

Abstract
Papillomaviruses have a strict tropism for epithelial cells and they are fully reliant on cellular differentiation for completion of their life cycles, resulting in the production of progeny virions. Thus, a permissive environment for full viral replication in vitro wherein virion morphogenesis occurs under cooperative viral and cellular cues requires the cultivation of epithelium. Presented in the first section of this unit is a protocol for growing differentiating epithelial tissues, whose structure and function mimics many important morphological and biochemical aspects of normal skin. The technique, pioneered by Asslineau and Pruniéras (Asselineau and Prunieras 1984) and modified by Kopan et al. (Kopan et al. 1987), involves growing epidermal cells atop a dermal equivalent consisting of live fibroblasts and a collagen lattice. Epithelial stratification and differentiation ensues when the keratinocyte-dermal equivalent is placed at the air-liquid interface. The apparent floating nature of the cell-matrix in this method led to the nickname “raft” cultures. The general technique can be applied to normal low passage keratinocytes, to cells stably transfected with papillomavirus genes or genomes, as well as keratinocytes established from neoplastic lesions. However, infectious papillomavirus particles have only been isolated from organotypic epithelial cultures initiated with cells that maintain oncogenic human papillomavirus genomes in an extrachomosomal replicative form. The second section of this unit is dedicated to a virion isolation method that minimizes aerosol and skin exposure to these human carcinogens. Although the focus of the protocols is on the growth of tissues that yields infectious papillomavirus progeny, this culture system facilitates the investigation of these fastidious viruses during their complex replicative cycles, and raft tissues can be manipulated and harvested at any point during the process. Importantly, a single step virus growth cycle is achieved in this process, as it is unlikely that progeny virions are released to initiate subsequent rounds of infection.
Keywords: papillomavirus, organotypic epithelial tissue culture, air-liquid interface, keratinocyte, virion isolation
Introduction
Understanding the biology of productive papillomavirus infections has lagged behind that of many other viruses because of the complex papillomavirus replicative cycle, which has been challenging to faithfully recapitulate in the laboratory. Furthermore, human papillomavirus virions are produced at relatively low levels in clinical lesions, and thus, obtaining virus stocks for the study of experimental infections has been hampered. An infectious papillomavirus particle is composed of the ≈8 kb circular genome, condensed with cellular histones and encapsidated by 360 copies of the major capsid protein, L1, and 12-72 copies of the L2 minor capsid protein (Buck et al. 2008). Such particles can be assembled in undifferentiated monolayer cell cultures via co-transfection of viral genomes and plasmids that express L1 and L2 proteins, which spontaneously assemble to nonspecifically encasidate appropriately-sized genomes (Buck et al. 2004, Pyeon et al. 2005) (cite unit on this protocol, if present). In contrast, the organotypic raft tissue culture system affords the opportunity for the papillomavirus genetic material and gene products to interact with cellular components and the ensuing intracellular and microenvironment that accompany epithelial differentiation. Thus, virion morphogenesis in raft tissues occurs in a regulated cellular milleu that better resembles the epithelium in vivo. The ex vivo nature of the raft system is more amenable to experimental manipulation compared to host tissue xenografts in rodents that provide a permissive replicative environment for some papillomavirus genotypes. We have recently summarized the advantages and limitations to the various methods for obtaining infectious papillomavirus particles (Ozbun and Kivitz 2011).
The complete life cycle of a human papillomavirus (HPV) was first achieved in the Laimins' laboratory over 20 years ago. Initially, McCance and co-workers adapted the organotypic epithelial tissue culture system of (Asselineau and Prunieras 1984) as modified by (Kopan et al. 1987), to study HPV-transformation (McCance et al. 1988). Meyers et al. later showed the production of virions in raft cultures grown from cervical cells that harbored extrachromosomal HPV31 (subtype b) (Meyers et al. 1992). Currently, the raft system's ability to yield papillomavirus virions is limited to papillomavirus genotypes that are replication-competent and capable of conferring a cellular growth advantage.
This unit is comprised of complementary protocols for the culture of epithelial tissues (Basic Protocol 1) and the isolation of human papillomavirus virions (Basic Protocol 2). Non-transformed keratinocytes are fastidious in their growth requirements. Although they can be cultivated in monolayer cultures grown in fully defined keratinocyte serum-free media, we find the cells grow more robustly in epithelial medium (“E Medium”) containing 5% heat-inactivated fetal calf serum and co-cultured with mitomycin-C treated fibroblast feeder cells (irradiated fibroblasts can also be used). This is especially true for low passage keratinocytes and those that maintain extrachromosomal genomes. Therefore, the recipe for E Medium is provided as a Support Protocol 1.
CAUTION: Oncogenic human papillomaviruses are considered Biosafety Level 2 (BSL-2) pathogens. Follow all appropriate guidelines and regulations for the use and handling of pathogenic microorganisms. See UNIT 1A.1 and other pertinent resources (APPENDIX 1B) for more information. Additionally, during the tissue extraction and virion isolation protocol, when aerosols might be generated, we recommend a modified BSL-2+ handling, wherein personnel should be HPV-vaccinated, and be fitted with a HEPA filter 3M model 8233 respirator (N100 particulate). Additionally, all equipment and disposables coming into contact with the virus extract or tissue cultures should be decontaminated with a 1:128 dilution of Vesphene®, 10% chlorine bleach, or 70% ethanol. Papillomavirus infectivity has been shown to be sensitive to 70% ethanol (Roden et al. 1997). Physical manipulations should be confined to biosafety cabinets and disposable materials should be placed into a biohazard bag to be sealed before removing to the biohazard waste containers for autoclaving. Centrifuge tubes and other reusable lab-ware should be decontaminated in the same manner or by placing in a closed container, while in the biosafety cabinet, to be autoclaved before washing. Spills that are potentially contaminated with virus should immediately be decontaminated using either 1:128 Vesphene® solution or 70% ethanol for 30 minutes before rinsing.
BASIC PROTOCOL 1: Preparation and Growth of Organotypic Epithelial Tissue Cultures
This experimental system is commonly referred to as the raft culture system owing to the illusion that the tissues float atop the culture media. The technique is as much art as science, and with practice, the procedure can be used to grow literally hundreds of rafts at a time. In brief, live mouse or human fibroblasts are mixed with type 1 collagen to form a gelatinous lattice that will act as a dermal counterpart. Keratinocytes seeded atop the lattice are grown to full confluence in submerged cultures. The matrix is then lifted to a wire support grid that provides additional structure for the air-liquid interface and this platform allows diffusion of nutrients from the growth media below to the keratinocytes on the apical surface. The keratinocytes proximal to the dermal equivalent at the air-liquid interface continue to divide, and the cells stratify and differentiate as evidenced by biochemical and morphological changes that peak over a 10- to 20-day period. In various iterations, this system has been used to examine numerous aspects of skin biology and to yield functional skin for toxicity screening and potential replacement grafting (Zhang and Michniak-Kohn 2012). As papillomavirus vegetative genome replication and robust expression of late viral genes are intimately linked to epithelial differentiation, the organotypic epithelial raft tissue culture system also provides a permissive environment for the compete viral life cycle (Meyers et al. 1992). The general technique can be applied to normal low passage keratinocytes, to cells stably transfected with papillomavirus genes or genomes, as well as keratinocytes established from neoplastic lesions. However, infectious papillomavirus particles have only been isolated from organotypic epithelial cultures initiated with cells that maintain oncogenic human papillomavirus genomes in an extrachomosomal replicative form. Importantly, only a single step virus growth cycle is achieved in this process, as it is unlikely that progeny virions are released to initiate subsequent rounds of infection in adjacent cells.
The preparation and growth of organotypic epithelial cultures can be broken chronologically into 4 stages (see Figure 1A).
Figure 1.
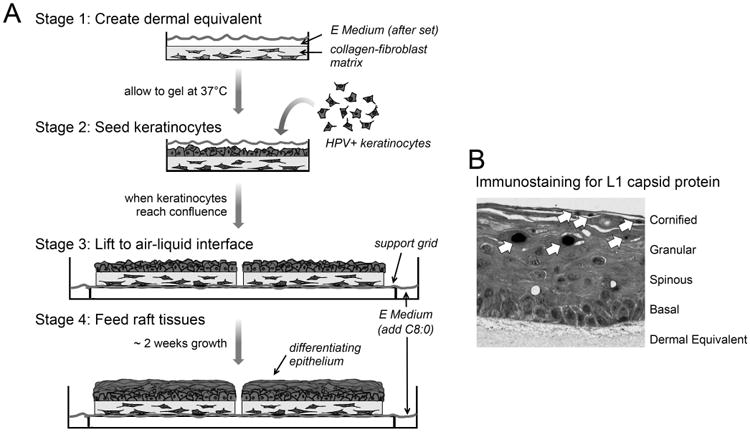
The growth of organotypic raft epithelial tissue culture can be broken into four stages. In stage 1, collagen-fibroblast dermal equivalents are prepared in 6-well plates and allowed to gel at 37°C. In stage 2, keratinocytes that maintain extrachromosomal HPV genomes are seeded atop the dermal equivalent submerged in growth medium. In stage 3, when the keratinocytes have reached 100% confluence, each dermal equivalent with keratinocytes is lifted to the air-liquid interface on wire grids contained in 100-mm cell culture plates. In stage 4, the raft tissues at the air-liquid interface are fed with E medium containing 5% FCS, 10 unit/ml nystatin and freshly diluted 10 μM C8:0. Tissues are fed by diffusion up through the dermal equivalent; the epithelium stratifies and differentiates over ∼12-16 days post lifting. See Basic Protocol 1 for specifics. Cross-section of a CIN 612-9E raft following 14-days at the air-liquid interface, treated with 10μM C8:0 every other day, and subjected to heat shock (90 min, 43°C, CO2 incubator) on days 6, 8, and 10 post lifting. The tissue was fixed in 10% neutral buffered formalin, paraffin imbedded, and 4 μm thick cross-sections were prepared. Immunostaining reveals the L1 major capsid protein in the granular and cornified layers of the epithelium. The layers of the epithelium are indicated at the right.
Materials
Human or mouse fibroblast “feeder” cells (e.g., J2 3T3 fibroblasts at early passage; estimate about 3 to 6 rafts per confluent 100 mm dish of fibroblasts, depending upon the confluence)
Keratinocytes that maintain extrachromosomal HPV genomes (e.g., CIN 612-9E cells (Meyers et al. 1992); estimate 2 to 6 rafts per near confluent 100 mm dish of cells, depending upon cell line)
Rat Tail Type 1 Collagen, concentration 3.8 to 4.0 mg/ml; store at 4°C (BD Biosciences, cat. no. 354236)
Reconstitution Buffer (Add 11.0 g NaHCO3 and 23.85 g HEPES to 100 ml 0.162 N NaOH. When dissolved bring final volume to 500 ml with 0.162 N NaOH, filtersterilize, and store in aliquots at -20°C)
10× Dulbecco's Modified Eagle's Media (DMEM), powder, high glucose, no pyruvate, no bicarbonate (Life Technologies-Gibco®, cat. no. 12100-061; fully dissolve an amount suitable for 1 liter in 100 ml warm ultrapure water. Filtersterilize and store in 20-50 ml aliquots at -20°C)
10 N NaOH
E medium containing 5% heat inactivated fetal calf serum (FCS) and 10,000 units/ml nystatin suspension (see Support Protocol 1 for recipe)
10 μg/ml EGF (BD Biosciences, cat. no. 354001; dissolved in sterile ultrapure dH2O)
20 mM 1,2-dioctanoyl-sn-glycerol (C8:0; Calbiochem, cat. no. 317505; in 100% EtOH)
70% EtOH (for dipping forceps and spatulas if needed)
Flat-bottom 6-well plates, sterile, with lids
50-ml graduated conical polypropylene centrifuge tubes with screw-cap lids, sterile
5- and 10-ml graduated plastic, individually wrapped pipets, sterile
100-mm tissue culture plates
Stainless steel screen grids 3.063 in. diameter, 40 mesh, 0.01 wire (Johnson screens®, Houston, TX; cat. no. JDE 244934; sterile, autoclaved 40 min on dry cycle; see Support Protocols 2 for preparation)
5-ml test tube (12 × 75 mm)
2-liter Pyrex® beakers
9-inch forceps, individually wrapped, sterile
9-inch stainless steel spatula/spoon tool (e.g. Fisher, cat. no. S50789A; individually wrapped, sterile)
Bunsen burner in cell culture cabinet (to flame 70%-EtOH treated forceps and spatulas, if needed)
Stage 1: Preparing Collagen-Fibroblast Dermal Equivalents
To maintain sterility, conduct all steps in a cell culture cabinet.
It is important that all cell lines have tested negative for mycoplasma.
Each dermal equivalent will comprise 3 ml of sterile collagen mix containing ≈5 × 105 live (not mitomycin C-treated) J2 fibroblasts. To account for loss, plan to make enough mix to pour at least two extra collagen-fibroblast matrixes.
- Calculate the volumes of collagen, reconstitution buffer, 10× DMEM, and number of fibroblasts needed for the number of raft tissues desired. The following table may be useful:
Collagen mix components Volume of each (3 ml) Calculate the total volume needed for the number of rafts desired 8 parts collagen type 1 2.4 ml —— ml 1 part Reconstitution buffer 0.3 ml —— ml 1 part 10× DMEM 0.3 ml —— ml J2 3T3 fibroblasts 5 × 105 cells —— × 106 cells Remove the live J2 fibroblasts from cell culture plates using standard trypsinization techniques. Add trypsinized cells to a 50-ml conical tube with 5-10 times the volume of normal, serum-containing medium to quench the trypsin activity.
Count the cells and remove the unwanted volume of cells.
Centrifuge the cells at 1000 RPM for 5 min. Siphon off the liquid to leave the cell pellet.
To prepare the Collagen Mix, place the pelleted J2 cells and any needed additional 50-ml sterile tubes on ice. Make sure that each solution is ice-cold as collagen will gel at warmer temperatures.
Resuspend fibroblasts in Reconstitution Buffer to 1.5 × 106 cells per ml per the calculations in step 3 above (final will be 1.5 × 105 cells per ml in the collagen mix).
Mix the 10× DMEM well to re-suspend any precipitate that reformed, and add the correct volume to the cells in reconstitution buffer, mixing well with the pipette.
If making more than 16 rafts, divide the fibroblast solution with Reconstitution Buffer and DMEM into multiple tubes to accommodate the volume of collagen needed.
10. Collagen is viscous, so to minimize the loss of this expensive reagent, use a single 10-ml pipet to deliver all of the collagen needed, even if multiple 50-ml tubes are required.
11. After all the collagen has been added to the tubes, add 1.0 to 2.0 μl of 10 N NaOH per ml of the final total Collagen Mix to the side of the tube and immediately mix well (avoiding bubbles).
pH adjustment is best accomplished by adding the NaOH and mixing one tube at a time. The solutions can be mixed efficiently by securely capping the tube and gently inverting until a homogenous solution (color) is achieved. Alternatively, the 10-ml pipet can be used to gently mix and/or pipet up and down on ice until completely mixed. A salmon pink color is desired after adding the NaOH. If the solution is more orange than pink, add more 10 N NaOH until the pink color is obtained. Repeat on each additional tube.
12. With a single 10-ml pipet, transfer 3 ml of collagen/fibroblast mix into each well of a 6-well plate. Avoid forming bubbles while pipetting.
13. Be certain that the collagen-fibroblast mix covers the entire plate.
Bubbles present atop the collagen should be pulled to the perimeter of the dish and aspirated with a Pasteur pipet.
14. Gently move the 6-well dishes containing the collagen-fibroblast matrix solution into a 37°C incubator; allow the collagen to solidify for 0.5 to 1.5 hours.
15. After the collagen has formed an opaque gel, gently add 2 ml of E media containing 5% FCS and 10 units/ml nystatin to each dish and return to the 37°C, CO2 incubator until ready for use.
The collagen-fibroblast matrixes can be kept under media at 37°C in the CO2 incubator until ready for keratinocyte seeding. We recommend seeding keratinocytes within 48 hrs after matrixes are poured.
Stage 2: Seeding of Epithelial Cells onto the Dermal Equivalents
Continue all steps in a cell culture cabinet.
16. If collagen-fibroblast matrixes are incubated ≥16 hrs, replace the media with 2 ml fresh E media containing 5% FCS and 10 units/ml nystatin. If incubated <16 hrs, go to step 17.
Be careful not to place the aspirating pipet too close to the collagen when removing the old media; the collagen gel is easily damaged or dislodged by strong suction, especially if a vacuum pump is used for aspiration.
17. For cells of larger size (e.g., CIN 612-9E cells [≈20 μm diameter]), add 1 ml containing 1.0 × 106 epidermal cells to each plate. For smaller cells (primary human keratinocytes or NIKS cells) seed 1 ml containing 3.0 × 106 cells per ml.
18. Move the plates to the 37°C, CO2 incubator.
When moving the plates, the motion may cause the media to swirl and the cells to accumulate in the center. With the plates sitting on the incubator shelf, sharply (but without splashing media), move the plate side-to-side 3-4 times in one direction; repeat in the perpendicular direction to disperse the cells.
19. Allow the cells to settle and attach to the collagen (16-24 h).
20. Thereafter, replace the media with fresh E medium containing 5% FCS and 10 units/ml nystatin.
If growing HPV-negative cells that are EGF dependent, feed with E medium containing 5% FCS plus 5 ng/ml EGF. This reduces ridge formation and encourages keratinocyte spreading so that complete confluence can be achieved.
21. When the keratinocyte monolayers have reached confluence, the collagen-fibroblast matrixes with keratinocytes are ready to be transferred to the air-liquid interface (see below).
22. Transfer the beaker containing the cool sterile grids to a cell culture cabinet (see Support Protocol 2 for instruction on preparing the grids).
23. Maintaining sterile technique, use sterile forceps to move one grid to a 100-mm cell culture dish so that the flat surface is raised on its feet above the lower half of the dish.
24. Cover the dish and repeat as needed to obtain one grid/dish for each two collagen-fibroblast matrixes with keratinocytes.
Do not be concerned if the grids are a bit moist. This will actually aid in the first addition of media. Grids that are very dry may cause increased surface tension with the media, and this could cause bubbles to form under grids that are not completely flat and level.
Stages 3-4: Lifting the Dermal Equivalent with Keratinocytes to the Air-Liquid Interface and Growth of the Epithelial Raft Tissues
Continue all subsequent steps in a cell culture cabinet.
25. Move the 6-well plates containing collagen-fibroblast matrixes with keratinocytes to the cell culture cabinet.
This can be done in groups of 6-well plates. Do not be too concerned about leaving the collagen matrix plugs out of the CO2 incubator as the increase in pH will actually make the rafts a bit more sturdy and easier to lift.
26. Carefully remove the media from each of the collagen-fibroblast matrixes.
27. Hold the plate up at a 30-45° angle. Using the scoop-end of the sterile spatula, work one at a time to release a collagen-fibroblast matrix from the rim of a plate (see Figure 2).
Figure 2.
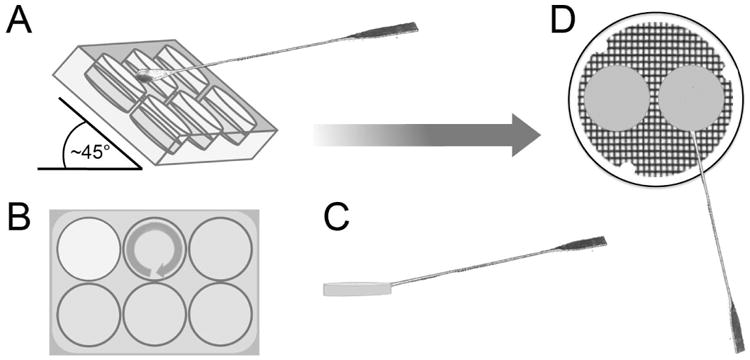
Lifting the dermal equivalent with confluent keratinocytes to the air-liquid interface. A, After removing the growth media from atop the collagen gels on a plate, hold the plate at an angle to best facilitate the release and removal of the gel. B, Insert the spoon-end of the spatula/spoon tool at the bottom of a gel. In a single motion, move the spatula/spoon tool around the gel and when reaching the bottom, slide the spoon under the center of the gel. C, Slowly lift the gel out and move it to a sterile grid in a 100-mm cell culture dish, D. Place the gel flat on the grid and, keeping the spoon under the gel, smooth out the edges to the extent possible. Two rafts can be placed on each grid.
Beginning at the bottom of each well, gently slide the spatula around the perimeter of the gel to end at the bottom in one continuous motion. Without removing the scoop, slide it under the collagen-fibroblast matrix from the bottom and slowly lift out the matrix.
28. Move the gel to the grid in the 100-mm dish keeping the scoop under the gel and using the scoop to spread out the gel edges to maintain the shape as much as possible.
29. Repeat steps 3-4 until all gels are on grids, 1-2 rafts per grid.
To ensure proper nutritional exposure, it is important to have no more than 2 rafts per grid.
The first two-day period of growth will utilize E media containing 5% FCS and 10 units/ml nystatin. The tissues will be fed every other day thereafter with E media containing 5% FCS, 10 units/ml nystatin, and freshly supplemented with a final concentration of 10 μM C8:0 (v/v).
As EGF inhibits differentiation, it is never added to the media after rafts are lifted to the air-liquid interface.
30. Add media to each plate until the underside of the grid is entirely filled making certain that no bubbles are trapped under the grid and that no media covers any part of the collagen-fibroblast matrixes with keratinocytes.
Excess media in the plate can slosh onto the tissues when moving plates to and from the incubator. Note the volume needed for each plate. This will aid in making sufficient growth media containing fresh 10 μM C8:0 for subsequent feeding. Optimal media volume for two days of growth is 10-15 ml.
31. When replacing the growth media, again, make certain that no bubbles are trapped under the grid and that no media are touching any part of the raft above the grid.
The air-liquid interface supports the creation of an oxidative gradient important for differentiation. Any media contacting the sides or top of the tissue will destroy this gradient and inhibit differentiation.
32. Media containing fresh 10 μM C8:0 should be replaced every-other-day for a total of 12-16 days at the air-liquid interface.
We have found that longer growth (i.e., greater than 12 days) does not necessarily result in greater virus production. The reason for this is unclear.
33. During growth, the tissues can be subjected to any variety of treatments or labeling afforded to cells in culture. By 8 days post lifting, the epithelial tissues should begin to appear dry atop the collagen matrix.
We have found subjecting the rafts to 90 min heat-shock treatments at 43°C in a CO2 incubator increases capsid protein accumulation in the nucleus and enhances virion production (Song et al. 2010).
The final number of stratified cells on a 35 mm epithelial organotypic culture should be 10 to 20-fold more than originally seeded.
34. The tissue edges tend to attach to the grid and may be difficult to dislodge. To harvest the tissues for HPV virion isolation, a sterile scalpel should be used to gently score a break in the tissue, which will promote the ability to scrape the epithelial tissue away from the collagen.
To aid in the virion harvest step, avoid harvesting the collagen matrix as much as possible. If good differentiation has been achieved, the collagen matrix can often be pulled out from under the epithelial tissue, which then can be scraped from the grid.
35. Place up to 50 organotypic tissues (without collagen) in a single sterile 50-ml conical polypropylene centrifuge tube (no freezing medium is required as the virions are quite stable). These can be stored at -80°C for months prior to virion isolation in Protocol 2 below.
36. It is prudent to harvest at least one (or half of one or more) representative tissue for histology to verify the level of differentiation achieved.
This is best accomplished by transferring the entire thickness of tissue and collagen matrix to a histology cassette.
37. If tissues are to be harvested for protein and/or nucleic acids, it is also best to remove only the epithelial tissue as above, avoiding the collage-fibroblast matrix, by scraping and/or peeling the tissues away from the collagen.
38. Loosely stack the used stainless steel screen grids into a 2-liter Pyrex® beaker. When finished, cover the top with foil and move to a fume hood. Fill the beaker with chromic acids cleaning solution until the grids are covered.
39. Treat the grids for 4 hours, and then process as described in steps 24-31 above.
40. If any grids are warped, reshape them before proceeding with the autoclaving. Discard any grids that begin to rust.
BASIC PROTOCOL 2: HPV Virion Isolation from Epithelial Tissues
The following protocol for extracting papillomavirus particles from raft tissues was modified from Favre et al. to include increased biological containment (Favre et al. 1975, Ozbun 2002). Sterility should be maintained to the extent possible. Briefly, the epithelial tissues devoid of the collagen matrix are broken apart with small glass beads inside a sealed homogenizer. The cellular debris and beads are removed by centrifugation at low speed, leaving the 50 nm virus particles in the supernatant. Viral genome-containing particles are then pelleted by ultra-centrifugation in a swinging bucket rotor based on a sedimentation coefficient of 296S–300S (Crawford and Crawford 1963). The virus pellets are then suspended in a small volume of buffer using a Dounce homogenizer. Any remaining debris is removed by centrifugation and the viral supernatants removed to a sterile tube. Further purification by density centrifugation is optional. Viral stocks can be stored at -80°C.
Materials
Solution A: 27.6 g NaH2PO4 • H2O per liter (0.2 M)
Solution B: 53.65 g Na2HPO4 • 7H2O per liter (0.2 M)
5 M NaCl (filtersterile)
1 M NaCl (filtersterile)
150 mM NaCl (filtersterile)
0.5 M EDTA
0.1 M Na-Phosphate Buffer, pH 8 (13.25 ml Solution A; 236.75 ml Solution B; 250 ml H2O)
0.1 M Na-Phosphate Buffer pH 7.4 (47.5 ml Solution A; 202.5 ml Solution B; 250 ml H2O)
Virus Extraction Buffer 1 - 1 M NaCl/50 mM Na-Phosphate Buffer pH 8.0 (100 ml of 5 M NaCl; 250 ml of 0.1 M Na-Phosphate Buffer, pH 8; 150 ml of dH2O; filtersterile)
Virus Extraction Buffer 2 - 50 mM NaCl/100 mM EDTA/50 mM Na-Phosphate Buffer pH 7.4 (25 ml of 1 M NaCl; 100 ml of 0.5 M EDTA; 250 ml of 0.1 M Na-Phosphate Buffer, pH 7.4; 125 ml of dH2O; filtersterile)
Vesphene 1:128 dilution
70% Ethanol
10% Chlorine Bleach
Absorbent bench paper
Autoclavable containers with lids: one for consumables and waste; one for permanent lab items (e.g., Nalgene 6910-0618 Polypropylene Autoclavable Instrument Tray)
HEPA mask 3M model 8233 respirator (N100 particulate)
BeadBeater homogenizer (BioSpec Model 1107900) www.biospec.com
Glass Beads, 1.0 mm (BioSpec, cat. no. 11079110) (autoclaved in a beaker)
BeadBeater Chambers:
350 ml polycarbonate chamber (use as 2nd containment, BioSpec, cat. no. 110792)
50 ml chamber (for ≈10-75 raft tissues, BioSpec, cat. no. 110803)
15 ml chamber (for 1-5 raft tissues, BioSpec, cat. no. 110803)
Stainless-steel 15 ml (1 TBS) measuring spoon (autoclaved)
38-ml screw top Nalgene™ Oak Ridge High-Speed Centrifuge Tubes (sterile)
Ultracentrifuge tubes (Beckman cat. no. 344058); sterilize with 70% EtOH 10 min, rinse 3× with sterile ddH2O.
High speed centrifuge
SW27 (swinging bucked) ultracentrifuge rotor
Ultracentrifuge
2-ml Dounce Homogenizers
CAUTION REMINDER: Parts of this extraction and isolation protocol may generate aerosols and/or spills. Thus, the procedures are carried out in the cell culture hood, and/or sealed containers to the extent possible. We recommend the additional precautions of HPV vaccination, and PPE (disposable Tyvek lab jackets, gloves, protective eyewear, and a HEPA mask). Spills that are potentially contaminated with virus should immediately be decontaminated using either 1:128 Vesphene® solution or 70% ethanol for 30 minutes before rinsing.
Step 1: Extracting virions from the tissues
Handle all materials in a cell culture cabinet; neutralize any spills with 70% ethanol or 10% bleach.
1.Ensure Virus Extraction Buffer 1 is at 4° C before you begin and place all tubes on ice.
2.Assemble the following inside of the cell culture cabinet: an absorbent sheet of bench protection to catch spills; two autoclavable containers with lids -- one for consumables and waste, one for permanent lab items.
3.Rinse the appropriately sized BeadBeater chambers by filling them with 70% EtOH for 10 minutes, then rinsing 3-4 times with sterile water. Let them air dry in the cell culture cabinet. This protocol is optimized for extracting groups of ≈50 organotypic tissues each.
4.Fill the BeadBeater chamber half-full with sterile 1.0 mm glass beads.
Use an autoclaved measuring spoon to transfer the beads as the autoclaving causes the beads to stick to one another.
5. Place the 50 ml conical centrifuge tubes containing ≤50 epithelial tissues each on ice.
6. Resuspend the tissues in 30 ml of Virus Extraction Buffer 1, and carefully transfer the tissues and buffer into the BeadBeater chamber.
7. Fill the remaining space in the BeadBeater with sterile beads.
8. Grind tissues in BeadBeater for 2-3 min, then check the temperature. If the temperature is approaching 50°C, remove the chamber and place on ice to cool. Grind up to 2.5 min more until no tissue chunks remain.
9. Pour the ground tissue/beads into the 38-ml high-speed centrifuge tubes. A sterile funnel may useful to minimize spillage. Balance the tubes with buffer if needed.
At this point the infectious virions should be released from the cell nuclei and thus the solutions are likely to be infectious. Neutralize any spills with 70% ethanol.
10. Carefully tighten the screw-cap lids, and wipe the exterior of the tubes and lids with 70% ethanol and centrifuge at 8000 × g for 10 min, 4°C.
11. Decant the supernatant into a labeled 50 ml conical and keep on ice. Try to avoid decanting the beads. If the supernatant has frozen, let it sit for a few minutes.
12. Re-extract the tissue debris pellets/glass beads in another 30 ml of Virus Extraction Buffer 1.
If any tissue debris remains in the original storage 50 ml conical tubes, use part of the 30 ml volume of Virus Extraction Buffer to rinse the debris out into the 38-ml high-speed centrifuge tube containing the tissue debris pellets/glass beads. Cap the tube and shake to mix the beads and debris, then pour the pellet solution back into the BeadBeater to grind again.
13. Grind in BeadBeater again for 2-3 minutes, monitoring the temperature. Pour the ground tissue/beads back into the high-speed centrifuge tubes.
14. Carefully tighten the screw-cap lids, and wipe the exterior of the tubes and lids with 70% ethanol and centrifuge at 8000 × g for 10 min, 4°C.
15. Pool the supernatants and keep on ice.
If debris remains, another high-speed centrifugation can be performed. It is important to remove as many tissue fragments as possible as they will be pelleted with the virus in the next ultracentrifugation step.
Step 2: Concentrating the HPV virions from the supernatants
Handle all materials in a cell culture cabinet; neutralize any spills with 70% ethanol or 10% bleach.
16. Fill the pre-sterilized Beckman Ultraclear centrifuge tubes with the combined supernatants, and bring the volume to within 2-3 mm of the top of the tube using Virus Extraction Buffer 1 as needed.
Consult the manual for the ultracentrifuge to determine how closely the weights of the tubes must match. Typically, tubes should be balanced to within ≤0.1 g of each other using an electronic balance.
17. Carefully lower tubes into buckets and attach the lids securely.
18. Pellet virus particles at 130,000 × g (SW27 @27K rpm) for 1 h at 4°C. The break can be engaged.
This combination of speed and time is calculated to preferentially pellet the “heavier” DNA-containing particles.
19. Remove the swinging buckets to the bucket holder. Use tweezers to release the tubes out of the buckets.
Decontaminate the buckets by rinsing them with 70% ethanol and allowing them to air dry.
20. Decant the supernatant into a sterile 50 ml conical propylene tube.
If the pellet has not formed well, this step will allow the recovery of the virus by spinning again.
21. Carefully allow the excess fluid to drip from the virus pellet.
The color may be yellow, tan, or blackish (from inert carbon on the beads). Remember, the solutions are likely infectious and should be handled appropriately.
22. Add 0.5 ml of Virus Extraction Buffer 2 to the virus pellet in each ultracentrifuge tube.
23. Use a 5-ml plastic volumetric pipet to dislodge and solubilize the pellet, and move the solution to a 2-ml glass Dounce homogenizer.
The goal is to remove as much of the pellet as possible to the glass homogenizer, where the pellet will be more fully solubilized.
24. Rinse the ultracentrifuge tube with an additional 0.5 ml of Virus Extraction Buffer 2 pipetting up and down the sides to retrieve the remainder of the pellet. Transfer the slurry to the Dounce homogenizer containing the first 0.5 ml of virus suspension.
25. Carefully, use 10-15 strokes of the Dounce glass pestle to homogenize the pellet and buffer.
26. Transfer the homogenized virus suspension to a 1.5 or 2 ml microcentrifuge tube.
27. Wipe the exterior of the tubes and lids with 70% ethanol and pellet the remaining non-viral debris by centrifugation at 8000 × g for 10 min at 4°C.
28. Use a micropipet to transfer the viral suspension (supernatant) to a clean and sterile microcentrifuge tube. Keep the suspension on ice unless ready for long-term storage.
Depending upon the efficiency of the steps above, and how many tissues were pooled for extraction, additional virus may be recovered by a second extraction of the pellets (from step 28 above).
29. Add 0.5 to 1.0 ml of Virus Extraction Buffer 2 to the ultracentrifugation tubes (if desired), then transfer with any leftover chunks to the microcentrifuge tube containing the pellet.
30. Use a 1-ml micropipet or the 2 ml pestle to break up the pellet, then transfer with a Pasteur pipette to the 2 ml Dounce homogenizer and repeat steps 25 to 27 above.
31. If desired, pool the virus-containing supernatants and note the volume.
32. At this point, the virus stock can be quantified for viral-genome equivalents, or further purified by isopycnic cesium chloride density gradient centrifugation. See unit 15C.1 for steps for CsCl gradient centrifugation.
OptiPrep™ iodixanol density gradient solution is often used for purification of papillomavirus pseudovirions, however, we find that discontinuous CsCl gradients made from 4 ml light (1.25 g/ml) CsCl underlaid with 4 ml heavy (1.4 g/ml) CsCl provides much better virion purity (Surviladze et al. 2013). It must be noted that the yield of virions from the raft system is orders of magnitude lower than that from the transfection-based packaging systems described. Thus, loss of infectious particles increases dramatically with each purification step.
SUPPORT PROTOCOL 1: Epithelial Medium (E Medium) Preparation
Non-transformed keratinocytes have fastidious growth requirements. Although they can be cultivated as monolayer cultures grown in fully defined keratinocyte serum-free media, we find the cells grow more robustly in E Medium containing 5% heat-inactivated fetal calf serum and co-cultured with mitomycin-C treated fibroblast feeder cells (irradiated fibroblasts can also be used). This appears to be especially true for low passage keratinocytes and those that maintain extrachromosomal HPV genomes.
Materials
Ultrapure water (e.g., Milli-Q system from Millipore)
Ultrapure water (autoclaved)
Dulbecco's Modified Eagle's Media (DMEM), powder, high glucose, no pyruvate, no bicarbonate for 10 L (Life Technologies-Gibco®, cat. no.12100-061)
Hams F-12 Nutrient Mix powder, 10 × 1L (Life Technologies-Gibco®, cat. no.21700-075)
HEPES Ultra Pure ([4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] Life Technologies-Gibco®, cat. no.113440411)
10,000 U/ml Pen-Strep (Life Technologies-Gibco®, cat. no. 15140-022)
Adenine, suitable for cell culture (Sigma-Aldrich, cat. no. A-2786)
Insulin, suitable for cell culture (Sigma-Aldrich, cat. no. I-6634)
Apo-Transferrin, suitable for cell culture (Sigma-Aldrich, cat. no. T-1147)
3,3′,5-Triiodo-l-Thyronine, suitable for cell culture (“T3” Sigma-Aldrich, cat. no. T-6397)
Hydrocortisone, for cell culture (Sigma-Aldrich, cat. no. H-0888)
Cholera Toxin (Sigma-Aldrich #C-8052)
NaHCO3, cell culture grade (Gibco TC grade, cat. no. 118110-025)
12.1 N HCl (conc.)
10,000 unit/ml Nystatin Suspension (Sigma-Aldrich, cat. no. N-1638)
Sterile PBS (Appendix 2A)
20 liter Carboy
Graduated beakers
Graduated cylinders
Stericap PLUS sterile bottle-top 0.22 μm filter units (Millipore, cat. no. SCGPCAPRE)
Pyrex 1-liter bottles
Step 1: Stock Solutions
Make 250 ml of 1 M HEPES Buffer, pH 7.0 by dissolving 59.575 g in 200 ml ultrapure water. Use 10 N NaOH to adjust the pH to 7.0 and qs to 250 ml and filtersterilize.
Make the 0.4 mg/ml (w/v) Hydrocortisone Stock by first dissolving 100 mg of hydrocortisone in 20 ml of absolute ethanol to make a 5 mg/ml stock. Add 19.2 ml of this 5 mg/ml stock to 220.8 ml of sterile 1 M HEPES buffer. Mix completely. Store in 20-ml aliquots in 50-ml sterile conical tubes at -20°C. Makes 240 ml (12 × 20-ml aliquots).
Make the 0.18M Adenine by dissolving 2.43 g of Adenine plus 75 ml autoclaved ultrapure H2O. Add 1.5 ml 10 N HCl. Add additional drops of HCl (≈10 or so) until dissolved; qs to 100 ml with sterile ultrapure H2O. Store in 20-ml aliquots in 50-ml sterile conical tubes at -20°C. Makes 100 ml (5 × 20-ml aliquots).
Make the 5 mg/ml (w/v) Insulin stock by dissolving 500 mg of Insulin in 100 ml of 0.1 N HCl. Mix completely until dissolved. Store in 20-ml aliquots in 50-ml sterile conical tubes at -20°C. Makes 100 ml (5 × 20-ml aliquots).
Make the 5 mg/ml (w/v) apo-Transferrin stock by dissolving 1 g plus 200 ml of sterile PBS. Mix completely until dissolved. Store in 20-ml aliquots in 50-ml sterile conical tubes at -20°C. Makes 200 ml (10 × 20-ml aliquots).
Make 2 × 10-8 M 3,3′,5-Triiodo-l-Thyronine (T3) stock by first dissolving 13.6 mg plus 100 ml of 0.02 N NaOH (to make a 2 × 10-4 M stock). Add 0.1 ml of the 2 × 10-4 M stock to 9.9 ml sterile PBS, mix well (to make a 2 × 10-6 M stock). Further dilute 1.0 ml of the 2 × 10-6 M T3 stock by adding to 99 ml of sterile PBS. Mix well and store the stocks at -20°C. Makes 100 ml of the 2 × 10-8 M T3 stock (5 × 20-ml aliquots).
Make 1 × 10-7 M Cholera Toxin by adding 1 ml of sterile ultrapure water to the 1-mg vial to make a 1 × 10-5 M cholera toxin solution. Further dilute the 1 ml of the 1 × 10-5 M cholera toxin solution with 99 ml of sterile ultrapure water. Mix well and store in 20-ml aliquots in 50-ml sterile conical tubes in the dark at -20°C. Makes 100 ml (5 × 20-ml aliquots).
Step 2: Prepare E medium
8. Add 8.0 liters of ultrapure water to a 20-liter carboy.
9. Dissolve 1 bottle of DMEM (enough for 10 liters) and qs to 2 liters with ultrapure water; add to the carboy.
10. Mix ½ of a bottle of DMEM (enough for 5 liters) plus 5 × 1-liter packets of Hams F-12 (enough for 5 liters), plus 61.38 g of NaHCO3 and qs to 2 liters with ultrapure water; add to the carboy.
11. Mix 120 ml of PBS, 20 ml of 0.18M Adenine stock, 20 ml of 5 mg/ml (w/v) Insulin stock, 20 ml of 5 mg/ml (w/v) apo-Transferrin stock, 20 ml of 2 × 10-8 M T3 stock and qs to 2 liters with 1.8 liters of ultrapure water; add to the carboy.
12. Mix 2 × 100 ml bottles of Pen-Strep with 1.8 liters of ultrapure water; add to the carboy.
13. Mix 20 ml of the 0.4 mg/ml Hydrocortisone stock and 20 ml of the 1 × 10-7 M Cholera Toxin stock and qs to 2 liters with 1.8 liters of ultrapure water; add to the carboy.
14. Mix 6.25 ml of concentrated HCl by adding to >500 ml of ultrapure water; qs to 2 liters with of ultrapure water and add to the carboy.
15. Swirl the carboy to mix thoroughly and verify that the pH is 7.4 with accurate pH strips.
16. Sterilize the media by 0.22 μm filtration using a vacuum-line bottle-top filter unit into sterile Pyrex 1-liter bottles (≈950 ml aliquots). Store at 4°C in the dark.
17. Prior to use, add 50 ml of heat inactivated FCS (final 5%) and 10 ml of 10,000 units/ml nystatin suspension (final 10 units/ml).
Support Protocol 2: Preparing the Stainless Steel Screen Support Grids
Although this section may seem mundane, it is critical to create grids that are flat and with legs that provide level elevation of the platform in order to prevent the splashing of the tissues with media and air bubbles from being trapped under the grid. An even and flat grid will also facilitate media changing over the next 12-16 days.
Materials
Chromic acid cleaning solution (dissolve 400.0 g potassium dichromate in 4000.0 ml distilled water; move to 4-liter brown glass bottle. In a fume hood, slowly add 400.0 ml conc. sulfuric acid)
Stainless steel screen disks 3.063 in. diameter, 40 mesh, 0.01 wire (Johnson screens®, Houston, TX; cat. no. JDE 244934)
Oven-bake clay kit (e.g., Sculpey® Keepsake® ornament kit with 4 inch diameter casting ring, available at hobby stores)
2-liter Pyrex® beakers
-
Lay the screen disk on the grin-bending device (see below) and press the test tube into to indent the screen disk to create three equally-spaced legs of similar height (≈2.5-3 mm).
An efficient grid-bending device can be created by using the template in Figure 3, a 5-ml test tube, and an oven-bake clay ornament kit.
Looking across the horizontal surface, be certain the grid is a flat as possible. This is best adjusted by gently flexing the screen.
The flat grids with legs can be loosely stacked in a 2-liter Pyrex® beaker to within 3 cm of the top.
Under a fume hood, fill the beaker containing the grids with chromic acid cleaning solution until the grids are covered. Leave 1 hr.
Decant the acid solution back into the dark container. The acid can be reused until it turns dark brown.
Fill the beaker and rinse the grids 3-5 times with tap H2O in the fume hood to remove residual acid. Discard rinse per institutional safety precautions.
If possible, attach a hose to tap H2O and place the hose beneath the lowest grid in the beaker. Rise with tap H2O for 24 hr.
Using a tube connected to deionized H2O, place the hose beneath the lowest grid in the beaker and run the deionized H2O to rinse for an additional 1 to 2 hr.
Allow the grids to drain by simply tipping the beaker upside-down and leaving a few hours.
Right side the beaker containing the grids and place aluminum foil over the opening. Sterilize the grids in the beaker by autoclaving 40 min on dry cycle.
Figure 3.
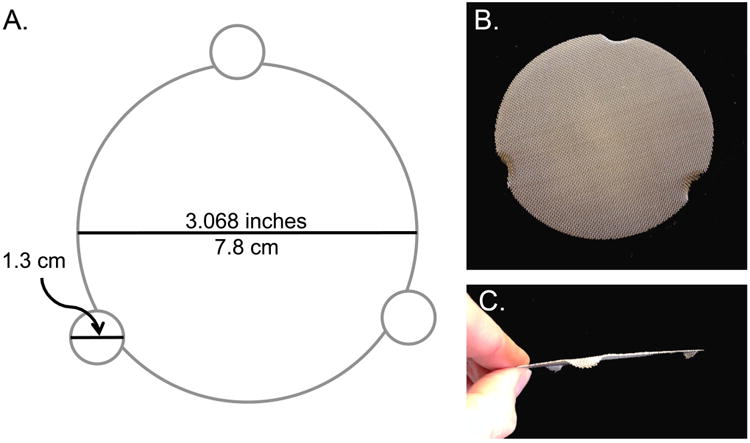
A. Template for creating an efficient grid-bending device. Copy the template above making certain the diameter is exactly the same as that of the wire grid. Cut out the template including the three notches. Follow the kit directions for preparing the clay in the plastic ring, making sure the surface is completely flat and level. Press a wire grid into the center of the clay to just make an imprint. Lay the template over the clay exactly covering the grid imprint, and gently drive the top of a 12 × 75 mm test tube straight into the clay at each of the three notches until the tube reaches the bottom surface, gently remove the clay from the hole. Carefully press the clay out of the circle form and be certain the notches are still aligned with the each other and the template and that the surface is completely level. Use the base of the test tube to be sure the holes are still properly formed. The clay can be remolded in the ring if needed. Bake the clay disk according to the kit directions. When cool, place an unbent grid onto the grid imprint of the hardened clay form and insert the base of the 12 × 75 mm test tube into each the three holes to bend the legs of the grid. Repeat to bend the needed number of grids. B, C. Top- and side-views (respectively) of a flat grid that is properly bent to have even legs.
Commentary
The regulation of the differentiation-dependent life cycles of papillomaviruses is incompletely understood, but requires stratified epithelium. The complex program of epithelial differentiation supports the later stages of the papillomavirus replicative or “vegetative” life cycle. Although there are other permutations of these protocols for growing stratified epithelium in the laboratory, the methods detailed herein give rise to a cellular environment that provide the cues and signals needed for the full papillomavirus replicative cycle and the growth of epithelium that biochemically and morphologically resembles HPV-infected skin in vivo.
Critical Parameters and Troubleshooting
Growth of cells
It is important that all cell lines have tested negative for mycoplasma. The fibroblasts and the keratinocytes should be in active growth phase prior to their use in the raft system. Thus, they should not be allowed to reach full confluence on the plates used to seed the rafts. In general, keratinocytes should never be allowed to become overly confluent during their culture as monolayers as they will begin to differentiate. This will inhibit their ability to attach upon subsequent passaging.
The best differentiation of raft tissues occurs using both J2 fibroblasts and keratinocytes that have not been subjected to long-term passaging in culture (Pourreyron et al. 2011). In general, fibroblasts should be used prior to passage 25 (ideally before passaged 20 times at a 1:10 split). Preferably, HPV-positive kerationcytes should never be split at greater than 1:6 upon passaging and should be used for rafts prior to 20 passages post cloning.
Preparation of the Collagen-Fibroblast Dermal Equivalents
Rat tail, type 1 collagen is typically solubilized in acetic acid and the solution will gel at room temperature. Thus, it is important to store and work with collagen solutions on ice. The pH of the final collagen matrix is critical to its function as a dermal equivalent; therefore, it is imperative to obtain collagen at a concentration of 3.8 to 4.0 mg/ml. This solution will have the appropriate consistency for making dermal equivalents and require a modest volume of NaOH to adjust the pH appropriately. Proper pH can be accurately assessed in the final solution by a light pink color and is adjusted with microliter volumes of 10 N NaOH. The collagen is viscous and a single, or limited number of, 10-ml plastic pipets should be used when transferring the collagen among vessels; this will avoid loss of this expensive reagent. Introduction of bubbles during mixing of the collagen-fibroblast suspension should be avoided. If bubbles appear prior to pipetting into the 6-well plates, allow the solution to sit on ice for a few minutes. Bubbles that form in the collagen plugs may be detrimental to kerationcyte differentation. If bubbles form at the top of the collagen plug after pipetting, use a Pasteur pipet to gently aspirate the bubble.
Seeding of Epithelial Cells onto the Dermal Equivalents
It is important that the keratinocytes be fully confluent on the dermal equivalents prior to lifting to the air-liquid interface. This can be improved in various ways: 1) increase the number of cells seeded; 2) allow the monolayers to grow extra days before lifting; 3) add 5-10 ng/ml of EGF to the submerged cultures to encourage proliferation and cell spreading. If more days are needed, be sure to change the media every two days. It is better to let the keratinocytes grow an extra day to ensure confluence then to lift the rafts prematurely.
Lifting Matrixes to the Air-Liquid Interface
When removing the media from the collagen matrixes, take care not to place the aspirating pipet too close to the collagen as the gel is easily damaged and/or dislodged from the plastic plate by strong suction. Dislodging the gel can render the matrix unusable if it flips, and/or it may be impossible to use the spatula to move the gel to the grid thereafter. If the matrix flips during the process, attempt to right the collage gel; typically, if this happens it may not be possible to discern which is the top edge with keratinocytes. An inverted microscope may be used to verify the keratinocytes are atop the matrix. Best effort should be used to flatten and spread the raft on the grid for subsequent growth.
The grid “legs” should be bent equally and such that 10 to 15 ml of media fill the space under the grid without extra room where bubbles can be trapped. Bubbles under the matrixes will prevent even feeding of the tissues. A volume of 10-ml or more of growth media are needed to provide adequate nourishment of the rafts for a 48-hr period. Grids can be checked for proper leg height and levelness at the time of bending the legs by placing in a 10-cm cell culture dish and pipetting water into the dish. Legs that differ in height will cause bubbles to be trapped under the grids and/or lead to swamping rafts with media during feeding.
Tissue Differentiation and Virus Morphogenesis
Failure of epithelial differentiation could be due to a number of issues, including (but likely not limited to) subconfluence of keratinocytes prior to lifting, wetting of the epithelium, poor quality (old or mycoplasma-infested) fibroblasts or keratinocytes, keratinocytes that are not differentiation capable due to transformation, deficient growth media, inferior quality fetal calf serum. The histological examination of the resulting tissues should provide insight if poor differentiation is suspected. The heat shock steps we have included have consistently increased the yield of HPV31 from CIN 612-9E rafts by nearly 10-fold and should do so for other HPV types as well (Song et al. 2010).
Virion Isolation
Our protocol has been modified with special attention to preventing aerosols, particularly because of the increased knowledge of high-risk human papillomaviruses in the etiology of oropharyngeal squamous cell cancers (Gillison et al. 2012). It is essential that all the tissues are sufficiently minced by the beads to release virions. There is probably a trade-off between efficient virus extraction from too many rafts in Step 1 versus the loss of particles during the process of concentration in Step 2 (or gradient purification).
Anticipated Results
Growth of Organotypic Raft Epithelial Tissues
Visually, by 8-days post lifting to the air-liquid interface, the top of the tissues should begin to appear dry and may look yellowish. This signals very good growth and differentiation. If by 10- to 12-days post lifting this does not occur, good differentiation is unlikely to be achieved. This will prevent robust production of virions. See troubleshooting section for possible causes and remedies. When histological cross sections are prepared from full-term rafts, H & E staining should reveal 8-20 layers of cells, depending upon the cell lines used. If thin areas are seen interspersed with thicker areas, this may indicate that the keratinocytes were subconfluent prior to lifting to the air-liquid interface. Whereas normal keratinocytes will differentiate such that nuclei are absent in the upper layers, HPV-infected cells tend to maintain nuclei in the upper layers. These nuclei in or near the cornified layer will be filled with virus particles as evidenced by electron microscopy (Meyers et al. 1992, Meyers et al. 1997) and by immunostaining for capsid proteins (See Figure 1B). Immunostaining should reveal keratin 10 (K10) in the suprabasal layers. Staining for fillagrin should be evidenced in the granular and cornified layer. We have found this marker of differentiation correlates well with virion production.
HPV Virion Isolation from Epithelial Tissues
Based on qPCR quantification of viral genome equivalents (VGE) in isolated virion preparations from rafts, we consistently obtain HPV18 and HPV31 stocks yielding 1-3 × 107 VGE per raft (≤1 vge/cell or 4×105 vge per mg tissue). The amounts of HPV16 tend to be lower.
Estimated Time Requirements
Basic Protocol 1 will be completed over a few days, depending upon the growth of the cells. The time estimation is for the preparation of 10 rafts. For an experienced laboratorian performing this technique for the first time, it is recommended to begin with 10-20 rafts until comfortable with all the manipulations.
Preparing the dermal equivalents (Stage 1): After the J2 fibroblasts have been trypsinized, the hands-on time will be about 60-90 minutes.
Seeding of epithelial cells onto the dermal equivalents (Stage 2): After the keratinocytes have been trypsinized, the hands-on time will be 20-40 minutes.
Preparing the stainless steel support grids should take 15-20 minutes of hands-on time.
Lifting the rafts on to prepared grids in cell culture dishes (Stage 3) will take about 15-30 minutes.
Estimate 15-30 minutes to change the media (Stage 4) if feeding a total of 5 dishes. Harvesting a total of 10 rafts for virion isolation will take about 15-30 minutes. Estimate twice the time if also harvesting tissues for histology.
Basic Protocol 2: Tissue homogenization and virion isolation will take about 6-8 hours of time from start to finish. Handling of infectious materials can be tense and making certain all the containment, decontamination, and personal protective equipment are in place will make this easier.
Preparation and filtration of E medium will take about 4 hours if all the stock solutions are premade.
Acknowledgments
This work was supported by the following grants to M.A.O.: NIH R01CA85747, R01CA132136 and American Cancer Society Grants IRG-192 and RPG-00-276.
Literature Cited
- Asselineau D, Prunieras M. Reconstitution of ‘simplified’ skin: control of fabrication. Br J Dermatol Suppl. 1984;27:219–221. doi: 10.1111/j.1365-2133.1984.tb15608.x. [DOI] [PubMed] [Google Scholar]
- Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. Arrangement of L2 within the Papillomavirus Capsid. J Virol. 2008;82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck CB, Pastrana DV, Lowy DR, Schiller JT. Efficient intracellular assembly of papillomaviral vectors. J Virol. 2004;78:751–757. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford LV, Crawford EM. A comparative study of polyoma and papilloma viruses. Virology. 1963;21:258–263. doi: 10.1016/0042-6822(63)90265-4. [DOI] [PubMed] [Google Scholar]
- Favre M, Breitburd F, Croissant O, Orth G. Structural polypeptides of rabbit, bovine, and human papillomaviruses. J Virol. 1975;15:1239–1247. doi: 10.1128/jvi.15.5.1239-1247.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillison ML, Alemany L, Snijders PJF, Chaturvedi A, Steinberg BM, Schwartz S, Castellsagué X. Human papillomavirus and diseases of the upper airway: head and neck cancer and respiratory papillomatosis. Vaccine. 2012;30(Suppl 5):F34–54. doi: 10.1016/j.vaccine.2012.05.070. [DOI] [PubMed] [Google Scholar]
- Kopan R, Traska G, Fuchs E. Retinoids as important regulators of terminal differentiation: examining keratin expression in individual epidermal cells at various stages of keratinization. J Cell Biol. 1987;105:427–440. doi: 10.1083/jcb.105.1.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCance DJ, Kopan R, Fuchs E, Laimins LA. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc Natl Acad Sci U S A. 1988;85:7169–7173. doi: 10.1073/pnas.85.19.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers C, Frattini MG, Hudson JB, Laimins LA. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science. 1992;257:971–973. doi: 10.1126/science.1323879. [DOI] [PubMed] [Google Scholar]
- Meyers C, Mayer TJ, Ozbun MA. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J Virol. 1997;71:7381–7386. doi: 10.1128/jvi.71.10.7381-7386.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbun MA. Infectious human papillomavirus type 31b: purification and infection of an immortalized human keratinocyte cell line. J Gen Virol. 2002;83:2753–2763. doi: 10.1099/0022-1317-83-11-2753. [DOI] [PubMed] [Google Scholar]
- Ozbun MA, Kivitz MP. The Art and Science of Obtaining Virion Stocks for Experimental Human Papillomavirus Infections. In: Gaston K, editor. Small DNA Tumor Viruses. Norfolk U.K.: Horizon Scientific and Caister Academic Press; 2011. pp. 19–35. [Google Scholar]
- Pourreyron C, Purdie KJ, Watt SA, South AP. Methods in Molecular Biology (Clifton, NJ) Vol. 731. Springer; 2011. Feeder layers: co-culture with nonneoplastic cells; pp. 467–470. [DOI] [PubMed] [Google Scholar]
- Pyeon D, Lambert PF, Ahlquist P. Production of Infectious Human Papillomavirus Independently of Viral Replication and Epithelial Cell Differentiation. Proc Natl Acad Sci U S A. 2005;102:9311–9316. doi: 10.1073/pnas.0504020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden RBS, Lowy DR, Schiller JT. Papillomavirus is resistant to desiccation. J Infect Dis. 1997;176:1076–1079. doi: 10.1086/516515. [DOI] [PubMed] [Google Scholar]
- Song H, Moseley P, Lowe SL, Ozbun MA. Inducible heat shock protein 70 enhances HPV31 viral genome replication and virion production during the differentiation-dependent life cycle in human keratinocytes. Virus Res. 2010;147:113–122. doi: 10.1016/j.virusres.2009.10.019. [DOI] [PubMed] [Google Scholar]
- Surviladze Z, Sterk RT, De Haro SA, Ozbun MA. Cellular Entry of Human Papillomavirus Type 16 Involves Activation of the PI3K/Akt/mTOR Pathway and Inhibition of Autophagy. J Virol. 2013;87:2508–2517. doi: 10.1128/JVI.02319-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Michniak-Kohn BB. Tissue engineered human skin equivalents. Pharmaceutics. 2012;4:26–41. doi: 10.3390/pharmaceutics4010026. [DOI] [PMC free article] [PubMed] [Google Scholar]