

Abstract
Chronic lymphocytic leukemia B cells express auto/xeno antigen-reactive antibodies that bind to self-epitopes and resemble natural IgM antibodies in their repertoire. One of the antigenic structures recognized is oxidation-induced malonedialdehyde that is present on low-density lipoprotein, apoptotic blebs, and on certain microbes. The poor-prognostic stereotyped subset #1 (Clan I IGHV genes-IGKV1(D)-39) express IgM B-cell receptors that bind oxidized low-density lipoprotein. In this study, we have used for the first time this authentic cognate antigen for analysis of downstream B-cell receptor-signal transduction events, since it is more faithful to B-cell physiology than anti-IgM. Multivalent oxidized low-density lipoprotein showed specific binding to subset #1 IgM/IgD B-cell receptors, whereas native low-density lipoprotein did not. The antigen binding induced prompt receptor clustering followed by internalization. However, the receptor-signal transduction was silenced, revealing no Ca2+ mobilization or cell-cycle entry, while phosphorylated extracellular-regulated kinase 1/2 basal levels were high and could not be elevated further by oxidized low-density lipoprotein. Interestingly, B-cell receptor responsiveness was recovered after 48-h culture in the absence of antigen in half of the cases. Toll-like receptor 9-ligand was found to breach the B-cell receptor-signaling incompetence in 5 of 12 cases pointing to intra-subset heterogeneity. Altogether, this study supports B-cell receptor unresponsiveness to cognate self-antigen on its own in poor-prognostic subset #1 chronic lymphocytic leukemia, indicating that these cells proliferate by other mechanisms that may override B-cell receptor silencing brought about in a context of self-tolerance/anergy. These novel findings have implications for the understanding of chronic lymphocytic leukemia pathobiology and therapy.
Introduction
Molecular and functional studies support the concept that chronic lymphocytic leukemia (CLL) cells are antigen-experienced. Alternatively, CLL may be derived from a specialized subset of self-replenishing innate B1-like cells that produce natural IgM antibodies (Abs) without apparent antigen exposure early in life.1–3 From an immunogenetic perspective, perhaps the strongest evidence for antigen involvement is the existence of different subsets of cases with highly similar or ‘stereotyped’ immunoglobulins (Igs) in their B-cell receptors (BcRs),4–7 indicating selection by antigen in CLL ontogeny.8,9 Several self and exogenous antigens (i.e. non-muscle myosin, vimentin, cofilin, oxLDL, bacterial polysaccharides, fungal β-glycans) are considered to be involved in the initiation and/or progression of CLL,2,10–13 in synergy with microenvironment factors,14–17 that signal via innate immune receptors, such as Toll-like receptors (TLRs).18
Within stereotyped subsets, similarities between different cases extend from BcR Ig sequences to biological and prognostic features, clinical presentation, and even outcome. For example, patients assigned to subset #4 (IGHV4-34/IGKV2-30) are relatively young at diagnosis,7 uniformly express mutated IgG-switched Igs with pronounced intraclonal diversifications,7–9,19 display distinctive gene expression and microRNA profiles,20–22 and follow an indolent disease course.7,23 In contrast, patients belonging to subset #1 (Clan I IGHV/IGKV1(D)-39) uniformly express unmutated BcR Igs, exhibit a distinctive gene expression signature with several differentially expressed transcripts involved in BcR signaling, apoptosis regulation, cell proliferation and oxidative processes,24 and suffer from a poor prognosis even when compared to other patients with unmutated IGHV genes.7,23
Recent studies from our group and others showed that CLL Abs may recognize oxidation-specific neoepitopes on lipoproteins and apoptotic cells. It is worthy of note that these structures may also share molecular identity with epitopes on infectious pathogens (molecular mimicry).2,11,25 In mice and men, natural germ-line Abs bind to these oxidation-specific epitopes and IgM anti-oxLDL Abs are frequently found in the circulation of healthy persons.1 For this reason, we have proposed that CLL cells may be derived from self-reactive natural Ab-producing B cells that are part of the innate first-line defence.2,3
In addition to natural Abs, there are numerous mechanisms to protect against the presence of oxidation-induced self-antigens and microbes. These include TLRs, scavenger receptors (SRs), i.e. CD5, CD6, CD36, complement, C-reactive protein, mannose binding protein,1 and they constitute a link between innate and adaptive immune responses. In particular, TLR stimulation provides a signal that synergizes with BcR triggering,26 and T cells help to amplify human B-cell responses to antigen.27 Hence, stimulation with TLR agonists increases the expression of co-stimulatory molecules, which in turn raise the surface expression of activation markers such as CD25 and CD86.28–30
Overall, CLL cells display TLR expression profiles similar to those of memory cells, supporting the assumption that they are antigen-experienced.20,21,31 As we recently showed, however, subgroups of CLL cases, especially those belonging to subsets with stereotyped BcRs, exhibited differential responses to immune stimulation through the TLRs, and these responses may extend to cell proliferation control, apoptosis, B-cell anergy, or TLR tolerance.21
Stereotyped subset #1 is the largest subset among the IGHV-unmutated CLL with frequencies of approximately 2.5–3%.4,6,7,23 We recently found that IgM from subset #1 CLL cells bind to oxidized phospholipids,2 and more specifically to malondialdehyde (MDA), a major degradation product of unsaturated lipids reacting with reactive oxygen species. The discovery of subset #1 antigenic targets enabled us to study the contribution of a specific and natural (polyvalent) antigen in triggering proliferation and/or differentiation of CLL cells.
In the present work, we hypothesized that purified oxLDL, a multivalent cognate T-independent antigen (containing several MDA epitopes), could induce a full proliferative response on its own in this aggressive subset; this was based on previous studies showing that antigen alone could induce proliferation.2,13,32 By analyzing BcR-signaling events, we found, however, that oxLDL alone was not sufficient for induction of Ca2+-flux or elevation of phosphorylated extracellular regulated kinase 1/2 (pERK1/2), indicating that cell proliferation in this progressive CLL subset is brought about by a constellation of factors superimposed on BcR signaling.
Methods
Patients
Peripheral blood mononuclear cells (PBMCs) were collected from 12 consenting CLL patients diagnosed according to revised guidelines of the National Cancer Institute Working group. All patients were either untreated or had been off therapy for at least six months before sampling. Patients’ clinical data are shown in Table 1. The study was approved by the local ethics committee (Diary n. M30-05).
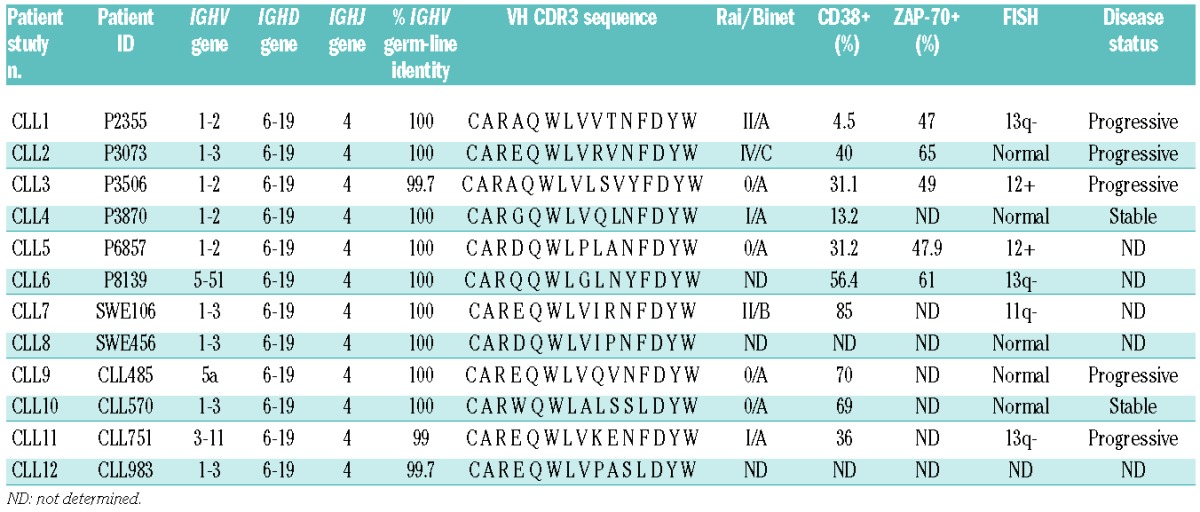
Table 1.
Characteristics of the subset #1 CLL patient cases.
Antibodies and flow cytometry
Study Ab reagents are listed in Online Supplementary Table S1. Antigens or Abs were incubated for 30 min on ice. Live CLL cells were always detected using anti-CD5 and anti-CD19 Abs gated on annexinV-negative cells. Cell surface markers were analyzed in a Gallios flow-cytometer using Kaluza software v.1.2 (Beckman Coulter, Brea, CA, USA).
Cell cultures
PBMC (45–98% CD5+/CD19+) or MACS microbead negatively-selected (Miltenyi Biotec, Auburn, CA, USA) CD5+/CD19+ cells (≥98% CD5+/CD19+) were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) (Invitrogen) for 2–3 days. Cells were stimulated with 25 μg/mL oxLDL, 5 μg/mL CpG-oligodeoxynucleotide (CpG), 25 μg/mL native-LDL (nLDL), 10 μg/mL goat anti-IgM F(ab´)2, or 50 ng/mL PMA.
PCR amplification and sequence analysis of IGHV-IGHD-IGHJ rearrangements
PCR amplification and sequence analysis of IGHV-IGHD-IGHJ rearrangements and assignment to stereotyped subsets were performed as previously described.4
LDL and modifications
nLDL was isolated by sequential ultracentrifugation from pooled healthy donor plasma and MDA modification was performed as previously described.33 In this paper, MDA-LDL is termed oxLDL.
Competition oxLDL ELISA
Ab binding to oxLDL-coated MicroFluor plates (Dynatech Laboratories, Chantilly, VA, USA) was performed as detailed previously,2 using primary CLL IgM Abs (<3 μg/mL), oxLDL or nLDL competitors in various concentration, and anti-IgM alkaline phosphatase-conjugate (ALP) and LumiPhos530 substrate (Lumigen, Southfield, MI, USA).
Immunofluorescence
Cells were incubated with 50 μg/mL oxLDL-biotin or anti-IgM-biotin for 30 min on ice, washed and incubated at 37°C for 0 and 60 min. Cells were fixed on microscope slides with 4% paraformaldehyde (PFA), permeabilized with 0.1% saponin, and incubated with Streptavidin-Alexa488.
Intracellular Ca2+-flux
Intracellular Ca2+-flux was measured as described, using a threshold of 5% or more responding cells.34 Cells were stimulated with 20 μg/mL goat anti-human IgM F(ab´)2, 50 μg/mL oxLDL, 10 μg/mL CpG. Data were acquired in a BD FACSCalibur (BD Biosciences, San José, CA, USA) and analyzed with FlowJo software v.7.2.4 (Tree Star, Ashland, OR, USA).
Analysis of proliferation by BrdU-incorporation
Magnetic-bead selected CLL cells (≥98% CD5+/CD19+) or CLL PBMC (45–98% CD5+/CD19+) were stimulated as described for 56 h followed by addition of 10 μM 5-bromo-2′-deoxyuridine (BrdU) for 16 h. Medium alone was used as control. Cells were stained with anti-BrdU mAb (BD Biosciences). Cells were analyzed in a BD FACSCalibur using Kaluza software.
IgM ELISA and cytokine assay
Cell supernatants were analyzed for 10 different cytokines, IL1β, IL-2, IL-4, IL5, IL6, IL8, IL10, TNF-α, IFN-γ, and GM-CSF, using Luminex Human Cytokine TenPlex-kit (Invitrogen), and for secreted IgM using ELISA as described.2
Western blot
CLL cells were cultured in the presence or absence of 10 μg/mL anti-human IgM F(ab′)2 or 50 μg/mL oxLDL for indicated time points. Cell lysates were separated by gel electrophoresis and analyzed for pERK1/2 (Thr202/Tyr204) and total ERK1/2. Densitometric analysis of ERK-specific bands was performed using ImageLab software v.4.1 (Bio-Rad, Hercules, CA, USA). OD ratios for pERK/total ERK were calculated.
Results
Subset #1 cells bind and internalize oxLDL through the clonotypic BcR
We previously showed that monoclonal IgM Abs from CLL subset #1 bind to oxidized phospholipids.2 Here specificities of IgM Abs from CLL subset #1 patients (n=6) were further evaluated in competition ELISA. All Abs showed specific binding to oxLDL, whereas none bound to nLDL with the exception of CLL6, which showed partial affinity for nLDL (Figure 1A). The avidity (e.g. accumulated strength of multiple binding site affinities) of the subset #1 IgM Abs was in the range 1–10 pM as estimated from IC50 values. Furthermore, flow analysis of oxLDL surface-binding to subset #1 CLL patient cells (n=5) showed a mean of 79% positive cells (range 49–96%), whereas nLDL (included as specificity control) did not bind (Figure 1B). Surface BcR-oxLDL-interaction was further verified in a kinetic live cell analysis with oxLDL-biotin, which showed potent binding at onset (T0h) followed by cross-linking of BcR and receptor uptake at 60 min (Figure 1C). We next explored whether oxLDL also bound to SRs, previously reported to be expressed in a fraction of B cells including CLL cells.35,36 To this end, we first determined sIgM levels; these were low (mean MFI=2.3) compared with normal B cells (mean MFI=17) as was to be expected in CLL (Figure 1D and Online Supplementary Figure S1). The expression of sIgM strongly correlated in a linear fashion with oxLDL binding sites (R2=0.9482; P=0.0051) (Online Supplementary Figure S2A) indicating that oxLDL almost exclusively binds to BcRs and not to other receptors in these cells. Secondly, receptor-inhibition analysis showed that anti-IgM Ab (anti-μ+anti-κ), and anti-IgD Ab (anti-δ+anti-κ) (Online Supplementary Figure S2B) blocked oxLDL binding sites in a linear fashion in the range of 0.01–10 μg/106 cells. Scavenger receptor CD36 expression was analyzed and found to be absent in subset #1 cells, as determined with 2 different Abs (Figure 1E), and oxLDL binding was not blocked by anti-CD36 Abs. Preliminary data show that scavenger receptors SR-B1 and SR-PSOX were expressed at low levels in subset #1 cells (data not shown). Taken together these results indicate that oxLDL preferentially binds to BcRs and that binding to SRs is negligible. It has been suggested that SRs can be induced by TLR-ligands;37 however, we found no changes in SR (CD36 or SR-B1) expression after 24-h stimulation of subset #1 cells with oxLDL + CpG (data not shown).
Figure 1.

Subset #1 CLL cells bind and internalize oxLDL through BcR. (A) Specificity of secreted subset #1 CLL IgM Abs tested in competition ELISA. B/B0 indicates ratio of IgM bound in presence of competitor (oxLDL or nLDL) divided by IgM bound without competitor. (B) Live subset #1 CLL cells bind oxLDL. Cells were incubated with biotinylated oxLDL and Streptavidin-Alexa488. Streptavidin-Alexa488 (Ctrl) and nLDL were used as controls in flow cytometry. Gating was performed on CD5+/CD19+/annexinV-cells. (C) BcR clustering and internalization by oxLDL. (Left) At time T0 subset #1 CLL cells after pre-incubation for 30 min on ice with oxLDL-biotin. (Middle) BcR-oxLDL receptor internalization after 60 min at 37°C. (Right) Negative control Streptavidin-conjugate only. Cells were examined in a Zeiss Axiovert 200 fluorescence microscope equipped with a 63X planar objective and images were collected with an AxioCam MRm CCD camera (Carl Zeiss, Heidelberg, Germany). (D) IgM surface expression of 8 subset #1 CLL cases. Only CD5+/CD19+/annexinV-cells were analyzed. (E) Surface expression of CD36 (stained with anti-CD36-PE, clone CD38 from BD) in viable CD5+/CD19+ cells was analyzed by flow cytometry in 4 subset #1 CLL cases. Healthy donor PBMC served as positive control. The flow diagrams represent one of nine experiments performed on subset #1, non-subset #1, and normal B cells, including staining with different CD36 Abs (clones CD38 and 336213 from BD and BioLegend, respectively).
oxLDL-BcR-ligation does not induce Ca2+-flux in subset #1 CLL cells
BcR-signaling competence (at the single cell level) was determined by intracellular Ca2+ mobilization after exposure to oxLDL; surrogate antigen control was anti-IgM F(ab′)2. Cognate antigen oxLDL did not induce a detectable level of Ca2+-flux in any of the subset #1 patients’ cells (Figure 2A and Online Supplementary Figure S3). Anti-IgM F(ab’)2 response was also very low; in 3 of 7 cases, less than 5% of cells released Ca2+ (termed non-responders), and in 4 of 7 cases, 5% or more (but less than 20% of cells in this study) were positive (termed responders). CpG, alone or in combination with cognate antigen oxLDL also failed to induce Ca2+-flux. The Ca2+ ionophore, ionomycin, was used as positive control showing signaling competence in all included cases. Data in Figure 2B illustrate, as an example, one of the 7 cases represented in Figure 2A; all 7 cases can be found in Online Supplementary Figure S3. A CLL non-subset #1 clone (KJ) was used as a positive control (to check the stimulatory capacity of anti-IgM F(ab´)2), showing 45% responding (Ca2+-releasing) cells.
Figure 2.
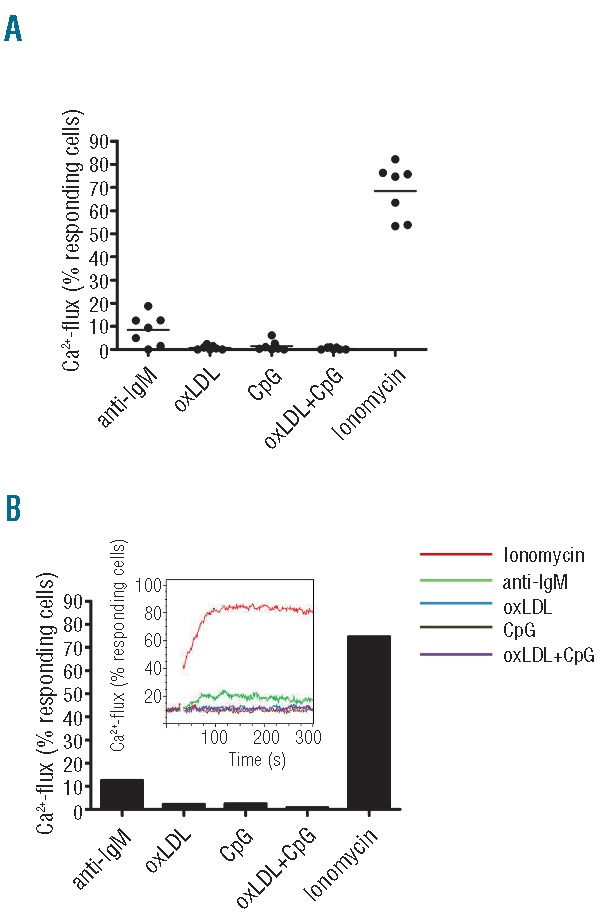
oxLDL ligation does not induce Ca2+-flux. (A) Subset #1 lymphocytes from 7 CLL cases were analyzed for Ca2+-release. Cells were loaded with the Ca2+-sensitive dye Fluo-4-AM and analyzed by flow cytometry before and after addition of oxLDL (50 μg/mL), anti-IgM F(ab´)2 (20 μg/mL), and/or CpG (10 μg/mL). The calcium ionophore ionomycin was used as a positive control. The dot plot shows percent of responding cells of total viable cells. (B) The Ca2+-mobilization data of CLL2 (1 of the 7 CLL cases included in A) is shown for clarity. The diagrams show percent responding cells of total viable cells. The insert shows flow image for Ca2+-flux responses. (See Online Supplementary Figure S2 for all flow images).
Molecular mechanisms maintaining BcR unresponsiveness to oxLDL in subset #1
The subset #1 cell unresponsiveness to oxLDL and anti-IgM prompted us to analyze this condition in detail. Purified (≥98% CD5+/CD19+) leukemic B cells from 9 patients were stimulated with oxLDL (50 μg/mL, 5 min, 37°C) or anti-IgM (10 μg/mL, 5 min, 37°C) and analyzed for ERK1/2 phosphorylation by Western blot. Constitutive (high) phosphorylation of ERK1/2 at basal (ex vivo) level was observed in all 9 subset #1 cases (Figure 3A). The cells could not be stimulated further with oxLDL (n=7) or anti-IgM (n=9) (Figure 3A). There was no significant difference between pERK/ERK ratios in stimulated cells and ratios in unstimulated cells (P=0.8).
Figure 3.
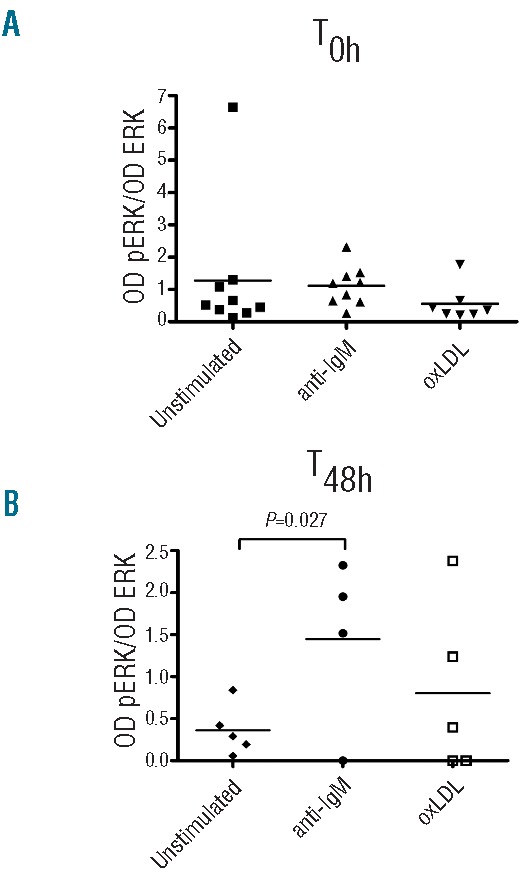
BcR responsiveness at start (0 h) and after 48 h of in vitro culture. (A) Subset #1 CLL cells were analyzed for phosphorylation of ERK1/2 by Western blot analysis at basal level (T0h), and (B) after 48-h in vitro culture (T48h). Cells were maintained in RPMI 1640 medium with 10% FCS (unstimulated control) or stimulated at both time points with 50 μg/mL oxLDL or 10 μg/mL anti-IgM F(ab´)2 for 5 min. Densitometric analyses of all the Western blot analyzed samples are shown and data are presented as optical density (OD) ratio of pERK/OD of total ERK1/2. Individual CLL cases and mean values are shown.
We next evaluated whether the BcR-responsiveness could be recovered after 48 h in culture (without antigen). Leukemic ex vivo cells from 5 patients were kept in culture for 48 h after which they were either unstimulated (n=5), or stimulated with anti-IgM (n=4) or oxLDL (n=5), and again analyzed for ERK1/2 phosphorylation (Figure 3B). As previously reported for anergic cells, ERK1/2 phosphorylation was decreased after in vitro culture,38 and 3 of 5 of the cultured leukemic cells also regained their ability to respond to BcR-triggering (Figure 3B), indicating that continuous autoantigen occupancy may be critically implicated in attenuated BcR triggering, similar to reports in other contexts.39 The elevated level of pERK1/2 after anti-IgM exposure was statistically significant (P=0.027), whereas the elevation seen after oxLDL-stimulation was not (Figure 3B). The data plotted as fold increase of pERK/total ERK showed a 2.5–3.0-fold increase after the in vitro relaxation time (‘antigen wash-out time’) (Online Supplementary Figure S4A and B).
oxLDL does not induce cell cycle entry in subset #1 CLL cells but TLR9 can breach BcR-silence in a proportion of cases
Since oxLDL is a multivalent low-affinity T-independent antigen, we hypothesized that it would induce efficient proliferative drive. However, we found that oxLDL failed to induce cell cycle entry of subset #1 cells and neither did anti-IgM F(ab´)2 (Figure 4A). CpG induced a low level of proliferation, in line with previously reported data40 (Figure 4A), and we observed no additive effect of oxLDL + CpG (Figure 4A) and no significant pERK1/2 elevation after anti-IgM, anti-IgM+CpG, oxLDL or oxLDL + CpG stimulation in 4 patients (Figure 4B). WB image from CLL7 exemplifies the data (Figure 4C). The data from these 4 patient cases are also included in the group of 9 cases presented in Figure 3 (T0h).
Figure 4.
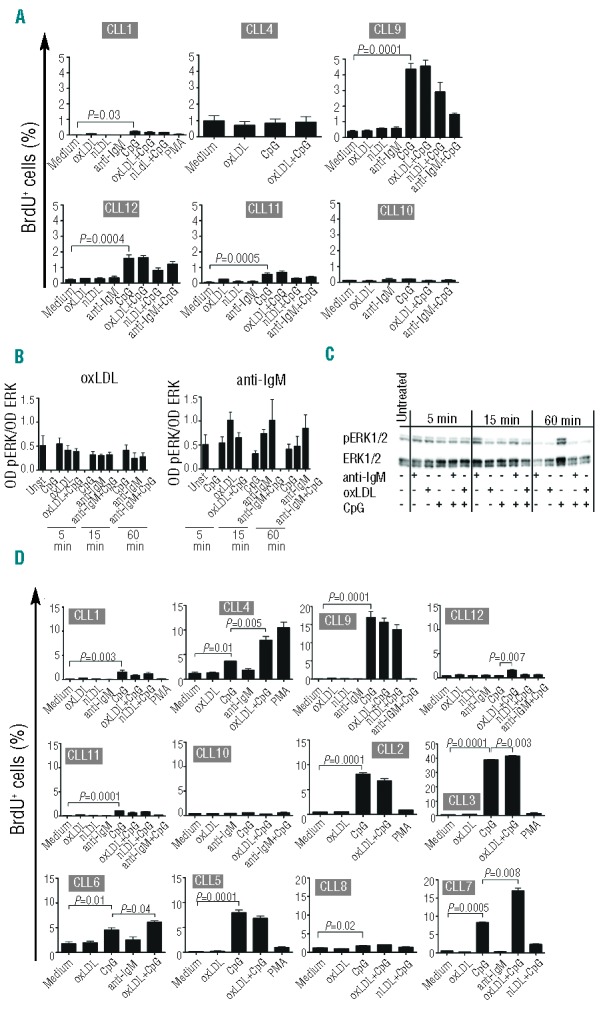
oxLDL does not induce cell cycle entry. (A) Isolated subset #1 CLL cells (≥98% CD5+/CD19+) from 6 patients were cultured in the presence or absence of oxLDL (25 μg/mL), nLDL (25 μg/mL), anti-IgM F(ab´)2 (10 μg/mL), PMA (50 ng/mL) and/or CpG (5 μg/mL) for 72 h. BrdU was added to cells during the last 16 h followed by development with anti-BrdU Ab. (B) Isolated subset #1 CLL cells (≥98% CD5+/CD19+) from 4 patients were untreated or stimulated as indicated for 5, 15, and 60 min and analyzed for ERK1/2 phosphorylation and total ERK1/2 protein by immunoblotting. Densitometric analyses are shown and data are presented as optical density (OD) of pERK/OD of total ERK ratio. (C) Western blot analysis of phosphorylated ERK1/2 and total ERK1/2 in one representative case (CLL7). (D) CLL subset # 1 cells (PBMC fraction with 45–98% CD5+/CD19+) from 12 patients were cultured as in A. B-cell proliferation was determined in (A) and (D) as BrdU-positive cells in flow cytometry. CD5+/CD19+-gated cells were analyzed. Data are expressed as mean ± SEM.
In a second series of proliferation/BrdU-incorporation experiments, we analyzed 12 subset #1 cases using PBMCs containing a low fraction of T cells, which may contribute to B-cell proliferation. However, there was no response to oxLDL alone (Figure 4D), and nor to anti-IgM or nLDL. In 10 of 12 cases, as expected, a significant proliferative response to CpG was detected.40 Interestingly, we found that TLR9/CpG ligation could breach the silent state in 5 of the 12 cases (CLL3, CLL4, CLL6, CLL7, CLL12) (Figure 4D), where we observed a low, but significant, augmentation of response to oxLDL in combination with CpG. However, since 7 of 12 cases were not affected, these results reveal intra-subset heterogeneity.
IgM, IL6, and IL10 secretion in subset #1 CLL cells after oxLDL and CpG stimulation
IgM released from the 12 subset #1 cases analyzed above were also determined. Antigen alone did not induce IgM release, whereas CpG triggered secretion in 11 of 12 cases. The combination of oxLDL and CpG augmented IgM release in 4 of 11 cases only (Online Supplementary Figure S5).
Ten different cytokines (IL1β, IL-2, IL-4, IL5, IL6, IL8, IL10, TNF-α, IFN-γ and GM-CSF) were analyzed in 3 subset #1 cases after 72-h stimulation with oxLDL ± CpG. OxLDL alone did not generate any cytokines, whereas CpG induced prompt and significant IL6 and IL10 release from all 3 cases. Finally, combined treatment with CpG and oxLDL had no significantly different effects compared to CpG alone (Online Supplementary Figure S6). No significant release of the other 8 cytokines was found after stimulation as compared to unstimulated cells, except for IL4 that was induced by CpG (data not shown).
CD25 and CD86 expression after TLR9 activation
Expression of the co-stimulatory molecules CD25 and CD86 was analyzed after activation of TLR9 with or without BcR engagement. After addition of oxLDL alone, there was a significant increase in CD86 expression (Online Supplementary Figure S7A) in only 2 of 5 cases (CLL1 and CLL4). This was not observed with the control antigen nLDL. CD86 expression was also significantly further increased in these 2 cases when stimulating cells via TLR9 with CpG (P=0.001 for CLL1 and CLL4) together with oxLDL, but not nLDL, compared with TLR-ligand alone (Online Supplementary Figure S7A). Expression of CD25 was not affected by oxLDL exposure, yet it was increased significantly by CpG (Figure 5B and Online Supplementary Figure S7).
Figure 5.
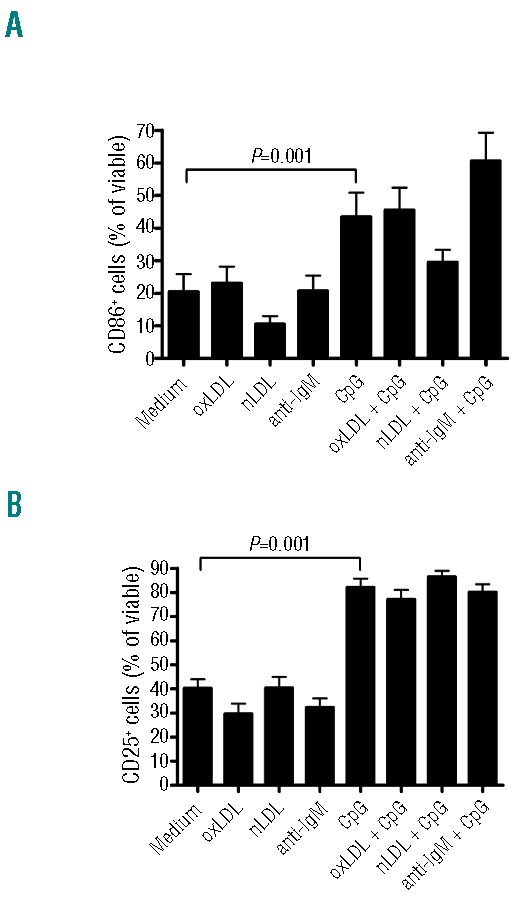
Surface expression of CD25 and CD86 after BcR and TLR stimulation. CLL subset #1 cells from 5 patients were cultured in the presence or absence of oxLDL (25 μg/mL), nLDL (25 μg/mL), anti-IgM F(ab′)2 (10 μg/mL), and/or CpG (5 μg/mL) for 24 h. Surface expression of CD86 (A) and CD25 (B) was measured with flow cytometry. Only annexinV negative cells were analyzed. Data are expressed as mean ± SEM.
Taken together, CpG was the only stimulant that significantly induced CD25 and CD86 in subset #1 CLL cells (Figure 5A and B).
Discussion
In the present study, we explored the potential role of antigen stimulation in the clonal dynamics of CLL using a natural multivalent (auto)antigen oxLDL shown to interact with good avidity (IC50 1–10 pM) with the BcR of stereotyped subset #1 CLL. This is one of the largest subsets (2.5–3% of the entire cohort)4,7 and the patients have a poor prognosis with short time to treatment.
Whereas most previous studies have investigated BcR-signaling in CLL using surrogate antigen (anti-IgM and/or anti-IgD),34,38 we explored BcR-to-cognate antigen interaction, which we consider more faithful than the in vivo situation. The antigen was analyzed alone or in combination with TLR ligands on the grounds that TLRs exert co-stimulatory effects on the BcR. Cognate antigen has the advantage over surrogate antigens (anti-IgM or IgD) in functional studies of CLL cells since it allows for multiple (low-affinity) interactions with both sIgM, sIgD, and potentially with B-cell SRs such as SR-B1, SR-PSOX and CD36. Receptor-inhibition analysis confirmed that oxLDL preferentially binds to sIgM and sIgD in stereotyped subset #1 cells, whereas SRs did not interact with oxLDL as judged by the low or absent expression and lack of anti-SR blocking capacity. It is worthy of note that oxLDL-binding is not unique to subset #1 CLLs and binding to IgM from certain non-subset #1 CLL e.g. IGHV1-69UM, IGHV3-30.3UM/subset#32, IGHV1-2UM, IGHV3-21UM, IGHV3-30M, as well as CLL subsets #6, #8, and #9 has been reported,2,25,41 underscoring the importance of these natural Abs in innate immunity.1
The main finding of this study is that the multivalent cognate antigen oxLDL can bind, cluster and internalize BcR of subset #1 CLL cells. Yet on its own this is not translated into proliferative signals due to a silenced BcR pathway, as evidenced by absence of Ca2+-flux, elevated basal ERK1/2 phosphorylation, and lack of proliferative antigenic drive.
Interestingly, half of the subset #1 cases regained BcR responsiveness (to anti-IgM or oxLDL) after 48-h culture without antigen, indicating that continuous BcR occupancy by self-antigen may be implicated in the induction of self-tolerance in subset #1. However, due to the relatively limited number of cases, more extended studies should be performed, including testing of different ‘antigen-washout’ times, as well as analysis of other self-antigen in different CLL subsets, in order to draw any definitive conclusions. Of note, concurrent stimulation of TLR9 by CpG was found to breach BcR silence in a sizable proportion of cases, highlighting a potential mechanism of proliferative drive that, combined with other mechanisms, e.g. high constitutive NF-κB activity or NOTCH1 mutations, recently reported to be frequent in subset #1,42 may underlie the clinical aggressiveness of CLL subset #1.
Previous studies by Stevenson’s group have shown that CLL cells are heterogeneous in their ability to respond by Ca2+-release after stimulation via the BcR (responders and non-responders).34 Although responses tended to be associated with IGHV-unmutated status, still there were several non-responders also among IGHV-unmutated cases.34 Non-responsiveness appeared to be isotype specific, since cases that failed to signal via sIgM were capable of signaling via sIgD.34 In the present study, we compared oxLDL-triggered effects with those triggered by anti-IgM and found that 4 of 7 (57%) subset #1 clones actually respond to anti-IgM (range 5–20% positive cells), although all were negative with oxLDL. Thus, cognate antigen responses are less pronounced than anti-IgM responses, which may be explained by the fact that natural cognate antigens such as oxLDL bind with lower affinity to IgM and IgD, than anti-IgM, which binds with high affinity to epitopes outside the V-region of sIgM.
IgM from stereotyped subset #1 CLL cases thus resemble natural Abs that exhibit a stable and restricted repertoire against diverse structures such as phospholipids, carbohydrates, glycoproteins, and nucleic acids.43 The natural Abs are encoded by germ-line IGHV and IGHV/V genes, with no or few mutations. The relatively low antigen-binding affinities are largely compensated by the pentameric structure of secreted IgM molecules, and the apparent polyreactivity may often reflect the ubiquitous nature of the common structures they recognize, as exemplified by the oxidation-specific epitopes.1
Observations of B-cell anergy in CLL subgroups have been extended to separate subsets of CLL,28,38 and recently stereotyped subset #4, which is clinically indolent, seemed to present B-cell anergy.44 BcR unresponsiveness in a context of B-cell anergy is brought about by a condition in which self-reactive B cells are silenced upon chronic exposure to low-affinity autoantigens in vivo. Anergized B cells are characterized by low sIgM as a result of constant BcR internalization and recycling, elevated basal intracellular Ca2+ concentration, and subsequent constitutive activation of ERK1/2.45 Eventually the BcR-signaling components are re-directed to generate a block through various alteration processes.45,46 The state of paralysis may be recovered by exogenous or endogenous factors overriding the lack of response and this is relevant for understanding CLL clonal dynamics. Indeed, co-signals from exogenous microbes, or, alternatively, aberrant signals via endogenous innate receptors such as NOTCH1, may circumvent normal controls. Importantly, BcR synergizes with TLRs for efficient triggering of both normal and CLL B cells,26 while TLR signaling, which induces differentiation, has been reported to breach B-cell anergy,45 as was also the case in roughly half of the CLL cases of the present study (Figure 4D). Recent studies on PRDM1/Blimp-1, a regulator of plasmacytic differentiation, suggest that the reduced differentiation capacity seen in CLL may be a consequence of anergy.47 Most likely, therefore, a combination of several signals (including environmental/microbial TLR-ligands) are required to surpass a critical threshold for allowing S-phase entry in these CLL cells.
The functional significance of silenced or dampened BCR signaling may lie in the observation that CLL B cells (at least the unmutated CLL cases with stereotyped IgM) resemble innate B1-like cells as far as the expression of natural IgM Abs is concerned. In this cell population, it is possible that endogenous antigen binding may provide the essential survival signal, adequate to prevent clonal deletion, but not strong enough to induce Ab production and cell cycling.48 For this second signal, macrophage and stromal cell factors in the microenvironment seem to be crucial, which may also be relevant for the still elusive CLL precursor.49
In conclusion, we provide evidence for signaling incompetence with antigen alone in a unique cognate BcR-ligand setting relevant for CLL subset #1, a large and clinically aggressive stereotyped CLL subset. Our data show that self-antigens alone favor efficient maintenance of unresponsiveness. The observed BcR-signaling incompetence in the face of progressive disease suggests that other mechanisms may override the blocked BcR pathway, eventually promoting B-cell proliferation. The data presented in this study have important implications not only for an improved understanding of CLL biology, but also for ongoing and future treatment strategies targeting BcR signaling.50
Acknowledgments
We are grateful to Ms. Sirpa Reinikko and Dr. Sohvi Hörkkö for fruitful suggestions and their generosity in providing LDL.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by funding from Cariplo Foundation, Milan, Italy (K.S.), the Swedish Research Council, Swedish East Gothia Cancer Foundation, Linköping University Hospital Funds, and Swedish Cancer Association n. 3171-B04-16XBB. GSD.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009; 119(5):1335–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanemo Myhrinder A, Hellqvist E, Sidorova E, Söderberg A, Baxendale H, Dahle C, et al. A new perspective: molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood. 2008; 111(7):3838–48. [DOI] [PubMed] [Google Scholar]
- 3.Rosén A, Murray F, Evaldsson C, Rosenquist R. Antigens in chronic lymphocytic leukemia--implications for cell origin and leukemogenesis. Semin Cancer Biol. 2010;20(6):400–9. [DOI] [PubMed] [Google Scholar]
- 4.Agathangelidis A, Darzentas N, Hadzidimitriou A, Brochet X, Murray F, Yan XJ, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood. 2012;119(19): 4467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darzentas N, Hadzidimitriou A, Murray F, Hatzi K, Josefsson P, Laoutaris N, et al. A different ontogenesis for chronic lymphocytic leukemia cases carrying stereotyped antigen receptors: molecular and computational evidence. Leukemia. 2010;24(1): 125–32. [DOI] [PubMed] [Google Scholar]
- 6.Murray F, Darzentas N, Hadzidimitriou A, Tobin G, Boudjogra M, Scielzo C, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood. 2008;111(3):1524–33. [DOI] [PubMed] [Google Scholar]
- 7.Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood. 2007;109(1):259–70. [DOI] [PubMed] [Google Scholar]
- 8.Sutton LA, Kostareli E, Hadzidimitriou A, Darzentas N, Tsaftaris A, Anagnostopoulos A, et al. Extensive intraclonal diversification in a subgroup of chronic lymphocytic leukemia patients with stereotyped IGHV4–34 receptors: implications for ongoing interactions with antigen. Blood. 2009;114(20):4460–8. [DOI] [PubMed] [Google Scholar]
- 9.Kostareli E, Sutton LA, Hadzidimitriou A, Darzentas N, Kouvatsi A, Tsaftaris A, et al. Intraclonal diversification of immunoglobulin light chains in a subset of chronic lymphocytic leukemia alludes to antigen-driven clonal evolution. Leukemia. 2010; 24(7):1317–24. [DOI] [PubMed] [Google Scholar]
- 10.Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117(6):1781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu CC, Catera R, Zhang L, Didier S, Agagnina BM, Damle RN, et al. Many chronic lymphocytic leukemia antibodies recognize apoptotic cells with exposed nonmuscle myosin heavy chain IIA: implications for patient outcome and cell of origin. Blood. 2010;115(19):3907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309–12. [DOI] [PubMed] [Google Scholar]
- 13.Hoogeboom R, van Kessel KP, Hochstenbach F, Wormhoudt TA, Reinten RJ, Wagner K, et al. A mutated B cell chronic lymphocytic leukemia subset that recognizes and responds to fungi. J Exp Med. 2013;210(1):59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14(3):276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bäckman E, Bergh AC, Lagerdahl I, Rydberg B, Sundström C, Tobin G, et al. Thioredoxin, produced by stromal cells retrieved from the lymph node microenvironment, rescues chronic lymphocytic leukemia cells from apoptosis in vitro. Haematologica. 2007;92(11):1495–504. [DOI] [PubMed] [Google Scholar]
- 16.Nilsson J, Söderberg O, Nilsson K, Rosén A. Thioredoxin prolongs survival of B-type chronic lymphocytic leukemia cells. Blood. 2000;95(4):1420–6. [PubMed] [Google Scholar]
- 17.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–63. [PubMed] [Google Scholar]
- 18.Muzio M, Fonte E, Caligaris-Cappio F. Toll-like Receptors in Chronic Lymphocytic Leukemia. Mediterr J Hematol Infect Dis. 2012;4(1):e2012055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Potter KN, Mockridge CI, Neville L, Wheatley I, Schenk M, Orchard J, et al. Structural and functional features of the B-cell receptor in IgG-positive chronic lymphocytic leukemia. Clin Cancer Res. 2006;12(6):1672–9. [DOI] [PubMed] [Google Scholar]
- 20.Arvaniti E, Ntoufa S, Papakonstantinou N, Touloumenidou T, Laoutaris N, Anagnostopoulos A, et al. Toll-like receptor signaling pathway in chronic lymphocytic leukemia: distinct gene expression profiles of potential pathogenic significance in specific subsets of patients. Haematologica. 2011;96(11):1644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ntoufa S, Vardi A, Papakonstantinou N, Anagnostopoulos A, Aleporou-Marinou V, Belessi C, et al. Distinct innate immunity pathways to activation and tolerance in subgroups of chronic lymphocytic leukemia with distinct immunoglobulin receptors. Mol Med. 2012;18:1281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papakonstantinou N, Ntoufa S, Chartomatsidou E, Papadopoulos G, Hatzigeorgiou A, Anagnostopoulos A, et al. Differential microRNA profiles and their functional implications in different immunogenetic subsets of chronic lymphocytic leukemia. Mol Med. 2013;19:115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bomben R, Dal Bo M, Capello D, Forconi F, Maffei R, Laurenti L, et al. Molecular and clinical features of chronic lymphocytic leukaemia with stereotyped B cell receptors: results from an Italian multicentre study. Br J Haematol. 2009;144(4):492–506. [DOI] [PubMed] [Google Scholar]
- 24.Del Giudice I, Chiaretti S, Santangelo S, Tavolaro S, Peragine N, Marinelli M, et al. Stereotyped subset #1 chronic lymphocytic leukemia: A direct link between BCR structure, function and patients’ prognosis. Am J Hematol. 2014;89(1):74–82. [DOI] [PubMed] [Google Scholar]
- 25.Catera R, Silverman GJ, Hatzi K, Seiler T, Didier S, Zhang L, et al. Chronic lymphocytic leukemia cells recognize conserved epitopes associated with apoptosis and oxidation. Mol Med. 2008;14(11–12):665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyper-responses to DNA-containing antigens. Immunity. 2008;28(6):799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol. 2006;36(4):810–6. [DOI] [PubMed] [Google Scholar]
- 28.Muzio M, Apollonio B, Scielzo C, Frenquelli M, Vandoni I, Boussiotis V, et al. Constitutive activation of distinct BCR-signaling pathways in a subset of CLL patients: a molecular signature of anergy. Blood. 2008;112(1):188–95. [DOI] [PubMed] [Google Scholar]
- 29.Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: upregulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. 2003;101(11):4500–4. [DOI] [PubMed] [Google Scholar]
- 30.Rozkova D, Novotna L, Pytlik R, Hochova I, Kozak T, Bartunkova J, et al. Toll-like receptors on B-CLL cells: expression and functional consequences of their stimulation. Int J Cancer. 2010;126(5):1132–43. [DOI] [PubMed] [Google Scholar]
- 31.Muzio M, Scielzo C, Bertilaccio MT, Frenquelli M, Ghia P, Caligaris-Cappio F. Expression and function of toll like receptors in chronic lymphocytic leukaemia cells. Br J Haematol. 2009;144(4):507–16. [DOI] [PubMed] [Google Scholar]
- 32.Zwick C, Fadle N, Regitz E, Kemele M, Stilgenbauer S, Buhler A, et al. Autoantigenic targets of B-cell receptors derived from chronic lymphocytic leukemias bind to and induce proliferation of leukemic cells. Blood. 2013;121(23):4708–17. [DOI] [PubMed] [Google Scholar]
- 33.Palinski W, Yla-Herttuala S, Rosenfeld ME, Butler SW, Socher SA, Parthasarathy S, et al. Antisera and monoclonal antibodies specific for epitopes generated during oxidative modification of low density lipoprotein. Arteriosclerosis. 1990;10(3):325–35. [DOI] [PubMed] [Google Scholar]
- 34.Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood. 2007;109(10):4424–31. [DOI] [PubMed] [Google Scholar]
- 35.Jordo ED, Wermeling F, Chen Y, Karlsson MC. Scavenger receptors as regulators of natural antibody responses and B cell activation in autoimmunity. Mol Immunol. 2011;48(11):1307–18. [DOI] [PubMed] [Google Scholar]
- 36.Rutella S, Rumi C, Puggioni P, Barberi T, Di Mario A, Larocca LM, et al. Expression of thrombospondin receptor (CD36) in B-cell chronic lymphocytic leukemia as an indicator of tumor cell dissemination. Haematologica. 1999;84(5):419–24. [PubMed] [Google Scholar]
- 37.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11(2):155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apollonio B, Scielzo C, Bertilaccio MT, Ten Hacken E, Scarfo L, Ranghetti P, et al. Targeting B-cell anergy in chronic lymphocytic leukemia. Blood. 2013;121(19):3879–88, S1–8. [DOI] [PubMed] [Google Scholar]
- 39.Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol. 2005;6(11):1160–7. [DOI] [PubMed] [Google Scholar]
- 40.Decker T, Schneller F, Sparwasser T, Tretter T, Lipford GB, Wagner H, et al. Immunostimulatory CpG-oligonucleotides cause proliferation, cytokine production, and an immunogenic phenotype in chronic lymphocytic leukemia B cells. Blood. 2000;95(3):999–1006. [PubMed] [Google Scholar]
- 41.Que X, Widhopf GF, 2nd, Amir S, Hartvigsen K, Hansen LF, Woelkers D, et al. IGHV1–69-encoded antibodies expressed in chronic lymphocytic leukemia react with malondialdehyde-acetaldehyde adduct, an immunodominant oxidation-specific epitope. PLoS One. 2013;8(6):e65203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strefford JC, Sutton LA, Baliakas P, Agathangelidis A, Malcikova J, Plevova K, et al. Distinct patterns of novel gene mutations in poor-prognostic stereotyped subsets of chronic lymphocytic leukemia: the case of SF3B1 and subset #2. Leukemia. 2013;27(11):2196–9. [DOI] [PubMed] [Google Scholar]
- 43.Baumgarth N, Tung JW, Herzenberg LA. Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin Immunopathol. 2005;26(4):347–62. [DOI] [PubMed] [Google Scholar]
- 44.Ntoufa S, Papakonstantinou N, Apollonio B, Gounari M, Anagnostopoulos A, Belessi C, et al. B-Cell Anergy Underlies Indolent Clinical Behavior Of CLL Stereotyped Subset #4. Blood. 2013;122(21). [DOI] [PubMed] [Google Scholar]
- 45.Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol. 2007;7(8):633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, et al. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood. 2012;119(26):6278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duckworth A, Glenn M, Slupsky JR, Packham G, Kalakonda N. Variable induction of PRDM1 and differentiation in chronic lymphocytic leukemia is associated with anergy. Blood. 2014;123(21):3277–85. [DOI] [PubMed] [Google Scholar]
- 48.Baxendale HE, Johnson M, Stephens RC, Yuste J, Klein N, Brown JS, et al. Natural human antibodies to pneumococcus have distinctive molecular characteristics and protect against pneumococcal disease. Clin Exp Immunol. 2008;151(1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caligaris-Cappio F, Bertilaccio MT, Scielzo C. How the microenvironment wires the natural history of chronic lymphocytic leukemia. Semin Cancer Biol. 2014;24:43–8. [DOI] [PubMed] [Google Scholar]
- 50.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]