

Abstract
Myelodysplastic syndromes (MDS) are clonal hematopoietic stem cell disorders characterized by peripheral cytopenias and ineffective hematopoiesis. MDS is an example of an age-related malignancy and its increasing prevalence and incidence can be attributed to a greater life expectancy in developed countries. Although frequently encountered in hematology/oncology clinics, MDS may constitute a diagnostic challenge especially with equivocal bone marrow morphology. Certain syndromes of bone marrow failure (BMF) may mimic MDS and formulating a correct diagnosis is vital for adequate prognostication as well as therapeutic approaches. Metaphase karyotyping (MK) is a very important diagnostic tool and marker of prognosis and can be an indicator of response to certain therapies. Unfortunately chromosomal abnormalities may only be found in approximately 50% of patients with MDS. In this review, we discuss the diagnostic approaches to patients with pancytopenia with a particular focus on the growing number of somatic mutations through new molecular testing.
Keywords: Bone marrow failure, Myelodysplastic syndrome, Aplastic anemia, Large granular lymphocytosis, Paroxysmal nocturnal hemoglobinuria, Molecular markers, SNP-arrays, Somatic mutations
Introduction
Myelodysplastic syndromes (MDS) comprise a heterogeneous group of clonal hematopoietic stem cell disorders characterized by peripheral blood cytopenias and ineffective hematopoiesis [1]. MDS is part of a wider spectrum of bone marrow failure (BMF) conditions that clinically and pathologically may share features. Despite distinct underlying etiology of the specific BMF, similar phenotypic characteristics may hamper the correct diagnosis, especially at early stages. Familiarity with various MDS mimics, their etiology, natural history and prognosis is essential during diagnostic work-up, prognostication and subsequent therapeutic decision-making [1-4].
Disorders categorized as BMF include MDS (including hypocellular MDS [hMDS]), aplastic anemia (AA), inherited BMF syndromes (IBMFS), large granular lymphocytosis (LGL), pure red cell aplasia (PRCA), and paroxysmal nocturnal hemoglobinuria (PNH) (Figure 1). Some also consider myeloproliferative neoplasms (MPN) in this arena. BMF may be classified in multiple ways: by bone marrow cellularity (cellular or hypocellular), clonality (neoplastic or non-clonal), etiology (inherited or acquired) and manifestation (acute or chronic). Additionally, some degree of overlap among BMF exists, which can complicate making the diagnosis. AA frequently occurs with a detectable PNH clone, MDS may overlap with MPN or LGL and the correct diagnosis may remain unclear even after a thorough evaluation. Given that the management paradigm differs depending on the diagnosis, the evaluation should focus on distinguishing conditions such as MDS, AA or PNH, whose therapies are distinctly different. Currently, several therapeutic options are available for MDS patients including hypomethylating agents (HA) and immunomodulators (IMIDs); thus, identifying the correct diagnosis is crucial. More importantly, hematopoietic stem cell transplantation (HSCT), the only curative therapy for MDS, can be considered as a vital option for select patients. These aforementioned therapies have potential serious side effects and thus it is essential that patients without MDS avoid their toxicity, and those patients with lower-risk (and potentially self-limited conditions) are not unnecessarily directed to HSCT.
Figure 1. Bone Marrow Failure Syndromes.
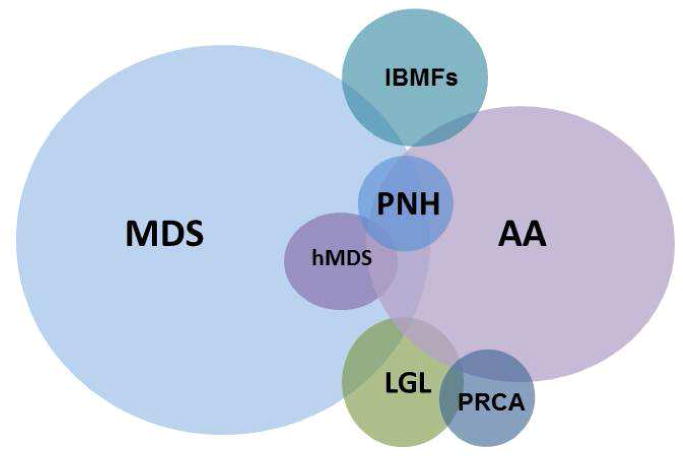
In this review we will focus on diagnostic strategies, which include standard hematopathological techniques, with a particular emphasis on the growing field of molecular diagnostics. While early diagnosis of BMF syndromes may be challenging, appropriate application of these newly available tests may facilitate timely diagnosis resulting in early introduction of therapies.
Diagnostic Approach to Marrow Failure
Virtually all patients with BMF present with peripheral cytopenias affecting at least one lineage. It can be challenging to distinguish relatively benign BMF from clonal malignant conditions[5]. After non-malignant causes of cytopenias such as nutritional deficiencies or bone marrow toxins are excluded, the diagnostic approach to the suspected BMF disorder begins with a detailed history and physical exam followed by review of peripheral blood and bone marrow mophology along with flow cytometry and MK (Table 1). More in-depth testing, including an assortment of molecular techniques, should be considered through referral to BMF centers. Repeat peripheral blood and bone marrow examinations are often helpful in making the correct diagnosis and in assessing the tempo of disease progression, and should be considered when the clinical picture changes.
Table 1. Diagnostic Workup for Bone Marrow Failure.
| Diagnostic test/tool | Application | |
|---|---|---|
| History | Onset of cytopenias | Inherited vs. acquired BMF |
|
| ||
| Medications/supplements Exposure | Reversible etiology | |
|
| ||
| Transfusion requirements | Disease severity | |
|
| ||
| Family history | Cytopenias | IBMFS |
|
| ||
| Hematologic malignancies | Inherited predisposition (e.g. GATA2 mutations) | |
|
| ||
| Interstitial lung disease, idiopathic liver failure | Telomeropathies | |
|
| ||
| Physical exam | Height | IBMFS |
|
| ||
| Skeletal abnormalities (limbs) | FA | |
|
| ||
| Skin and nails abnormalities | DKC | |
| Premature greying | ||
|
| ||
| Peripheral blood | Complete blood count with differential | Diagnosis, severity |
|
| ||
| Reticulocytes count | BMF vs. RBC destruction | |
|
| ||
| Metabolic panel Vitamin levels, copper | Metabolic derangement Nutritional deficiencies | |
|
| ||
| Hepatitis panel | Acquired AA | |
|
| ||
| Flow cytometry | FLAER assay for PNH LGL clone | |
|
| ||
| Telomere lengths | DKC | |
|
| ||
| Chromosomal breakage test | FA | |
|
| ||
| FISH | Clonal abnormalities/prognosis | |
| SNP array | ||
| Somatic mutations | ||
|
| ||
| Bone marrow | Aspirate and biopsy | Diagnostic |
|
| ||
| Iron stain/ringed sideroblasts | Sideroblastic anemia, nutritional deficiencies, RARS | |
|
| ||
| Flow cytometry | Phenotypic abnormalities, CD34 count | |
|
| ||
| Metaphase karyotyping | Clonal abnormalities, prognosis | |
| FISH | ||
| SNP-array | ||
| Somatic mutations | ||
|
| ||
| Colony forming assay | Hematopoietic cells defect vs.extrinsic etiology | |
|
| ||
BMF Bone marrow failure
AA Aplastic anemia
RARS Refractory anemia with ringed sideroblasts
IBMFS Inherited bone marrow failure syndromes
RBC Red blood cells
FISH Fluorescence in situ hybridization
SNP Single nucleotide polymorphism
FA Fanconi anemia
DKC Dyskeratosis congenita
PNH Paroxysmal nocturnal hemoglobinuria
FLAER Fluorescent aerolysin
Diagnostic Evaluation of MDS
MDS frequently presents as unilineage or multilinage cytopenias due to a hematopoietic stem cell (HSC) defect, leading to ineffective hematopoiesis and the propensity to malignant transformation in approximately 30% of patients. A peripheral blood morphology is often helpful in assessing dysplasia in myeloid cells. Hypogranulation, hypo- or hypersegmentation and the presence of pseudo Pelger-Huet cells are frequently observed in MDS and usually not appreciated in other disorders such as AA. A bone marrow biopsy with aspirate to assess marrow cellularity, dysplasia, number of blasts and underlying fibrosis is requisite. Additional flow cytometry and MK are also vital [6]. In approximately 90% of patients with MDS, the bone marrow examination is overtly hypercellular and dysplasia affecting erythroid or megakaryocytic lineages can be observed [1]. A finding of excess blasts (≥5%) is supportive of MDS or of an evolving acute leukemia. Bone marrow fibrosis may be an indicator of underlying MPN or of an overlap MDS/MPN. The confirmation of clonal hematopoiesis by MK is invaluable in cases with mild or very subtle dysplasia. Additionally, MK is the single most important predictor of survival. Some non-random chromosomal abnormalities are virtually diagnostic of a myeloid neoplasm [del(7/7q), del(5q), inv(3)] [7-9]. Others, like del(Y) may be seen in healthy elderly males with a significance that is not entirely clear [10, 11]. Given the fact that MDS is a very heterogeneous group of diseases, even in cases with established diagnosis, morphologic discrepancies still exist and correct classification of MDS subtypes may be problematic [12].
The diagnosis of MDS may be difficult in patients with a normal or non-informative karyotype, especially when frank dysplasia or characteristic morphologic markers like ring sideroblasts or increased blasts are not appreciated. As immediate therapy is not indicated at early stages of the disease, such patients can be closely monitored and repeat evaluation of the bone marrow can be performed at the time of worsening cytopenias, or at approximately 6 months from initial examination.[6] Increased numbers of CD34+ myeloid blasts may aid the diagnosis of MDS, especially in cases with a hypocellular marrow [13]. Blasts can be assessed either by multicolor flow cytometry or immunohistochemistry [14]. The latter test would allow for more precise quantification of CD34+ cells as hemodilution artifact is not a major problem in core biopsy evaluation, but may result in spuriously lower CD34+ cells during aspirate analysis.
Finally, novel diagnostic tools including array-based karyotyping and massive parallel sequencing may prove useful in equivocal cases and are discussed in detail below [15-17]. In most cases, these techniques can provide objective evidence of clonal hematopoiesis, prognostic stratification and serve as markers of disease monitoring.
Distinguishing MDS from other BMF syndromes (MDS mimics)
Evaluation of cytopenias in younger patients (acquired vs. inherited BMFs)
When evaluating the etiology of cytopenias in younger patients (usually <30 years old), one should strongly consider the possibility of an inherited condition. Often, acquired hypocellular conditions like AA may be challenging to distinguish from IBMFS [2, 3]. A detailed medical history, including prior complete blood counts and family history, can be particularly illustrative. Thorough physical examination may identify skeletal, skin or nail anomalies indicating the presence of an inherited syndrome. In some instances however, when phenotypic distinction is problematic, genotyping may be the only available alternative. Most IBMFS are caused by well-defined germline mutations: DNA repair (Fanconi anemia [FA]), mutations involving telomeres maintenance (dyskeratosis congenita [DKC]), ribosomopathies (Schwachman-Bodian-Diamond syndrome [SDBS], Diamond-Blackfan anemia [DBA]), mutations in thrombopoietin receptor (congenita amegakaryocytic thrombocytopenia), or other rare causes [18, 19]. The confirmation of underlying mutations is required to make a diagnosis of an IBMFS and referral to specialized center is usually recommended.
DKC is another inherited BMF characterized by germline defect in telomere maintenance leading to premature telomere shortening [20]. DKC, however is not the only condition associated with short telomeres [21]. The definition of clinically significant short telomere length varies in the literature due to methodology, but it is accepted that length below the first percentile is significant and can confirm the inherited form of marrow aplasia like DKC [22]. Given the significant variation of critical telomere length in BMF literature, its use as a precise diagnostic test remains a challenge. Widely accepted methods to determine telomere length (TL) in peripheral blood leukocytes include flow cytometry based fluorescence in situ hybridization (Flow-FISH) and polymerase chain reaction assay (PCR), with the former being the only test certified for clinical use [23-26]. Confirmation of short telomeres, although helpful, cannot definitively distinguish inherited from acquired forms of BMF [21]. Decreased TL has been described in a subset of patients with acquired AA [27, 21, 24, 28]. The exact role and consequences of short telomeres in acquired AA, though, are not entirely clear. The prognostic value of TL and its use to guide therapeutic options is limited at present though active investigations are ongoing [23]. Telomeres have also been studied in MDS, with TL found to be only mildly reduced as compared to age-matched controls [29, 30]. In summary, TL is a useful test in patients in whom the diagnosis of DKC is suspected. In such cases the TL is dramatically reduced reaching the level below 1st percentile and confirmation of presence of telomerase mutations is imperative.
Evaluation of cytopenias in in the context of hypocellular marrow (AA and hMDS)
Most patients with MDS have normal or hypercellular marrow at the time of diagnosis. However, 5-20% of patients may initially present with decreased marrow cellularity [31, 32]. The diagnosis in such patients may be challenging, as the presentation closely resembles AA. The diagnosis is even more difficult in cases without obvious bone marrow dysplasia, especially since mild erythroid dysplasia can be also seen in some cases of AA [31]. The evidence of clonal hematopoiesis as determined by cytogenetic studies is considered diagnostic of MDS. Unfortunately, 40-60% of MDS patients have normal or non-informative MK. Non-informative results may be particularly problematic in this group of patients given their deeply aplastic marrow and frequently limited amount of cells available for analysis. The limited amount of diagnostic material may also hamper the morphological evaluation of the bone marrow. Appropriate therapy is guided by morphological and cytogenetic evidence of clonal hematopoiesis and differs between AA and MDS patients. Although immunosuppressive therapy is effective in some patients with hMDS, an accurate diagnosis has major prognostic implications [33, 34].
Newer molecular techniques, discussed later in this review, may be an important adjunct in diagnostically challenging cases. In one study, single nucleotide polymorphism array (SNP-A) karyotyping applied along with MK in patient with AA and hMDS resulted in improved detection of chromosomal lesions allowing for better distinction between AA and hMDS. The fact that some clonal abnormalities including copy-neutral loss of heterozygosity (CN-LOH) were found in AA patients may raise the question whether the correct diagnosis was made. It is however possible that some degree of clonality exists in AA due to heavily depleted HSC pool, especially that some of the lesions may subside after immunosuppressive therapy [35]. Whether the detected lesions are associated with an increased risk of malignant transformation is yet to be determined.
It is well accepted that an MDS clone originates from an immature HSC and the level of CD34+ progenitors correlates with clonal expansion and disease progression [36, 37]. Conversely, in AA hematopoietic stem and progenitor cells are profoundly depleted due to immune attack [38, 39]. Thus, quantitative analyses of CD34+ hematopoietic progenitors may be helpful in distinguishing AA from hMDS. In one single center study, patients with a normal or increased CD34+ cells were found to have clonal cytogenetic abnormalities, suggesting the diagnosis of hMDS rather than AA. The opposite was true for patients with a low percentage of CD34+ cells who were ultimately diagnosed with AA and responded to immunosuppression [13].
Evaluation of cytopenias in the context of normo- and hypercellular marrow (LGL, PRCA and inflammatory diseases)
The majority of MDS cases are characterized by normo- or hypercellular bone marrow. Unlike hMDS, the differential diagnosis of a hypercellular marrow with no clonal abnormalities is relatively broad and includes PRCA, LGL, marrow suppression associated with systemic inflammation, and other immune-mediated cytopenias. Patients with clonal hematopoiesis due to, for instance, PNH may also present with cytopenias and a hypercellular marrow, but PNH is relative easy to rule-out using flow cytometric analysis (absence of membrane CD55 and CD59 in PNH cells). Distinguishing MDS from PRCA, LGL and inflammatory diseases is more challenging [40].
Unlike MDS, in which a block in differentiation and ineffective hematopoiesis is largely attributed to cell-intrinsic defects, cytopenias associated with PRCA, LGL and inflammatory diseases are usually driven by cell extrinsic immune attack. Functional in vitro colony forming assays are helpful in establishing the correct diagnosis. As expected, colony growth in vitro is preserved in cases of immune mediated attack (PRCA, inflammatory diseases). Additionally, robust colony formation is a predictor of response to immunosuppressive therapy [41-44]. Burst forming units-erythroid (BFU-E) assays can also be used to exclude MDS in cases where dysplasia or markers of clonal hematopoiesis are equivocal. BFU-E growth above 40 colonies per 105 marrow mononuclear cells virtually eliminates MDS as an underlying etiology [42].
LGL associated with other BMFs
LGL can overlap with other acquired BMF such as PRCA, MDS and AA. The pathophysiology of LGL cells in this setting is not entirely clear, but may signal an immune system response to clonal cells. It is important to determine whether the LGL clone is an underlying cause of pancytopenia or rather a co-existing condition. In some instances, even though LGLs are seen on peripheral blood smear or flow cytometric analysis they remain polyclonal and are likely reactive. Given its overlap with other BMF disorders, when LGLs are present, looking for other underlying bone marrow disorder like MDS is important. Using STAT3 mutational status to distinguish LGL is not helpful because these mutations are also detected in AA and MDS when associated with LGL, as well as in classical LGL phenotype (Felty's syndrome) of isolated neutropenia and rheumatoid arthritis.[45, 46] STAT3 mutations have also been reported in PRCA [47]. The presence of LGL cells concurrently with MDS correlated with poor response to IST as compared to “pure” LGL [48]. Better response to IST was, however, observed in PRCA associated with an LGL clone [41, 42]. The identification of concurrent LGL cells, although important, may not warrant LGL-directed therapies.
Markers of clonal hematopoiesis in the diagnosis and prognosis of MDS
Chromosomal abnormalities
MK remains the diagnostic gold standard and is invaluable in cases with peripheral cytopenias, absence of increased bone marrow blasts and discrete or no dysplasia. Unfortunately, the test is informative in only 40-60% MDS patients. In addition to its diagnostic value, MK has been the single most important predictor of survival and, in some cases, response to targeted therapies [49]. For example, lenalidomide has been shown to be effective in patients with 5q deletion [50, 51]. Methodology of MK requires bone marrow aspirates in order to obtain adequate bone marrow material containing viable cells. Additionally, the test is non-informative in approximately 10% of patients due to difficulties in cell culture. The detection rate of relatively small copy number changes is limited by the resolution of the microscopy and the lesions <1 million base pairs are frequently missed. Finally CN-LOH which is frequently seen in MDS, cannot be detected by MK.
Given these limitations, there has been a search for more sensitive and precise cytogenetic methods. Microarray-based assays have proved to be a useful diagnostic and prognostic tool. SNP-A are used concurrently with MK in a variety of hematological malignancies. The high resolution allows for detection of small, submicroscopic copy number changes in patients with unremarkable MK. Moreover, SNP-A allow for detection of CN-LOH, which is a frequent genetic aberration in cancer and remains undetectable by traditional MK. SNP-A is particularly useful in instances of inaspirable bone marrow. In such patients, diagnosis can be facilitated using SNP-A performed on DNA isolated from peripheral blood. Analogically, useful results can be obtained in patients with non-informative MK due to poor cell growth. In a study of 174 patients with MDS, MDS/MPN and secondary leukemias, using SNP-A, clonal chromosomal aberrations were detected in 75% cases and CN-LOH was seen in 20-30% of patients [52]. SNP-A also allowed for detection of additional copy number changed and CN-LOH in 119 patients with low-risk MDS [53]. In addition to being a complementary diagnostic test, chromosomal abnormalities detected by SNP-A are an important prognostic marker [52, 17, 54].
Somatic mutations
Since the introduction of powerful genetic tools like next-generation sequencing, many new somatic mutations have been identified in MDS. Genetic defects tend to affect genes responsible for epigenetic modifications, RNA splicing machinery and tyrosine kinase signaling. These are helpful molecular markers of clonal hematopoiesis that are also important prognostically (Table 2).
Table 2. Somatic Mutations in MDS.
| Mutation | Frequency | Prognostic impact | Comments | |
|---|---|---|---|---|
| DNA methylation | ||||
| TET2 | 20-30% MDS 20-43% MDS/MPN |
Unfavorable in CMML | More favorable in AML | |
| DNMT3A | 3-8% | Unfavorable | ||
| IDH1/2 | <5% | Unfavorable | IDH1 associated with worse prognosis | |
|
| ||||
| Chromatin modification | ||||
| ASXL1 | 17% MDS 50% MDS/MPN |
Unfavorable | ||
| EZH2 | 6% | Unfavorable | ||
|
| ||||
| Spliceosome machinery | ||||
| SF3B1 | 30% | Favorable | Frequent in RARS and RCMD-RS | |
| U2AF1 | 10% | Unfavorable | ||
| SRSF2 | 15% | Unfavorable | ||
| ZRSR2 | 10% | Unknown | ||
|
| ||||
| RAS signaling | ||||
| KRAS | <2% | Unknown | ||
| PTPN11 | <1% | Unknown | ||
| NF1 | <1% | Unknown | ||
|
| ||||
| Cohesin | ||||
| STAG2 | 5% | Unknown | ||
| RAD21 | <1% | Unknown | ||
| SMC3 | <1% | Unknown | ||
|
| ||||
| Others | ||||
| BCOR/BCROL1 | 4% | Unfavorable | ||
| ETV6 | 3% | Unfavorable | ||
| RUNX1 | 10-15% | Unfavorable | ||
| p53 | 10-15% | Unfavorable | ||
| JAK2 | 3% | Unknown | Frequent in RARS-t | |
| c-CBL | 1% | Unfavorable | Frequent in CMML | |
| MPL | 1% | Unknown | Associated with ET and MF | |
| SETBP1 | 4-25% | Unfavorable | Frequent in aCML and CMML | |
RARS Refractory anemia with ringed sideroblasts
RCMD-RS Refractory cytopenias with multilineage dysplasia with ringed sideroblasts
RARS-T Refractory anemia with ringed sideroblasts associated with thrombocytosis
CMML Chronic myelomonocytic leukemia
aCML Atypical chronic myeloid leukemia
ET Essential thrombocythemia
PV Polycythemia vera
Epigenetic modifiers
TET2
TET2 (Ten-Eleven translocation 2) gene is a putative tumor suppressor gene localized on chromosome 4q24. Loss of function mutations have been described in both myeloid and lymphoid malignancies as well as in healthy elderly individuals with clonal hematopoiesis. TET2 mutations are present in approximately 20-30% of patients with MDS, 20-43% MDS/MPN (including CMML) and 25%-40% of patients with secondary leukemia. Moreover, TET2 mutations are frequent in patients with normal MK, making them convenient indicators of clonal hematopoiesis in equivocal cases [16]. Data regarding the prognostic significance of TET2 mutations remain divisive. In one study by Kosmider et al. mutated TET2 was a favorable prognostic marker with 5-year survival of 76.9% in TET2 mutated group vs. 18.3% in the TET2 wild-type group [55]. Several studies however, showed no difference in survival based on TET2 mutation status [56, 57]. Although TET2 may seem to be a promising molecular marker facilitating diagnosis, mutations in this gene can be observed in up to 5% of elderly individuals with clonal hematopoiesis as determined by X chromosome inactivation studies and no apparent hematological abnormalities [58]. The exact risk of transformation in such patients is unknown and it is unclear whether TET2 alone or additional genetic events are necessary for development of overt hematologic malignancy. Consequently, mutations in TET2 appear to be an early genetic event likely predisposing to clonal evolution and eventually malignant transformation. As such, it may persist in antecedent clone even after successful therapy, which precludes its use as a marker of response or residual disease.
DNMT3A
Another example of a gene frequently mutated in hematologic malignancies and involved in epigenetic regulation via DNA methylation is DNA methyltransferase 3A (DNMT3A). Although more frequent in de novo AML, mutations in this gene have been identified in about 3-8% of MDS patients [59, 60]. Single institution experience suggests that mutated DNMT3A is a prognostic factor for both worse overall survival and higher rates of progression to leukemia [60]. Recent data suggest the DNMT3A is an initial genetic event occurring in pre-leukemic stem cells and may be present in <0.1% of otherwise healthy population. HSC clones harboring DNMT3A mutations can be also detected in post-remission samples [61]. Whether the presence of DNMT3A mutations in complete remission samples is a predictor of disease relapse remains to be seen.
IDH1 and IDH2
Two genes encoding isocitrate dehydrogenase (IDH1 and IDH2) were found to be frequently mutated in AML and to a lesser degree in MDS and MDS/MPN with a frequency of <5% [62-64]. IDH1/2 mutations are mutually exclusive with TET2 mutations indicating their functional redundancy and involvement in the same pathologic pathway. Also, similarl to TET2, IDH1 mutations were more frequently encountered in patients with normal karyotype. Unlike in AML, where IDH mutations are favorable prognostic markers, IDH1 but not IDH2 mutations seem to be an indicator of inferior outcome in MDS [65, 66].
Genes involved in chromatin modification
ASXL1
Loss of function mutations in additional sex comb-like 1 (ASXL1) gene involved in polycomb-group repressive complex 1 (PRC1) have been described in a variety of human malignancies including MDS. Approximately 17% of MDS and up to 49% of MDS/MPN patients harbor ASXL1 mutations [16, 67-69]. Frameshift and nonsense mutations are also an independent marker of adverse overall survival and shorter time to AML progression both in MDS and MDS/MPN [16, 70, 68]. Although ASXL1 seems to be a marker or inferior outcome, it does not contribute to leukemic transformation and progression [70]. Also, no association has been found between ASXL1 mutational status and response to lenalidomide or hypomethylating agents [71, 72]. Mutations in ASXL1 may also serve as an individualized marker of minimal residual disease (MRD) in patient undergoing allogeneic HSCT, although the presence of mutation did not predict survival in the setting of BMT [73].
EZH2
Enhancer of Zest homolog 2 (EZH2) located on the long arm of chromosome 7 encodes a histone methyltransferase subunit of PRC2 complex. Loss-of-function mutations have been observed in approximately 6% or MDS patients [74, 75]. The presence of EZH2 mutations was mostly seen in lower-risk patients and was strongly associated with decreased overall survival in multivariate model [16, 76].
Spliceosome machinery
Genes encoding proteins involved in mRNA splicing (SF3B1, ZRSR2, SRSF2 and U2AF1) were found to be the most frequently mutated group of genes in MDS [77-80]. Mutations in SF3B1 affected approximately 30% of patients and were nearly pathognomonic for dysplasia associated with the presence of ringed sideroblasts [77, 78, 81]. Interestingly, mutations in U2AF1, ZRSR2 and SRSF2 were seen in MDS cases without ring sideroblasts. SF3B1 mutations were found to be an independent predictor of better overall survival and lower risk of transformation to AML [82]. Although affecting the same pathway, mutations in U2AF1 and SRSF2 were associated with worse overall survival and shorter progression to AML [79, 80]. It is now well accepted that mutations affecting mRNA spicing machinery are likely to be early/initiating events in disease evolution and are present in virtually all malignant cells. The presence of point mutations as detected by PCR may also serve as a molecular marker of MRD after chemotherapy or hematopoietic stem cell transplant [79, 80, 83].
Other somatic mutations
RUNX1
Mutations affecting transcription factors have been reported in both de novo AML and in nearly 10% of MDS cases.[84] RUNX1 mutations were also described as a germline variant in familial platelets disorder and myelodysplasia with a propensity to leukemic transformation [85].
c-CBL
Mutations in Casitas b-cell lymphoma (c-CBL) were found by candidate screening in area of CN-LOH spanning long arm or chromosome 7 [86]. CBL mutations were most prevalent in CMML (5% of cases), and sAML (nearly 10% of cases) but were relatively infrequent in MDS [87]. Mutations in c-CBL tend to arise in a subclone of malignant cells during disease progression. As such, they are an indicator of poor outcome.
Mutations in Ras superfamily (NRAS, KRAS, NF1, PTPN11), DNA repair (TP53), cohesion complex (STAG2, SMC3) and SETBP1 have been observed in subsets of MDS patients and their frequencies and prognostic impact have been mentioned in Table 2 [88-95].
As above, a number of recurrently mutated genes have been identified in MDS. The total number of mutations present in a patient with MDS has important prognostic value, even independent of currently used clinical prognostic information [96, 16]. We have reviewed the most commonly mutated genes, some associated with worse outcome and decreased leukemia-free survival [76]. Certain mutational combinations are also likely critical in MDS pathogenesis. Specifically, several groups have now examined the mutational allele frequency of each of the commonly mutated genes to estimate the temporal order of mutation acquisition in MDS and to understand the impact of subclonal mutations (defined as mutations not present in the entire proportion of malignant cells) on disease course [96]. Studies have found that the detection of these mutations had a significant impact on outcome even when not present in the dominant clone. From a diagnostic standpoint, this suggests that molecular tests might simultaneously provide both mutational data and copy number changes capable of supplanting recurrent cytogenetic testing in MDS. However, there are always limitations and caveats to this type of testing. Mutations in isolation without appropriate clinical context must be interpreted with some caution. As an MDS community, we have yet to realize the full clinical utility of these innovative markers. It is only the patient with MDS and a clonal abnormality and a mutation in ETV6 or TP53 to whom consideration should be given for very aggressive therapy for their high risk disease. SNP-A or genomic analyses are also useful in detecting genetic alterations that contribute to disease phenotype, but they are not in and of themselves disease-defining. Therefore, these should again be used with caution in isolation in diagnosing a bone marrow failure condition as we gain additional knowledge and experience in the field.
Conclusions
Establishing the correct diagnosis in patients with cytopenias can be challenging. Clinicians may encounter a diagnostic dilemma, particularly in patients with only mild cytopenias, no obvious dysplasia and lack of cytogenetic abnormalities as determined by MK. A detailed work-up (Table 1) can direct the clinician towards the best diagnostic path and help distinguish acquired from inherited bone marrow failure. New molecular tools like SNP-A and somatic mutation analysis prove to be a helpful adjunct tool especially in equivocal cases. Early studies also suggest that certain somatic mutations and chromosomal changes detected by SNP-A may have an important prognostic value but prospective clinical studies will be required to prove their role as a risk stratification marker.
Footnotes
Conflict of Interest: Dr. Lukasz P. Gondek and Dr. Amy E. DeZern each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
Contributor Information
Lukasz P. Gondek, Email: lgondek1@jhmi.edu, Department of Oncology, Division of Hematological Malignancies, Johns Hopkins, University, 1650 Orleans St, CRB1-290, Baltimore, MD, 21231, Phone: 410-502-5847, Fax: 410-614-3809.
Amy E. DeZern, Email: adezern1@jhmi.edu, Department of Oncology, Division of Hematological Malignancies, Johns Hopkins University, 1650 Orleans St, CRB1- 3M87, Baltimore, MD, 21231, Phone: 410-502-7208, Fax: 410-614-7279.
References
Papers of particular interest, published recently, have been highlighted as:
•Of importance
•• Of major importance
- 1.Tefferi A, Vardiman JW. Myelodysplastic syndromes. NEnglJMed. 2009;361(19):1872–85. doi: 10.1056/NEJMra0902908. [DOI] [PubMed] [Google Scholar]
- 2.Young NS. Acquired bone marrow failure Blood: Principles and Practice of Hematology. Philadelphia, PA: JB Lippincott; 1995. [Google Scholar]
- 3.Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. HematologyAmSocHematolEducProgram. 2007:29–39. doi: 10.1182/asheducation-2007.1.29. [DOI] [PubMed] [Google Scholar]
- 4.Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61(19):7233–9. [PubMed] [Google Scholar]
- 5.Swerdlow SH, Campo E, Harris NL, Jaffe E, Pileri SA, Stein H, et al. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer (IARC); 2008. [Google Scholar]
- 6•.Malcovati L, Hellstrom-Lindberg E, Bowen D, Ades L, Cermak J, Del Canizo C, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943–64. doi: 10.1182/blood-2013-03-492884. This is a consensus report on diagnosis and treatment of MDS from European LeukemiaNet and provides a thorough review of the evidence through its publication dat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haase D, Germing U, Schanz J, Pfeilstocker M, Nosslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110(13):4385–95. doi: 10.1182/blood-2007-03-082404. [DOI] [PubMed] [Google Scholar]
- 8.Sole F, Luno E, Sanzo C, Espinet B, Sanz GF, Cervera J, et al. Identification of novel cytogenetic markers with prognostic significance in a series of 968 patients with primary myelodysplastic syndromes. Haematologica. 2005;90(9):1168–78. [PubMed] [Google Scholar]
- 9.Schanz J, Steidl C, Fonatsch C, Pfeilstocker M, Nosslinger T, Tuechler H, et al. Coalesced multicentric analysis of 2,351 patients with myelodysplastic syndromes indicates an underestimation of poor-risk cytogenetics of myelodysplastic syndromes in the international prognostic scoring system. J Clin Oncol. 2011;29(15):1963–70. doi: 10.1200/JCO.2010.28.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pierre RV, Hoagland HC. Age-associated aneuploidy: loss of Y chromosome from human bone marrow cells with aging. Cancer. 1972;30(4):889–94. doi: 10.1002/1097-0142(197210)30:4<889::aid-cncr2820300405>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 11.Wiktor A, Rybicki BA, Piao ZS, Shurafa M, Barthel B, Maeda K, et al. Clinical significance of Y chromosome loss in hematologic disease. Genes Chromosomes Cancer. 2000;27(1):11–6. [PubMed] [Google Scholar]
- 12.Naqvi K, Jabbour E, Bueso-Ramos C, Pierce S, Borthakur G, Estrov Z, et al. Implications of discrepancy in morphologic diagnosis of myelodysplastic syndrome between referral and tertiary care centers. Blood. 2011;118(17):4690–3. doi: 10.1182/blood-2011-03-342642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsui WH, Brodsky RA, Smith BD, Borowitz MJ, Jones RJ. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia. 2006;20(3):458–62. doi: 10.1038/sj.leu.2404119. [DOI] [PubMed] [Google Scholar]
- 14.Leung W, Chen AR, Klann RC, Moss TJ, Davis JM, Noga SJ, et al. Frequent detection of tumor cells in hematopoietic grafts in neuroblastoma and Ewing's sarcoma. Bone Marrow Transplant. 1998;22(10):971–9. doi: 10.1038/sj.bmt.1701471. [DOI] [PubMed] [Google Scholar]
- 15.Maciejewski JP, Mufti GJ. Whole genome scanning as a cytogenetic tool in hematologic malignancies. Blood. 2008;112(4):965–74. doi: 10.1182/blood-2008-02-130435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. The New England journal of medicine. 2011;364(26):2496–506. doi: 10.1056/NEJMoa1013343. This large study describes novel somatic mutations in MDS. Authors identified somatic muations in a large cohort of 439 patients. Mutations in TP53, EZH2, ETV6, RUNX1, and ASXL1 were found to be predictors of poor overall survival independently of established risk factor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tiu RV, Gondek LP, O'Keefe CL, Elson P, Huh J, Mohamedali A, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011;117(17):4552–60. doi: 10.1182/blood-2010-07-295857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–22. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsangaris E, Klaassen R, Fernandez CV, Yanofsky R, Shereck E, Champagne J, et al. Genetic analysis of inherited bone marrow failure syndromes from one prospective, comprehensive and population-based cohort and identification of novel mutations. J Med Genet. 2011;48(9):618–28. doi: 10.1136/jmg.2011.089821. [DOI] [PubMed] [Google Scholar]
- 20.Vulliamy TJ, Walne A, Baskaradas A, Mason PJ, Marrone A, Dokal I. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis. 2005;34(3):257–63. doi: 10.1016/j.bcmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Du HY, Pumbo E, Ivanovich J, An P, Maziarz RT, Reiss UM, et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood. 2009;113(2):309–16. doi: 10.1182/blood-2008-07-166421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armanios M. Syndromes of telomere shortening. Annual review of genomics and human genetics. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calado RT, Cooper JN, Padilla-Nash HM, Sloand EM, Wu CO, Scheinberg P, et al. Short telomeres result in chromosomal instability in hematopoietic cells and precede malignant evolution in human aplastic anemia. Leukemia. 2012;26(4):700–7. doi: 10.1038/leu.2011.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheinberg P, Cooper JN, Sloand EM, Wu CO, Calado RT, Young NS. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. JAMA. 2010;304(12):1358–64. doi: 10.1001/jama.2010.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aubert G, Hills M, Lansdorp PM. Telomere length measurement-caveats and a critical assessment of the available technologies and tools. Mutat Res. 2012;730(1-2):59–67. doi: 10.1016/j.mrfmmm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gadalla SM, Cawthon R, Giri N, Alter BP, Savage SA. Telomere length in blood, buccal cells, and fibroblasts from patients with inherited bone marrow failure syndromes. Aging (AlbanyNY) 2010;2(11):867–74. doi: 10.18632/aging.100235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brummendorf TH, Maciejewski JP, Mak J, Young NS, Lansdorp PM. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood. 2001;97(4):895–900. doi: 10.1182/blood.v97.4.895. [DOI] [PubMed] [Google Scholar]
- 28.Calado RT, Young NS. Telomere diseases. NEnglJMed. 2009;361(24):2353–65. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poloni A, Serrani F, Berardinelli E, Maurizi G, Mariani M, Costantini B, et al. Telomere length, c-myc and mad-1 expression could represent prognosis markers of myelodysplastic syndrome. Leuk Res. 2013;37(11):1538–44. doi: 10.1016/j.leukres.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Rollison DE, Epling-Burnette PK, Park JY, Lee JH, Park H, Jonathan K, et al. Telomere length in myelodysplastic syndromes. Leuk Lymphoma. 2011;52(8):1528–36. doi: 10.3109/10428194.2011.568648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrett J, Saunthararajah Y, Molldrem J. Myelodysplastic syndrome and aplastic anemia: distinct entities or diseases linked by a common pathophysiology? Semin Hematol. 2000;37(1):15–29. doi: 10.1016/s0037-1963(00)90027-1. [DOI] [PubMed] [Google Scholar]
- 32.Bennett JM, Orazi A. Diagnostic criteria to distinguish hypocellular acute myeloid leukemia from hypocellular myelodysplastic syndromes and aplastic anemia: recommendations for a standardized approach. Haematologica. 2009;94(2):264–8. doi: 10.3324/haematol.13755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dameshek W. Riddle: What do aplastic anemia, paroxysmal nocutrnal hemoglobinuria (PNH) and “hypoplastic” leukemia have in common? (Editorial) Blood. 1967;30(2):251–4. [PubMed] [Google Scholar]
- 34.Young NS. The problem of clonality in aplastic anemia: Dr. Dameshek's riddle, restated. Blood. 1992;79:1385–92. [PubMed] [Google Scholar]
- 35.Afable MG, 2nd, Wlodarski M, Makishima H, Shaik M, Sekeres MA, Tiu RV, et al. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. 2011;117(25):6876–84. doi: 10.1182/blood-2010-11-314393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woll PS, Kjallquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. 2014;25(6):794–808. doi: 10.1016/j.ccr.2014.03.036. [DOI] [PubMed] [Google Scholar]
- 37.Della Porta MG, Malcovati L, Boveri E, Travaglino E, Pietra D, Pascutto C, et al. Clinical relevance of bone marrow fibrosis and CD34-positive cell clusters in primary myelodysplastic syndromes. J Clin Oncol. 2009;27(5):754–62. doi: 10.1200/JCO.2008.18.2246. [DOI] [PubMed] [Google Scholar]
- 38.de Planque MM, van Krieken JH, Kluin-Nelemans HC, Colla LP, van der Burgh F, Brand A, et al. Bone marrow histopathology of patients with severe aplastic anaemia before treatment and at follow-up. Br J Haematol. 1989;72(3):439–44. doi: 10.1111/j.1365-2141.1989.tb07729.x. [DOI] [PubMed] [Google Scholar]
- 39.Young NS, Maciejewski J. The pathology of acquired aplastic anemia. N Engl J Med. 1997;336(19):1365–72. doi: 10.1056/NEJM199705083361906. [DOI] [PubMed] [Google Scholar]
- 40.Leguit RJ, van den Tweel JG. The pathology of bone marrow failure. Histopathology. 2010;57(5):655–70. doi: 10.1111/j.1365-2559.2010.03612.x. [DOI] [PubMed] [Google Scholar]
- 41.Charles RJ, Sabo KM, Kidd PG, Abkowitz JL. The pathophysiology of pure red cell aplasia: implications for therapy. Blood. 1996;87(11):4831–8. [PubMed] [Google Scholar]
- 42.DeZern AE, Pu J, McDevitt MA, Jones RJ, Brodsky RA. Burst-forming unit-erythroid assays to distinguish cellular bone marrow failure disorders. Exp Hematol. 2013;41(9):808–16. doi: 10.1016/j.exphem.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fureder W, Paulitsch-Buckingham A, Rabitsch W, Jager E, Schwarzinger I, Sperr WR, et al. Evaluation of treatment responses and colony-forming progenitor cells in 50 patients with aplastic anemia after immunosuppressive therapy or hematopoietic stem cell transplantation: a single-center experience. Wien Klin Wochenschr. 2014 doi: 10.1007/s00508-013-0484-2. [DOI] [PubMed] [Google Scholar]
- 44.Geissler K, Hinterberger W, Bettelheim P, Haas O, Lechner K. Colony growth characteristics in chronic myelomonocytic leukemia. Leuk Res. 1988;12(5):373–7. doi: 10.1016/0145-2126(88)90055-0. [DOI] [PubMed] [Google Scholar]
- 45.Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120(15):3048–57. doi: 10.1182/blood-2012-06-435297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905–13. doi: 10.1056/NEJMoa1114885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishida F, Matsuda K, Sekiguchi N, Makishima H, Taira C, Momose K, et al. STAT3 gene mutations and their association with pure red cell aplasia in large granular lymphocyte leukemia. Cancer science. 2014;105(3):342–6. doi: 10.1111/cas.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saunthararajah Y, Molldrem JL, Rivera M, Williams A, Stetler-Stevenson M, Sorbara L, et al. Coincident myelodysplastic syndrome and T-cell large granular lymphocytic disease: clinical and pathophysiological features. BrJHaematol. 2001;112(1):195–200. doi: 10.1046/j.1365-2141.2001.02561.x. [DOI] [PubMed] [Google Scholar]
- 49••.Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised International Prognostic Scoring System (IPSS-R) for myelodysplastic syndromes. Blood. 2012 doi: 10.1182/blood-2012-03-420489. This report instituted a new (revised) prognostic scoring system for MDS that is now being implemented in the clinical setting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456–65. doi: 10.1056/NEJMoa061292. doi: 355/14/1456. [DOI] [PubMed] [Google Scholar]
- 51.List A, Kurtin S, Roe DJ, Buresh A, Mahadevan D, Fuchs D, et al. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med. 2005;352(6):549–57. doi: 10.1056/NEJMoa041668. doi: 352/6/549. [DOI] [PubMed] [Google Scholar]
- 52.Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, Maciejewski JP. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood. 2008;111(3):1534–42. doi: 10.1182/blood-2007-05-092304. doi: blood-2007-05-092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohamedali A, Gaken J, Twine NA, Ingram W, Westwood N, Lea NC, et al. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood. 2007;110(9):3365–73. doi: 10.1182/blood-2007-03-079673. doi: blood-2007-03-079673. [DOI] [PubMed] [Google Scholar]
- 54.Tiu RV, Gondek LP, O'Keefe CL, Huh J, Sekeres MA, Elson P, et al. New lesions detected by single nucleotide polymorphism array-based chromosomal analysis have important clinical impact in acute myeloid leukemia. J Clin Oncol. 2009;27(31):5219–26. doi: 10.1200/JCO.2009.21.9840. doi: JCO.2009.21.9840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs) Blood. 2009;114(15):3285–91. doi: 10.1182/blood-2009-04-215814. [DOI] [PubMed] [Google Scholar]
- 56.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–10. doi: 10.1182/blood-2009-02-205690. doi: blood-2009-02-205690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gaken J, Lea NC, et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood. 2010;116(19):3923–32. doi: 10.1182/blood-2010-03-274704. [DOI] [PubMed] [Google Scholar]
- 58.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–81. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ewalt M, Galili NG, Mumtaz M, Churchill M, Rivera S, Borot F, et al. DNMT3a mutations in high-risk myelodysplastic syndrome parallel those found in acute myeloid leukemia. Blood cancer journal. 2011;1(3):e9. doi: 10.1038/bcj.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25(7):1153–8. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–33. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Itzykson R, Kosmider O, Fenaux P. Somatic mutations and epigenetic abnormalities in myelodysplastic syndromes. Best Pract Res Clin Haematol. 2013;26(4):355–64. doi: 10.1016/j.beha.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 63.Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24(5):1094–6. doi: 10.1038/leu.2010.52. [DOI] [PubMed] [Google Scholar]
- 64.Yoshida K, Sanada M, Kato M, Kawahata R, Matsubara A, Takita J, et al. A nonsense mutation of IDH1 in myelodysplastic syndromes and related disorders. Leukemia. 2011;25(1):184–6. doi: 10.1038/leu.2010.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patnaik MM, Hanson CA, Hodnefield JM, Lasho TL, Finke CM, Knudson RA, et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia. 2012;26(1):101–5. doi: 10.1038/leu.2011.298. [DOI] [PubMed] [Google Scholar]
- 66.Thol F, Weissinger EM, Krauter J, Wagner K, Damm F, Wichmann M, et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica. 2010;95(10):1668–74. doi: 10.3324/haematol.2010.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145(6):788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- 68.Gelsi-Boyer V, Trouplin V, Roquain J, Adelaide J, Carbuccia N, Esterni B, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010;151(4):365–75. doi: 10.1111/j.1365-2141.2010.08381.x. [DOI] [PubMed] [Google Scholar]
- 69•.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–7. doi: 10.1038/leu.2013.336. Authors performed targeted sequencing of 104 genes in a large cohort of 944 patients with various subtypes of MDS. Somatic mutations were found in nearly 90% of patients. Incorporation of somatic mutation status of 14 selected genes into existing risk factors improved overall survival prediction of IPSS-R scoring system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen TC, Hou HA, Chou WC, Tang JL, Kuo YY, Chen CY, et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood cancer journal. 2014;4:e177. doi: 10.1038/bcj.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia. 2014;28(1):78–87. doi: 10.1038/leu.2013.269. [DOI] [PubMed] [Google Scholar]
- 72.Sugimoto Y, Sekeres MA, Makishima H, Traina F, Visconte V, Jankowska A, et al. Cytogenetic and molecular predictors of response in patients with myeloid malignancies without del[5q] treated with lenalidomide. Journal of hematology & oncology. 2012;5:4. doi: 10.1186/1756-8722-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fu Y, Schroeder T, Zabelina T, Badbaran A, Bacher U, Kobbe G, et al. Postallogeneic monitoring with molecular markers detected by pretransplant next-generation or Sanger sequencing predicts clinical relapse in patients with myelodysplastic/myeloproliferative neoplasms. Eur J Haematol. 2014;92(3):189–94. doi: 10.1111/ejh.12223. [DOI] [PubMed] [Google Scholar]
- 74.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–7. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 75.Makishima H, Jankowska AM, Tiu RV, Szpurka H, Sugimoto Y, Hu Z, et al. Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. 2010;24(10):1799–804. doi: 10.1038/leu.2010.167. [DOI] [PubMed] [Google Scholar]
- 76••.Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30(27):3376–82. doi: 10.1200/jco.2011.40.7379. Study involved 288 patients with low-risk MDS. The mutation status of 22 genes was assessed and incorporated into Low-Risk Prognostic Scoring System. EZH2 mutations identified in 29% of patients with low-risk MDS was an adverse risk factor in a multivariable model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77••.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–95. doi: 10.1056/NEJMoa1103283. Authors described SF3B1 mutations in a large cohort of 354 patients with MDS. SF3B1 was mutated in 20% of cases and was enriched for in patients with ring siderobalsts (65%). Mutated SF3B1 was aslo found to ba a favorable prognostic factor in MDS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 79.Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44(1):53–7. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203–10. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012;120(16):3173–86. doi: 10.1182/blood-2012-05-430876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118(24):6239–46. doi: 10.1182/blood-2011-09-377275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matsuda K, Ishida F, Ito T, Nakazawa H, Miura S, Taira C, et al. Spliceosome-related gene mutations in myelodysplastic syndrome can be used as stable markers for monitoring minimal residual disease during follow-up. Leuk Res. 2012;36(11):1393–7. doi: 10.1016/j.leukres.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 84.Harada Y, Harada H. Molecular mechanisms that produce secondary MDS/AML by RUNX1/AML1 point mutations. J Cell Biochem. 2011;112(2):425–32. doi: 10.1002/jcb.22974. [DOI] [PubMed] [Google Scholar]
- 85.Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–75. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 86.Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460(7257):904–8. doi: 10.1038/nature08240. doi: nature08240. [DOI] [PubMed] [Google Scholar]
- 87.Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol. 2009;27(36):6109–16. doi: 10.1200/JCO.2009.23.7503. doi: JCO.2009.23.7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Al-Kali A, Quintas-Cardama A, Luthra R, Bueso-Ramos C, Pierce S, Kadia T, et al. Prognostic impact of RAS mutations in patients with myelodysplastic syndrome. Am J Hematol. 2013;88(5):365–9. doi: 10.1002/ajh.23410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45(10):1232–7. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
- 90.Damm F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27(6):1401–3. doi: 10.1038/leu.2013.35. [DOI] [PubMed] [Google Scholar]
- 91.Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013;27(10):2100–2. doi: 10.1038/leu.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45(1):18–24. doi: 10.1038/ng.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45(8):937–41. doi: 10.1038/ng.2698. [DOI] [PubMed] [Google Scholar]
- 94.Thol F, Suchanek KJ, Koenecke C, Stadler M, Platzbecker U, Thiede C, et al. SETBP1 mutation analysis in 944 patients with MDS and AML. Leukemia. 2013;27(10):2072–5. doi: 10.1038/leu.2013.145. [DOI] [PubMed] [Google Scholar]
- 95.Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169–77. doi: 10.1182/blood-2012-11-469619. [DOI] [PubMed] [Google Scholar]
- 96•.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–27. doi: 10.1182/blood-2013-08-518886. quiz 99. Mutation analysis in 738 patients identified oncogenic mutations in nearly 80% of patients with MDS. Subclonal analysis also revealed the set of candidate driver mutations leading to clonal evolution. Leukemia-free survival was closely related to the number of driver mutations and deteriorated as the number of driver mutations increased. [DOI] [PMC free article] [PubMed] [Google Scholar]