
Figure 4.
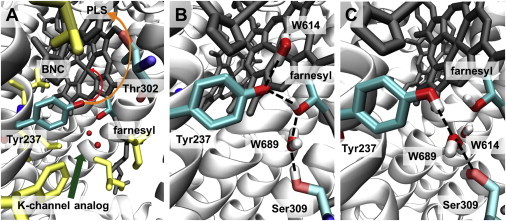
Different positions of Tyr-237 depending on its protonation state. (A) The crystal structure (PDB ID: 3S8F (6)) with the environment of Tyr-237. Tyr-237 at the end of the K-channel analog (green arrow) is close to or even part of the BNC (black stick model) and at a 5.0 Å distance from the OH group of farnesyl from heme a3. The putative PLS is above the BNC, with a few hydrophilic residues (e.g., Thr-302) in between. Hydrophobic residues around Tyr-237 are shown in yellow. The putative pathways of the chemical (red) and pumped (orange) protons above Tyr-237 are indicated by arrows, but the exact locations of the proton pathways are not known. (B) Typical snapshot of MD simulation with deprotonated Tyr-237 (Table 1, No. 1) at 47.0 ns. The deprotonated Tyr-237 remains at the crystal structure position with a stable H-bond from farnesyl of heme a3 (O-O distance 2.6 Å). The crystal water W614 moves above Tyr-237 and forms a stable H-bond (O-O distance 2.8 Å). H-bonds are indicated by dashed lines. Nonpolar hydrogens are not shown. (C) Typical snapshot of MD simulation with protonated Tyr-237 (Table 1, No. 2) at 64.0 ns. The H-bond between Tyr-237 and farnesyl of heme a3 is lost, and the water molecule W614 bridges Tyr-237 and Ser-309 (the O-O distances of Tyr-237 and Ser-309 with W614 are 2.8 Å and 2.6 Å, respectively).