
Figure 3.
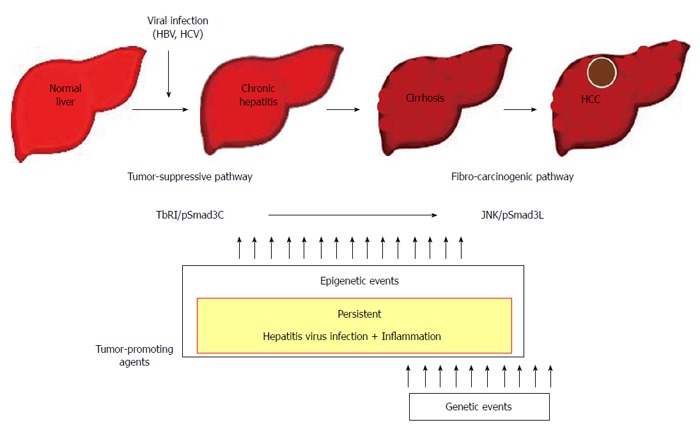
Contribution to fibro-carcinogenesis by tumor-promoting agents. Together, persistent hepatitis virus infection and chronic inflammation shift hepatocytic phospho-Smad3 signaling from the tumor-suppressive transforming growth factor-β (TGF-β) receptor type I (TβRI)/COOH-terminally phosphorylated Smad3 (pSmad3C) mode to the fibro-carcinogenic c-Jun N-terminal kinase (JNK)/linker phosphorylated Smad3 (pSmad3L) mode characteristic of median forebrain bundle, accelerating liver fibrosis while increasing risk of hepatocellular carcinoma (HCC). In the liver, persistent hepatitis virus infection and chronic inflammation contribute to fibro-carcinogenesis. In proportion to the severity of fibrosis, mitogenic genetic or epigenetic alterations can drive fibro-carcinogenesis via the pSmad3L pathway. Escaping the cytostatic action of pSmad3C is a critical step for progression to full malignancy in cancers, which must overcome multiple fail-safe genetic controls. HBV: Hepatitis B virus; HCV: Hepatitis C virus.