Elbers CC, Ferwerda B, Froment A, et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell. 2012;150:457–69. doi: 10.1016/j.cell.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalueza-Fox C, Gilbert MT. Paleogenomics of archaic hominins. Curr Biol. 2011;21:R1002–9. doi: 10.1016/j.cub.2011.11.021. [DOI] [PubMed] [Google Scholar]
- Lam HY, Clark MJ, Chen R, Natsoulis G, O’Huallachain M, et al. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol. 2012;30:78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laval G, Excoffier L. SIMCOAL 2.0: a program to simulate genomic diversity over large recombining regions in a subdivided population with a complex history. Bioinformatics. 2004;20:2485–7. doi: 10.1093/bioinformatics/bth264. [DOI] [PubMed] [Google Scholar]
- Leinonen T, McCairns RJ, O’Hara RB, Merila J. Q(ST)-F(ST) comparisons: evolutionary and ecological insights from genomic heterogeneity. Nat Rev Genet. 2013;14:179–90. doi: 10.1038/nrg3395. [DOI] [PubMed] [Google Scholar]
- Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–95. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A new test for detecting recent positive selection that is free from the confounding impacts of demography. Mol Biol Evol. 2011;28:365–75. doi: 10.1093/molbev/msq211. [DOI] [PubMed] [Google Scholar]
- Loftus RT, Ertugrul O, Harba AH, El-Barody MA, MacHugh DE, et al. A microsatellite survey of cattle from a centre of origin: the Near East. Mol Ecol. 1999;8:2015–22. doi: 10.1046/j.1365-294x.1999.00805.x. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Indap AR, Schmidt S, Boyko AR, Hernandez RD, et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–7. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. The power and promise of population genomics: from genotyping to genome typing. Nature reviews genetics. 2003;4:981–94. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–4. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McDougall I, Brown FH, Fleagle JG. Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature. 2005;433:733–6. doi: 10.1038/nature03258. [DOI] [PubMed] [Google Scholar]
- McEvoy B, Beleza S, Shriver MD. The genetic architecture of normal variation in human pigmentation: an evolutionary perspective and model. Hum Mol Genet. 2006;15(Spec No 2):R176–81. doi: 10.1093/hmg/ddl217. [DOI] [PubMed] [Google Scholar]
- McEvoy BP, Powell JE, Goddard ME, Visscher PM. Human population dispersal “Out of Africa” estimated from linkage disequilibrium and allele frequencies of SNPs. Genome Res. 2011;21:821–9. doi: 10.1101/gr.119636.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHenry HM. Origin and diversity of early hominin bipedalism. In: Reynolds SC, Gallagher A, editors. African Genesis: Perspectives on Hominin Evolution. Cambridge UK: Cambridge University Press; 2012. pp. 205–22. [Google Scholar]
- Moore LG, Shriver M, Bemis L, Hickler B, Wilson M, et al. Maternal adaptation to high-altitude pregnancy: an experiment of nature--a review. Placenta. 2004;25(Suppl A):S60–71. doi: 10.1016/j.placenta.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Moreau C, Bherer C, Vezina H, Jomphe M, Labuda D, Excoffier L. Deep human genealogies reveal a selective advantage to be on an expanding wave front. Science. 2011;334:1148–50. doi: 10.1126/science.1212880. [DOI] [PubMed] [Google Scholar]
- Narum SR, Hess JE. Comparison of F(ST) outlier tests for SNP loci under selection. Mol Ecol Resour. 2011;11(Suppl 1):184–94. doi: 10.1111/j.1755-0998.2011.02987.x. [DOI] [PubMed] [Google Scholar]
- Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. 1962;14:353–62. [PMC free article] [PubMed] [Google Scholar]
- Nei M, Maruyama T. Letters to the editors: Lewontin-Krakauer test for neutral genes. Genetics. 1975;80:395. doi: 10.1093/genetics/80.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R. Population genetic analysis of ascertained SNP data. Human genomics. 2004;1:218–24. doi: 10.1186/1479-7364-1-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3:e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Yamaguchi T, Watanabe C, Kawaguchi A, Haneji K, et al. Effects of an Asian-specific nonsynonymous EDAR variant on multiple dental traits. J Hum Genet. 2012;57:508–14. doi: 10.1038/jhg.2012.60. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Jensen JD, Stephan W, Stamatakis A. A critical assessment of storytelling: gene ontology categories and the importance of validating genomic scans. Mol Biol Evol. 2012;29:3237–48. doi: 10.1093/molbev/mss136. [DOI] [PubMed] [Google Scholar]
- Pennings PS, Hermisson J. Soft sweeps III: the signature of positive selection from recurrent mutation. PLoS Genet. 2006;2:e186. doi: 10.1371/journal.pgen.0020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–37. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–72. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Lunter G. Evolutionary biology: human brain gene wins genome race. Nature. 2006;443:149–50. doi: 10.1038/nature05154. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson N, Hancks DC, Myers S, Reich D, et al. Effects of cis and trans genetic ancestry on gene expression in African Americans. PLoS Genet. 2008;4:e1000294. doi: 10.1371/journal.pgen.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Pickrell JK, Coop G. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol. 2010;20:R208–15. doi: 10.1016/j.cub.2009.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranciaro A, Campbell MC, Hirbo JB, Ko W-Y, Froment A, et al. Genetic Origins of Lactase Persistence and the Spread of Pastoralism in Africa. 2013 doi: 10.1016/j.ajhg.2014.02.009. under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Guo X, Wang Y, Lohmueller KE, Rasmussen S, et al. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–8. doi: 10.1126/science.1211177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakumar A, Mousset S, Glemin S, Berglund J, Galtier N, et al. Detecting positive selection within genomes: the problem of biased gene conversion. Philos Trans R Soc Lond B Biol Sci. 2010;365:2571–80. doi: 10.1098/rstb.2010.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reader J. Africa: A Biography of the Continent. New York: Vintage; 1997. [Google Scholar]
- Reich D, Green RE, Kircher M, Krause J, Patterson N, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–60. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson PJ, Boyd R, Henrich J. Colloquium paper: gene-culture coevolution in the age of genomics. Proc Natl Acad Sci U S A. 2010;107(Suppl 2):8985–92. doi: 10.1073/pnas.0914631107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson A. Letters to the editors: Remarks on the Lewontin-Krakauer test. Genetics. 1975;80:396. doi: 10.1093/genetics/80.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald J, Akey JM. Genome-wide scans for loci under selection in humans. Hum Genomics. 2005;2:113–25. doi: 10.1186/1479-7364-2-2-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–8. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SA, Hartl DL. Population genetics of polymorphism and divergence. Genetics. 1992;132:1161–76. doi: 10.1093/genetics/132.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Durbin R. Revising the human mutation rate: implications for understanding human evolution. Nat Rev Genet. 2012;13:745–53. doi: 10.1038/nrg3295. [DOI] [PubMed] [Google Scholar]
- Scheinfeldt LB, Soi S, Thompson S, Ranciaro A, Woldemeskel D, et al. Genetic adaptation to high altitude in the Ethiopian highlands. Genome Biol. 2012;13:R1. doi: 10.1186/gb-2012-13-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeldt LB, Tishkoff SA. Living the high life: high-altitude adaptation. Genome Biol. 2010;11:133. doi: 10.1186/gb-2010-11-9-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebusch CM, Skoglund P, Sjodin P, Gattepaille LM, Hernandez D, et al. Genomic variation in seven Khoe-San groups reveals adaptation and complex African history. Science. 2012;338:374–9. doi: 10.1126/science.1227721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethupathy P, Hannenhalli S. A tutorial of the poisson random field model in population genetics. Adv Bioinformatics. 2008:257864. doi: 10.1155/2008/257864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–32. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, et al. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Human genomics. 2004;1:274–86. doi: 10.1186/1479-7364-1-4-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson TS, Yang Y, Huff CD, Yun H, Qin G, et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329:72–5. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- Spitze K. Population structure in Daphnia obtusa: quantitative genetic and allozymic variation. Genetics. 1993;135:367–74. doi: 10.1093/genetics/135.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapley J, Reger J, Feulner PG, Smadja C, Galindo J, et al. Adaptation genomics: the next generation. Trends Ecol Evol. 2010;25:705–12. doi: 10.1016/j.tree.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Steiper ME, Young NM. Primate molecular divergence dates. Mol Phylogenet Evol. 2006;41:384–94. doi: 10.1016/j.ympev.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, et al. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–44. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson DG, Kulathinal RJ, Singh RS. Mammalian sperm proteins are rapidly evolving: evidence of positive selection in functionally diverse genes. Mol Biol Evol. 2002;19:1973–80. doi: 10.1093/oxfordjournals.molbev.a004021. [DOI] [PubMed] [Google Scholar]
- Vargha-Khadem F, Gadian DG, Copp A, Mishkin M. FOXP2 and the neuroanatomy of speech and language. Nat Rev Neurosci. 2005;6:131–8. doi: 10.1038/nrn1605. [DOI] [PubMed] [Google Scholar]
- Vasey DE. An ecological history of agriculture: 10,000 B.C.-A.D. 10,000. xi. Ames: Iowa State University Press; 1992. p. 363. [Google Scholar]
- Vasseur E, Quintana-Murci L. The impact of natural selection on health and disease: uses of the population genetics approach in humans. Evolutionary Applications. 2013 doi: 10.1111/eva.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli BC, McDonald JH, Argyropoulos G, Destro-Bisol G, Froment A, et al. Evidence for balancing selection from nucleotide sequence analyses of human G6PD. Am J Hum Genet. 2002;71:1112–28. doi: 10.1086/344345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli BC, Tishkoff SA. Signatures of selection and gene conversion associated with human color vision variation. Am J Hum Genet. 2004;75:363–75. doi: 10.1086/423287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock MC. Evolutionary inference from QST. Mol Ecol. 2008;17:1885–96. doi: 10.1111/j.1365-294X.2008.03712.x. [DOI] [PubMed] [Google Scholar]
- Wyckoff GJ, Wang W, Wu CI. Rapid evolution of male reproductive genes in the descent of man. Nature. 2000;403:304–9. doi: 10.1038/35002070. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–17. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–8. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng K, Fu YX, Shi S, Wu CI. Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics. 2006;174:1431–9. doi: 10.1534/genetics.106.061432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Webb DM, Podlaha O. Accelerated protein evolution and origins of human-specific features: Foxp2 as an example. Genetics. 2002;162:1825–35. doi: 10.1093/genetics/162.4.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li WH. Human SNPs reveal no evidence of frequent positive selection. Mol Biol Evol. 2005;22:2504–7. doi: 10.1093/molbev/msi240. [DOI] [PubMed] [Google Scholar]