

Abstract
STUDY QUESTION
Are there abnormalities in gonadotrophin secretion, adrenal steroidogenesis and/or testicular steroidogenesis in brothers of women with polycystic ovary syndrome (PCOS)?
SUMMARY ANSWER
Brothers of women with PCOS have increased gonadotrophin responses to gonadotrophin releasing hormone (GnRH) agonist stimulation and alterations in adrenal and gonadal steroidogenesis.
WHAT IS KNOWN ALREADY
PCOS is a complex genetic disease. Male as well as female first-degree relatives have reproductive features of the syndrome. We previously reported that brothers of affected women have elevated circulating dehydroepiandrosterone sulfate levels.
STUDY DESIGN, SIZE, DURATION
This was a case–control study performed in 29 non-Hispanic white brothers of 22 women with PCOS and 18 control men.
PARTICIPANTS/MATERIALS, SETTING, METHODS
PCOS brothers and control men were of comparable age, weight and ethnicity. Adrenocorticotrophic hormone (ACTH) and GnRH agonist stimulation tests were performed. Gonadotrophin responses to GnRH agonist as well as changes in precursor-product steroid pairs (delta, Δ) across steroidogenic pathways in response to ACTH and GnRH agonist were examined.
MAIN RESULTS AND THE ROLE OF CHANCE
Basal total (T) levels did not differ, but dehydroepiandrosterone (DHEA) levels (0.13 ± 0.08 brothers versus 0.22 ± 0.09 controls, nmol/l, P = 0.03) were lower in brothers compared with control men. ACTH-stimulated Δ17-hydroxypregnenolone (17Preg)/Δ17-hydroxyprogesterone (17Prog) (7.8 ± 24.2 brothers versus 18.9 ± 21.3 controls, P = 0.04) and ΔDHEA/Δandrostenedione (AD) (0.10 ± 0.05 brothers versus 0.14 ± 0.08 controls, P = 0.04) were lower in brothers than in the controls. GnRH agonist-stimulated Δ17Prog/ΔAD (0.28 ± 8.47 brothers versus 4.79 ± 10.28 controls, P = 0.003) was decreased and luteinizing hormone (38.6 ± 20.6 brothers versus 26.0 ± 9.8 controls, IU/l, P = 0.02), follicle-stimulating hormone (10.2 ± 7.5 brothers versus 4.8 ± 4.1 controls, IU/l P = 0.002), AD (1.7 ± 1.4 brothers versus 0.9 ± 1.5 controls, nmol/l, P = 0.02) and ΔAD/ΔT (0.16 ± 0.14 brothers versus 0.08 ± 0.12 controls, P = 0.005) responses were increased in brothers compared with controls.
LIMITATIONS, REASONS FOR CAUTION
The modest sample size may have limited our ability to observe other possible differences in steroidogenesis between PCOS brothers and control men.
WIDER IMPLICATIONS OF THE FINDINGS
Decreased ACTH-stimulated Δ17Preg/Δ17Prog and ΔDHEA/ΔAD responses suggested increased adrenal 3β-hydroxysteroid dehydrogenase activity in the brothers. Decreased Δ17Prog/ΔAD and increased ΔAD/ΔT responses to GnRH agonist stimulation suggested increased gonadal 17,20-lyase and decreased gonadal 17β-hydroxysteroid dehydrogenase activity in the brothers. Increased LH and FSH responses to GnRH agonist stimulation suggested neuroendocrine alterations in the regulation of gonadotrophin secretion similar to those in their proband sisters. These changes in PCOS brothers may reflect the impact of PCOS susceptibility genes and/or programming effects of the intrauterine environment.
STUDY FUNDING/COMPETING INTEREST(S)
This research was supported by P50 HD044405 (A.D.), K12 HD055884 (L.C.T.), U54 HD034449 (A.D., R.S.L.) from the National Institute of Child Health and Development. Some hormone assays were performed at the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core that is supported by U54 HD28934 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Partial support for some of the clinical studies was provided by UL1 RR025741 and UL1 TR000150 (Northwestern University Clinical and Translational Sciences Institute) from the National Center for Research Resources, National Institutes of Health, which is now the National Center for Advancing Translational Sciences. The authors have no conflict of interest to declare.
Keywords: polycystic ovary syndrome, steroidogenesis, steroidogenic enzymes, male phenotype
Introduction
Polycystic ovary syndrome (PCOS) is a common endocrine disorder that affects ∼7% of reproductive age women (Knochenhauer et al., 1998; Diamanti-Kandarakis et al., 1999). It is characterized by hyperandrogenemia and disordered gonadotrophin secretion (Diamanti-Kandarakis and Dunaif, 2012). Familial aggregation is well established and twin studies demonstrate strong evidence for heritability, suggesting a genetic contribution to its pathogenesis (Givens, 1988; Legro et al., 1998; Vink et al., 2006). PCOS is a non-Mendelian complex genetic trait reflecting the interaction of multiple susceptibility genes as well as environmental factors, and several genetic susceptibility loci have been reproducibly mapped (Urbanek et al., 2007; Shi et al., 2012). Consistent with these observations, male as well as female first-degree relatives have reproductive and metabolic abnormalities (Legro et al., 1998, 2002a,b; Sir-Petermann et al., 2002; Yildiz et al., 2003; Sam et al., 2006).
We have shown that the underlying reproductive phenotype in PCOS families is hyperandrogenemia. Adrenal as well as ovarian androgen levels are increased in the sisters of affected women suggesting a defect in the regulation of steroidogenic pathways common to the gonad and the adrenal glands (Legro et al., 1998). Brothers of affected women also have dehydroepiandrosterone sulfate (DHEAS) elevations that are significantly positively correlated with those in their proband sisters (Legro et al., 2002b). This finding suggests that brothers have the same defect in adrenal steroidogenesis as their proband sisters. Other investigators have demonstrated increased 17-hydroxyprogesterone (17Prog) responses to long-acting GnRH agonist stimulation in brothers of women with PCOS, suggesting dysregulation of testicular 17α-hydroxylase activity (Sir-Petermann et al., 2004). We performed this study to test the hypothesis that alterations in testicular or adrenal steroidogenesis account for hyperandrogenemia in brothers of women with PCOS.
Materials and Methods
Study population
We studied 29 brothers, aged 18–47 years, of 22 non-Hispanic white women with PCOS and 18 unrelated control men of comparable age, body mass index (BMI) and ethnicity. A group of 46 women were also included as controls. All subjects were in excellent general medical health and had no history of abnormal growth or development. There was no history of infertility among the subjects in either group, but a number of subjects had not attempted to father children. Brothers were referred for participation by their proband sisters who had previously participated in our studies of PCOS women. We recruited control men through advertisements in the local media. Our cohort of PCOS brothers was young and healthy. Accordingly, we recruited control males of comparable health status, age and BMI. The control men had no personal history of hypertension, and no personal or first-degree family history of diabetes mellitus. Any potential control man with a female first-degree relative with a known history of PCOS was excluded. Nevertheless, it remains possible that control men carried PCOS susceptibility genes but that: (i) they lacked female relatives, (ii) PCOS was undiagnosed in their female relatives or (iii) the control male was unaware of the PCOS diagnosis in their female relatives. Accordingly, we assumed that 7% (the population prevalence of PCOS) of the control men had PCOS susceptibility genes.
The diagnosis of PCOS was made in the probands using the NIH criteria (Legro et al., 1998; Diamanti-Kandarakis and Dunaif, 2012) of elevated circulating testosterone (T) and/or bioavailable T (uT) levels associated with chronic oligomenorrhea, i.e. ≤6 menses per year. Women with non-classical 21-hydroxylase deficiency, hyperprolactinemia or androgen-secreting tumors were excluded by appropriate tests (Dunaif et al., 1996). The control women had regular menses every 27–35 days, no history of reproductive disorders, and no hirsutism (Ferriman–Gallwey score <8) (Hatch et al., 1981). The clinical and biochemical features of the probands and control women have been reported as part of previous studies (Legro et al., 1998, 2002a,b). Among the families studied, one had 3 brothers, 5 families had 2 brothers, and 16 families had 1 brother who volunteered to participate. Clinical and baseline reproductive hormone levels of the brothers have been reported as part of previous studies (Legro et al., 2002b; Sam et al., 2008a,b; Coviello et al., 2009).
Ethical approval
The study was approved by the Institutional Review Board of Northwestern University Feinberg School of Medicine and written informed consent was obtained from all subjects.
Study protocol
Neither brothers nor control men were taking any medications known to alter sex hormone metabolism or glucose homeostasis for at least 1 month prior to study. All subjects underwent a 2 h 75-g oral glucose tolerance test (OGTT) after a 3 day 300 g per day carbohydrate diet and an overnight fast (Sam et al., 2008a,b). Blood samples for glucose were obtained at 0 min and 120 min after the oral glucose load. All control men had normal glucose tolerance according to the American Diabetes Association criteria (American Diabetes Association, 2011). Nine brothers had impaired glucose tolerance but none had diabetes mellitus. Blood samples were obtained for inhibin B as a measure of spermatogenesis with the 0 min blood draw of the OGTT.
Between 1130 and 1230 h, each subject underwent an ACTH stimulation test during which 10 μg/m2 ACTH (Organon, Bedford, OH, USA) per m2 body surface area was injected iv over 2 min (Barnes et al., 1993). Venous blood was sampled prior to, 0 min, and 30 and 60 min following ACTH administration for 17-hydroxypregnenolone (17Preg), 17Prog, 11-deoxycortisol (11DC), cortisol, dehydroepiandrosterone (DHEA), DHEAS and androstenedione (AD) levels.
A GnRH agonist stimulation test was performed 60 min after completion of the ACTH stimulation test. Subjects were given 10 μg leuprolide acetate (Lupron, TAP Pharmaceutical Products, Inc., Lake Forest, IL, USA) per kg body weight sc (Rosenfield et al., 1996). Venous blood was sampled for luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels at times −30, −15, 0, 30 and 60 min and 4, 16, 20 and 24 h. Blood samples were drawn for 17Prog, AD and T levels at baseline and 4, 16, 20 and 24 h following GnRH agonist administration.
Assays
All steroid levels were measured by liquid chromatography/mass spectrometry/mass spectrometry (LC-MS/MS), except for DHEAS, which was measured by automated chemiluminescent competitive immunoassay on the IMMULITE® 2000 platform (Siemens Diagnostics, USA). Steroid assay performance parameters have been previously reported (Anderson et al., 2010). LH and FSH assays were also performed using the IMMULITE® chemiluminescent immunoassay system (Siemens Diagnostics, USA) (McCartney et al., 2007). Inhibin B assays were performed by ELISA (Ansh Labs, USA) (Kumar et al., 2013). T and bioavailable T (uT) levels in the PCOS probands and control women were measured as previously reported (Legro et al., 1998).
Calculations
Incremental precursor to product molar ratios were calculated to evaluate apparent activity of steroidogenic enzymes (Rosenfield et al., 1994). The stimulated hormone levels were maximal at the 60 min and 16 h time points during the ACTH and GnRH agonist stimulation tests, respectively. To calculate ΔPrecursor steroid (nmol/l)/ΔProduct steroid (nmol/l), the incremental change in a steroid hormone during the ACTH stimulation test was calculated by subtracting the baseline level from the stimulated level drawn 60 min after ACTH injection in nmol/l. For the GnRH agonist stimulation test, incremental change was calculated as the difference between the 16 h and baseline levels.
Data analysis
The sample size was calculated based on data in normal men to detect a 20% difference in GnRH agonist-stimulated peak T and 17Prog responses (Barnes et al., 1989) and in ACTH-stimulated DHEA levels (Griffing et al., 1985) between PCOS brothers and control males with 80% power and alpha set at 0.05. For analysis of biochemical data, the family unit was the case. Thus, data from brothers within the same family were averaged to yield one mean value per family. Data collected from one control subject during the ACTH stimulation test were excluded from analysis because there was no increase in cortisol at 60 min following a ACTH administration (210 nmol/L at baseline and 215 nmol/L at 60 min) strongly suggesting that ACTH was not administered due to a technical error. Data were log-transformed when necessary to achieve homogeneity of variance. Differences between groups were assessed by two-tailed unpaired t-tests or Mann–Whitney tests, if homogeneity of variance was not achieved.
Because there was a trend toward increased BMI in the PCOS brothers, linear regression or Spearman correlation, depending on the normality of the data, was performed on all end-points to determine whether there was a significant association with BMI. ANCOVA adjusting for BMI was performed when there was a significant association. Statistical analyses were performed using SAS 9·2 (SAS Institute, Inc., Cary, NC, USA). Categorical variables were compared by Fisher's exact test. Differences were considered to be significant at P = 0.05. Data are reported as the untransformed mean ± SD.
Results
Clinical features and baseline hormone levels
The clinical features and reproductive hormone levels of the PCOS proband sisters are summarized in Table I. By design, PCOS sisters had significantly higher T, uT, DHEAS and LH levels compared with control women of comparable age and BMI. Mean age and BMI did not differ in the brothers compared with control men by design: 31 ± 8 years in brothers versus 30 ± 7 years in controls (P = 0.79) and 28.9 ± 4.6 kg/m2 in brothers versus 26.3 ± 5.0 kg/m2 controls (P = 0.10). Inhibin B levels (182 ± 75 pg/ml brothers versus 221 ± 75 controls, P = 0.08) also did not differ suggesting that spermatogenesis was normal.
Table I.
Clinical features and reproductive hormone levels PCOS proband sisters.
| Proband sisters (n = 22) |
Control women (n = 46) |
P-value | |
|---|---|---|---|
| Age (years) | 29 ± 6 | 31 ± 5 | 0.15 |
| BMI (kg/m2) | 37.1 ± 8.8 | 34.5 ± 7.2 | 0.35 |
| T (nmol/l) | 2.5 ± 0.8 | 1.0 ± 0.4 | <0.0001 |
| uT (nmol/l) | 0.9 ± 0.5 | 0.2 ± 0.1 | <0.0001 |
| DHEAS (µmol/l) | 7.1 ± 3.3 | 4.3 ± 1.2 | <0.0001 |
| LH (IU/l) | 12 ± 9 | 5 ± 5 | <0.0001 |
| FSH (IU/l) | 9 ± 3 | 9 ± 4 | 0.93 |
BMI, body mass index; T, testosterone; uT, bioavailable testosterone; DHEAS, dehydroepiandrosterone sulfate; LH, luteinizing hormone; FSH, follicle-stimulating hormone.
ACTH stimulation test
Brothers had significantly lower 0 min ACTH DHEA (P = 0.03) levels than control men (Table II). ACTH-0 min 17 Preg, 17Prog, 11DC, AD, DHEAS and cortisol levels did not differ significantly between brothers and control men (Table II). 17Preg, DHEA, 17Prog, 11DC, AD, DHEAS and cortisol levels and incremental responses following ACTH stimulation did not differ between brothers and control men (Table II). The ACTH-stimulated Δ17Preg/Δ17Prog molar ratio (P = 0.04) and ΔDHEA/ΔAD molar ratio (P = 0.04) were significantly lower in brothers compared with control men, suggesting increased apparent 3β-hydroxysteroid dehydrogenase (3β-HSD) activity in brothers (Figs 1 and 3). There were no significant differences between the groups in incremental precursor to product molar ratios for 21-hydroxylase or 11β-hydroxylase activity. There was also no evidence of differences in 17,20-lyase activity in the Δ4 or Δ5 pathway (Figs 1 and 3).
Table II.
Hormone levels drawn at baseline and incremental change (Δ) in hormone resulting from ACTH administration.
| Hormone |
Controls n = 17 |
Brothers n = 22 |
P-value | |
|---|---|---|---|---|
| 17Preg (nmol/l) | Basal | 7.0 ± 4.5 | 4.4 ± 3.3 | 0.12a |
| Incremental Δ | 24.2 ± 9.6 | 21.2 ± 14.2 | 0.21 | |
| 17Prog (nmol/l) | Basal | 2.8 ± 1.1 | 2.6 ± 0.9 | 0.70 |
| Incremental Δ | 1.9 ± 1.0 | 2.5 ± 1.7 | 0.21 | |
| DHEA (nmol/l) | Basal | 0.22 ± 0.09 | 0.13 ± 0.08 | 0.03a |
| Incremental Δ | 0.23 ± 0.11 | 0.20 ± 0.13 | 0.53 | |
| AD (nmol/l) | Basal | 3.2 ± 1.0 | 3.3 ± 1.0 | 0.70a |
| Incremental Δ | 1.9 ± 0.9 | 2.4 ± 1.2 | 0.19 | |
| 11DC (nmol/l) | Basal | 0.70 ± 0.53 | 0.71 ± 0.68 | 0.75 |
| Incremental Δ | 1.68 ± 1.25 | 1.99 ± 1.88 | 0.84b | |
| Cortisol (nmol/l) | Basal | 248 ± 69 | 215 ± 92 | 0.22a |
| Incremental Δ | 397 ± 182 | 364 ± 108 | 0.57 | |
| DHEAS (µmol/l) | Basal | 5.5 ± 1.8 | 5.2 ± 2.6 | 0.40b |
17Preg, 17-hydroxypregnenolone; 17Prog, 17-hydroxyprogesterone; DHEA, dehydroepiandrosterone; AD, androstenedione; 11DC, 11-deoxycortisol; DHEAS, dehydroepiandrosterone sulfate.
aDenotes P-values adjusted for BMI (ANCOVA).
bDenotes two-group comparisons performed by Wilcoxon rank-sum test. All other two-group comparisons were assessed by two-tailed t-tests.
Figure 1.

Incremental precursor to product molar ratios across steroidogenic pathways during the ACTH stimulation test demonstrated increased apparent 3β-HSD activity in brothers of women with PCOS (black bars) (*P = 0.04 for Δ17Preg/Δ17Prog and **P = 0.04 for ΔDHEA/ΔAD) compared with control men (open bars). No differences were observed in apparent 17,20-lyase, 21-hydroxlyase or 11β-hydroxylase activity. Δ11DC/ΔCortisol were compared by Wilcoxon rank-sum test. All other comparisons were made by t-tests (17Preg, 17-hydroxypregnenolone; 17Prog, 17-hydroxyprogesterone; DHEA, dehydroepiandrosterone; AD, androstenedione; 11DC, 11-deoxycortisol).
Figure 3.
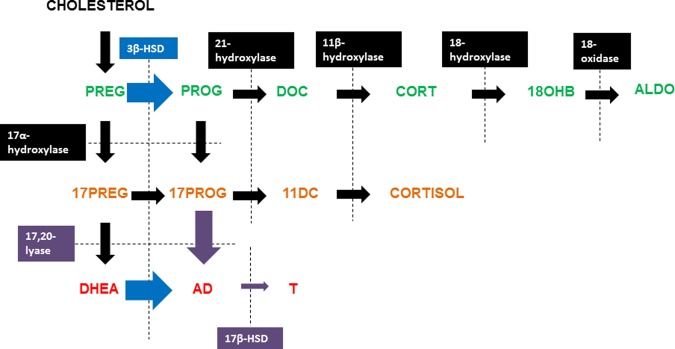
Summary of alterations in adrenal and gonadal steroidogenesis in PCOS brothers. Incremental precursor to product molar ratios across steroidogenic pathways during the ACTH stimulation test (blue arrows) demonstrated increased apparent 3β-HSD activity in brothers of women with PCOS compared with control men. Precursor to product molar ratios calculated from the GnRH agonist stimulation test (purple arrows) suggested PCOS brothers have increased gonadal 17,20-lyase activity and reduced 17β-HSD activity in comparison to control men (PREG, pregnenolone; PROG, progesterone; DOC, deoxycorticosterone; CORT, corticosterone; 18OHB, 18-OH-corticosterone; ALDO, aldosterone, 17PREG, 17-hydroxy pregnenolone; 17PROG, 17-hydroxyprogesterone; 11DC, 11-deoxycortisol; DHEA, dehydroepiandrosterone; AD, androstenedione; T, testosterone; 3β-HSD, 3β hydroxysteroid dehydrogenase; 17β-HSD, 17β hydroxysteroid dehydrogenase).
GnRH agonist stimulation test
ACTH 0 min 17Prog levels were similar to levels at the start of the GnRH agonist stimulation test (P = 0.91) suggesting that residual effects of ACTH did not confound the results of the GnRH agonist stimulation test. After adjusting for BMI, GnRH agonist 0 min T levels did not differ between the groups (Table III). Immediately following administration of a GnRH agonist, no differences were observed between groups in stimulated AD or 17Prog levels. There was no difference in the 17Prog incremental response to GnRH agonist, but the incremental AD response was higher in brothers than in controls (P = 0.02, Table III). There were no differences in GnRH agonist 0 min or GnRH agonist-stimulated T levels or incremental T responses after adjusting for BMI (Table III, Fig. 2).
Table III.
Hormone levels drawn prior to GnRH agonist administration and incremental change (Δ) in hormone levels resulting from GnRH agonist stimulation.
| Hormone |
Controls n = 18 |
Brothers n = 22 |
P-value | |
|---|---|---|---|---|
| LH (IU/l) | Basal | 2.7 ± 1.4 | 3.0 ± 1.7 | 0.55 |
| Incremental Δ | 26.0 ± 9.8 | 38.6 ± 20.6 | 0.02 | |
| FSH (IU/l) | Basal | 2.7 ± 1.5 | 3.2 ± 1.4 | 0.07b |
| Incremental Δ | 4.8 ± 4.1 | 10.2 ± 7.5 | 0.002 | |
| T (nmol/l) | Basal | 16.2 ± 5.5 | 11.6 ± 4.7 | 0.79a |
| Incremental Δ | 13.5 ± 6.5 | 11.1 ± 4.7 | 0.05a | |
| 17Prog (nmol/l) | Basal | 3.0 ± 1.3 | 2.5 ± 1.0 | 0.17 |
| Incremental Δ | 5.6 ± 3.5 | 5.2 ± 2.9 | 0.27 | |
| AD (nmol/l) | Basal | 3.9 ± 1.3 | 3.5 ± 1.1 | 0.39 |
| Incremental Δ | 0.9 ± 1.5 | 1.7 ± 1.4 | 0.02 | |
LH, luteinizing hormone; FSH, follicle-stimulating hormone; T, testosterone; 17Prog, 17-hydroxyprogesterone; AD, androstenedione.
aDenotes P-values adjusted for BMI (ANCOVA).
bDenotes two-group comparison performed by Wilcoxon rank-sum test. All other two-group comparisons were assessed by two-tailed t-tests.
Figure 2.

Baseline LH and FSH levels in brothers (black bars) and control men (open bars) were similar. LH and FSH responses to GnRH agonist stimulation were higher in brothers (*P = 0.02 for LH; **P = 0.004 for FSH). Lower Δ17Prog/ΔAD (†P = 0.003) in brothers during the GnRH agonist stimulation test suggests that they have increased gonadal 17,20-lyase activity compared with control men. Higher ΔAD/ΔT in brothers during the GnRH agonist stimulation test (‡P = 0.005) suggests that this group also has reduced 17β-HSD activity in comparison to control men. Comparison of baseline and stimulated FSH levels were made by Wilcoxon rank-sum tests. All other comparisons were made by t-tests (LH, luteinizing hormone; FSH, follicle-stimulating hormone; 17Prog, 17-hydroxyprogesterone; AD, androstenedione; T, testosterone).
The ΔAD/ΔT molar ratio was higher (P = 0.005) in brothers than in control men suggesting that brothers had reduced gonadal apparent 17β-hydroxysteroid dehydrogenase (17β-HSD) activity (Figs 2 and 3). The GnRH agonist-stimulated Δ17Prog/ΔAD ratio was lower (P = 0.003, Figs 2 and 3) in brothers than in control men, which suggested that gonadal apparent 17,20-lyase activity was increased in brothers.
GnRH agonist 0 min LH and FSH levels were similar between the two groups (Table III). However, stimulated LH and FSH levels were higher (P = 0.02 for LH; P = 0.004 for FSH, Fig. 2) in brothers than in control men, as were the incremental responses (Table III).
Discussion
Brothers of women with PCOS had evidence for alterations in adrenal and gonadal steroidogenesis, independent of age and weight. Baseline DHEA levels were lower in brothers. ACTH-stimulated Δ17Preg/Δ17Prog and ΔDHEA/ΔAD molar ratios were also decreased consistent with increased 3β-HSD activity in brothers compared with control men (Fig. 3). The GnRH agonist-stimulated ΔAD/ΔT molar ratio was increased and Δ17Prog/ΔAD ratio was reduced in brothers suggesting that they had reduced gonadal apparent 17βHSD activity and increased gonadal apparent 17,20-lyase activity (Fig. 3). Gonadotrophin responses to GnRH agonist stimulation were significantly increased in the brothers compared with control men suggesting that brothers also had alterations in neuroendocrine function.
The putative increase in 3β-HSD activity could account for the lower baseline DHEA levels in brothers. There are conflicting reports regarding 3β-HSD activity in women with PCOS. One study suggested increased 3β-HSD activity (Moran et al., 2004), while other studies have suggested that 3β-HSD activity is reduced in women with PCOS (Barnes et al., 1993; Carbunaru et al., 2004). There were no differences in cortisol or 11DC levels before or after ACTH stimulation, suggesting that glucocorticoid biosynthesis was normal in the PCOS brothers.
There were no differences in baseline or GnRH agonist-stimulated total or bioavailable T levels, after adjusting for BMI, suggesting that testicular T production was normal. GnRH agonist-stimulated ΔAD/ΔT molar ratios were increased and Δ17Prog/ΔAD ratios were reduced in brothers suggesting that they had increased gonadal apparent 17,20-lyase activity and reduced gonadal apparent 17βHSD activity. Increased protein expression of CYP17A1 has been reported in polycystic ovaries (Comim et al., 2013). Although no replicated variants have been identified in the CYP17A1 gene (Li et al., 2012), serine phosphorylation of the encoded protein, P450c17, increases its 17,20 lyase activity, which has been proposed as a post-translational mechanism regulating androgen biosynthesis (Tee and Miller, 2013).
Studies on the role of 17β-HSD in PCOS have been conflicting. Women with increased AD and estrone levels suggestive of ovarian 17β-HSD deficiency can present with a PCOS phenotype including hirsutism and oligo-amenorrhea (Pang et al., 1987; Toscano et al., 1990). In contrast, an activating polymorphism in the type 5 17β-HSD gene was found in a subset (∼10%) of women with PCOS (Qin et al., 2006), but this finding was not replicated in a larger cohort of affected women (Goodarzi et al., 2008). We also were unable to identify an association between genetic variants in the type 3 17β-HSD gene and PCOS (Moghrabi et al., 1998).
LH and FSH responses to GnRH agonist were significantly increased in brothers compared with control men. Total T levels at baseline and in response to GnRH agonist as well as inhibin B levels did not differ between the groups suggesting the testicular function was normal in brothers. Similar gonadotrophin responses to native GnRH have been documented in women with PCOS (Rebar et al., 1976). Stimulation with GnRH agonist also has resulted in increased LH release but blunted FSH responses in affected women, a pattern similar to that in normal men (Barnes et al., 1989). In women with PCOS, an increased frequency of pulsatile GnRH secretion is a putative contributor to enhanced LH release (Marshall and Eagleson, 1999). Increased pulsatile administration of native GnRH to GnRH-deficient men selectively increases LH release (Spratt et al., 1987). Thus, it is possible that the brothers also have neuroendocrine alterations in the regulation of gonadotrophin secretion similar to those observed in their proband sisters.
Variation in genes regulating ovarian and adrenal steroidogenesis or gonadotrophin secretion may account for the reproductive phenotype in brothers and their proband sisters with PCOS (Legro et al., 1998). Alternatively, some of the observed differences in adrenal and gonadal steroidogenesis could be the result of programming effects of the intrauterine environment. Prenatal androgen administration to pregnant rhesus macaques produces many of the phenotypic features of PCOS in female offspring, including abnormalities in adrenal steroidogenesis (Zhou et al., 2005). Prenatally androgenized male macaques have a metabolic phenotype similar to the one we have reported in brothers (Bruns et al., 2004; Sam et al., 2008a,b). Adult prenatally androgenized male sheep have reproductive changes including reduced sperm counts (Recabarren et al., 2008a) and germ cell number (Rojas-Garcia et al., 2010). Further, FSH responses to GnRH agonist are increased (Rojas-Garcia et al., 2010) but LH responses are unchanged (Recabarren et al., 2007) in these sheep. Leydig cell function appeared to be normal since circulating T levels and T responses to hCG were similar to control male sheep (Recabarren et al., 2008a; Rojas-Garcia et al., 2010).
The current study was not designed to have adequate power to determine if baseline DHEAS levels were increased in brothers, as we have previously reported (Legro et al., 2002b). Further, we did not examine the sulfatase enzyme that converts DHEA to DHEAS in this study. An increase in the activity of this enzyme could account for the reported increase in DHEAS levels in brothers (Legro et al., 2002b). Nevertheless, AD responses to GnRH agonist were increased in the brothers suggesting increased testicular androgen biosynthesis in response to LH. This finding supports the hypothesis that hyperandrogenemia is a PCOS male relative reproductive phenotype.
Our findings also differ from those of Sir-Petermann and colleagues who reported exaggerated 17Prog responses but no alterations in gonadotrophin secretion in response to GnRH agonist stimulation in their Chilean cohort of adult brothers of affected women compared with control men (Sir-Petermann et al., 2004). Ethnic differences may account for these discrepant findings. However, in contrast to their findings in brothers, the Chilean investigators (Recabarren et al., 2008b) found no differences in GnRH agonist-stimulated 17Prog responses during infancy, childhood or adulthood in the sons of women with PCOS. Such variations in the reproductive phenotype in PCOS brothers compared with sons may point to differing effects of genetic factors compared with programming effects of the intrauterine environment on gonadal steroidogenesis in these men.
Our study has potential limitations. GnRH agonist stimulation testing was performed 1 h following completion of the ACTH stimulation test, using a protocol similar to one validated in studies of the functional significance of polycystic ovaries (Mortensen et al., 2006, 2009). The tests were performed at the same time of day in all subjects and responsiveness (stimulated minus basal values), a parameter not known to have circadian changes, rather than basal values was the primary end-point. In addition, baseline 17Prog levels at the start of the ACTH stimulation test did not differ from those at the start of the GnRH agonist stimulation test. Accordingly, it is unlikely that the timing and sequence of stimulation testing impacted the study outcome. The failure to perform semen analyses to further characterize the reproductive phenotype of PCOS brothers is another limitation of our study. However, levels of inhibin B, which has been used as an endocrine marker of spermatogenesis (Pierik et al., 1998), did not differ in brothers. Further, no differences in sperm counts, motility or morphology were found in adult sons of women with PCOS compared with control men (Recabarren et al., 2008b). The proband sisters did not participate in the testing protocol since many previous studies have examined steroidogenic responses to ACTH and GnRH agonist in PCOS (Rebar et al., 1976; Barnes et al., 1989; Moran et al., 2004). Therefore, we could not compare responses in PCOS probands and to those in their brothers. However, we have previously reported that only baseline DHEAS levels are correlated in PCOS probands and their brothers (Legro et al., 2002b).
In summary, we have further characterized the reproductive phenotype in the brothers of women with PCOS. This phenotype includes evidence for abnormalities in both adrenal and gonadal steroidogenesis. We also found that brothers of affected women have alterations in the gonadotrophin responses to GnRH agonist similar to those found in affected women. Identification of reproductive traits shared by PCOS probands and their first-degree relatives suggests that these traits have a genetic basis. These observations provide insights into pathways that may be primarily disrupted in PCOS, such as androgen biosynthesis and the regulation of gonadotrophin secretion, since both male and female relatives have hyperandrogenemia and increased LH and FSH responses to GnRH agonist.
Authors’ roles
D.M.L. aided in data collection and data analysis, and drafted the manuscript. L.C.T. aided in data analysis and revised the manuscript. Y.S. aided in data collection and analysis. R.P aided in data collection. R.S.L. aided in study design and interpretation of data analysis and reviewed the manuscript. S.K.G., R.J.S. and R.L.T. performed the hormone assays. A.D. was the principal investigator for the study, provided oversight for all study activities and is the corresponding author for the manuscript.
Funding
This research was supported by P50 HD044405 (A.D.), K12 HD055884 (L.C.T), U54 HD034449 (A.D., R.S.L.) from the National Institute of Child Health and Development. Some hormone assays were performed at the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core that is supported by U54 HD28934 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Partial support for some of the clinical studies was provided by UL1 RR025741 and UL1 TR000150 (Northwestern University Clinical and Translational Sciences Institute) from the National Center for Research Resources, National Institutes of Health, which is now the National Center for Advancing Translational Sciences.
Conflict of interest
None declared.
Acknowledgements
The authors thank the women with PCOS and their brothers for participating in this study and the nurses of the Northwestern University General Clinical Research Center for their superb care of the study participants. We also thank Ajay Kumar, Bhanu Kalra, and Gopal Savjani of Ansh laboratories for performing the Inhibin B assays, as well as Elizabeth Podczerwinski and Barb Scheetz for coordinating this project.
References
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2011;34(Suppl 1):S62–S69. doi: 10.2337/dc11-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson H, Fogel N, Grebe SK, Singh RJ, Taylor RL, Dunaif A. Infants of women with polycystic ovary syndrome have lower cord blood androstenedione and estradiol levels. J Clin Endocrinol Metab. 2010;95:2180–2186. doi: 10.1210/jc.2009-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes RB, Rosenfield RL, Burstein S, Ehrmann DA. Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N Engl J Med. 1989;320:559–565. doi: 10.1056/NEJM198903023200904. [DOI] [PubMed] [Google Scholar]
- Barnes RB, Ehrmann DA, Brigell DF, Rosenfield RL. Ovarian steroidogenic responses to gonadotropin-releasing hormone agonist testing with nafarelin in hirsute women with adrenal responses to adrenocorticotropin suggestive of 3 beta-hydroxy-delta 5-steroid dehydrogenase deficiency. J Clin Endocrinol Metab. 1993;76:450–455. doi: 10.1210/jcem.76.2.8381802. [DOI] [PubMed] [Google Scholar]
- Bruns CM, Baum ST, Colman RJ, Eisner JR, Kemnitz JW, Weindruch R, Abbott DH. Insulin resistance and impaired insulin secretion in prenatally androgenized male rhesus monkeys. J Clin Endocrinol Metab. 2004;89:6218–6223. doi: 10.1210/jc.2004-0918. [DOI] [PubMed] [Google Scholar]
- Carbunaru G, Prasad P, Scoccia B, Shea P, Hopwood N, Ziai F, Chang YT, Myers SE, Mason JI, Pang S. The hormonal phenotype of Nonclassic 3 beta-hydroxysteroid dehydrogenase (HSD3B) deficiency in hyperandrogenic females is associated with insulin-resistant polycystic ovary syndrome and is not a variant of inherited HSD3B2 deficiency. J Clin Endocrinol Metab. 2004;89:783–394. doi: 10.1210/jc.2003-030934. [DOI] [PubMed] [Google Scholar]
- Comim FV, Teerds K, Hardy K, Franks S. Increased protein expression of LHCG receptor and 17alpha-hydroxylase/17–20-lyase in human polycystic ovaries. Hum Reprod. 2013;28:3086–3092. doi: 10.1093/humrep/det352. [DOI] [PubMed] [Google Scholar]
- Coviello AD, Sam S, Legro RS, Dunaif A. High prevalence of metabolic syndrome in first-degree male relatives of women with polycystic ovary syndrome is related to high rates of obesity. J Clin Endocrinol Metab. 2009;94:4361–4366. doi: 10.1210/jc.2009-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev. 2012;33:981–1030. doi: 10.1210/er.2011-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Kouli CR, Bergiele AT, Filandra FA, Tsianateli TC, Spina GG, Zapanti ED, Bartzis MI. A survey of the polycystic ovary syndrome in the Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab. 1999;84:4006–4011. doi: 10.1210/jcem.84.11.6148. [DOI] [PubMed] [Google Scholar]
- Dunaif A, Scott D, Finegood D, Quintana B, Whitcomb R. The insulin-sensitizing agent troglitazone improves metabolic and reproductive abnormalities in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1996;81:3299–3306. doi: 10.1210/jcem.81.9.8784087. [DOI] [PubMed] [Google Scholar]
- Givens JR. Familial polycystic ovarian disease. Endocrinol Metab Clin North Am. 1988;17:771–783. [PubMed] [Google Scholar]
- Goodarzi MO, Jones MR, Antoine HJ, Pall M, Chen YD, Azziz R. Nonreplication of the type 5 17beta-hydroxysteroid dehydrogenase gene association with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93:300–303. doi: 10.1210/jc.2007-1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffing GT, Allen J, Pratt H, Melby JC. Discordance of plasma DHEA-S, DHEA, and cortisol responses with various ACTH regimens. Metabolism. 1985;34:631–636. doi: 10.1016/0026-0495(85)90090-3. [DOI] [PubMed] [Google Scholar]
- Hatch R, Rosenfield RL, Kim MH, Tredway D. Hirsutism: implications, etiology, and management. Am J Obstet Gynecol. 1981;140:815–830. doi: 10.1016/0002-9378(81)90746-8. [DOI] [PubMed] [Google Scholar]
- Knochenhauer ES, Key TJ, Kahsar-Miller M, Waggoner W, Boots LR, Azziz R. Prevalence of the polycystic ovary syndrome in unselected black and white women of the southeastern United States: a prospective study. J Clin Endocrinol Metab. 1998;83:3078–3082. doi: 10.1210/jcem.83.9.5090. [DOI] [PubMed] [Google Scholar]
- Kumar A, Kalra B, Patel AS, Shah S. Development of a sensitive Inhibin B ELISA optimized to eliminate false positive results. Clin Chem. 2013;59:107. [Google Scholar]
- Legro RS, Driscoll D, Strauss JF, III, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci USA. 1998;95:14956–14960. doi: 10.1073/pnas.95.25.14956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legro RS, Bentley-Lewis R, Driscoll D, Wang SC, Dunaif A. Insulin resistance in the sisters of women with polycystic ovary syndrome: association with hyperandrogenemia rather than menstrual irregularity. J Clin Endocrinol Metab. 2002a;87:2128–2133. doi: 10.1210/jcem.87.5.8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legro RS, Kunselman AR, Demers L, Wang SC, Bentley-Lewis R, Dunaif A. Elevated dehydroepiandrosterone sulfate levels as the reproductive phenotype in the brothers of women with polycystic ovary syndrome. J Clin Endocr Metab. 2002b;87:2134–2138. doi: 10.1210/jcem.87.5.8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu F, Luo S, Hu H, Li XH, Li SW. Polymorphism T->C of gene CYP17 promoter and polycystic ovary syndrome risk: a meta-analysis. Gene. 2012;495:16–22. doi: 10.1016/j.gene.2011.12.048. [DOI] [PubMed] [Google Scholar]
- Marshall JC, Eagleson CA. Neuroendocrine aspects of polycystic ovary syndrome. Endocrinol Metab Clin North Am. 1999;28:295–324. doi: 10.1016/s0889-8529(05)70071-2. [DOI] [PubMed] [Google Scholar]
- McCartney CR, Blank SK, Prendergast KA, Chhabra S, Eagleson CA, Helm KD, Yoo R, Chang RJ, Foster CM, Caprio S, et al. Obesity and sex steroid changes across puberty: evidence for marked hyperandrogenemia in pre- and early pubertal obese girls. J Clin Endocrinol Metab. 2007;92:430–436. doi: 10.1210/jc.2006-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghrabi N, Hughes IA, Dunaif A, Andersson S. Deleterious missense mutations and silent polymorphism in the human 17beta-hydroxysteroid dehydrogenase 3 gene (HSD17B3) J Clin Endocrinol Metab. 1998;83:2855–2860. doi: 10.1210/jcem.83.8.5052. [DOI] [PubMed] [Google Scholar]
- Moran C, Reyna R, Boots LS, Azziz R. Adrenocortical hyperresponsiveness to corticotropin in polycystic ovary syndrome patients with adrenal androgen excess. Fertil Steril. 2004;81:126–131. doi: 10.1016/j.fertnstert.2003.07.008. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Rosenfield RL, Littlejohn E. Functional significance of polycystic-size ovaries in healthy adolescents. J Clin Endocrinol Metab. 2006;91:3786–3790. doi: 10.1210/jc.2006-0835. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Ehrmann DA, Littlejohn E, Rosenfield RL. Asymptomatic volunteers with a polycystic ovary are a functionally distinct but heterogeneous population. J Clin Endocrinol Metab. 2009;94:1579–1586. doi: 10.1210/jc.2008-2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang SY, Softness B, Sweeney WJ, III, New MI. Hirsutism, polycystic ovarian disease, and ovarian 17-ketosteroid reductase deficiency. N Engl J Med. 1987;316:1295–1301. doi: 10.1056/NEJM198705213162102. [DOI] [PubMed] [Google Scholar]
- Pierik FH, Vreeburg JT, Stijnen T, De Jong FH, Weber RF. Serum inhibin B as a marker of spermatogenesis. J Clin Endocrinol Metab. 1998;83:3110–3114. doi: 10.1210/jcem.83.9.5121. [DOI] [PubMed] [Google Scholar]
- Qin K, Ehrmann DA, Cox N, Refetoff S, Rosenfield RL. Identification of a functional polymorphism of the human type 5 17beta-hydroxysteroid dehydrogenase gene associated with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:270–276. doi: 10.1210/jc.2005-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebar R, Judd HL, Yen SS, Rakoff J, Vandenberg G, Naftolin F. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest. 1976;57:1320–1329. doi: 10.1172/JCI108400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recabarren SE, Lobos A, Figueroa Y, Padmanabhan V, Foster DL, Sir-Petermann T. Prenatal testosterone treatment alters LH and testosterone responsiveness to GnRH agonist in male sheep. Biol Res. 2007;40:329–338. [PubMed] [Google Scholar]
- Recabarren SE, Rojas-Garcia PP, Recabarren MP, Alfaro VH, Smith R, Padmanabhan V, Sir-Petermann T. Prenatal testosterone excess reduces sperm count and motility. Endocrinology. 2008a;149:6444–6448. doi: 10.1210/en.2008-0785. [DOI] [PubMed] [Google Scholar]
- Recabarren SE, Sir-Petermann T, Rios R, Maliqueo M, Echiburu B, Smith R, Rojas-Garcia P, Recabarren M, Rey RA. Pituitary and testicular function in sons of women with polycystic ovary syndrome from infancy to adulthood. J Clin Endocrinol Metab. 2008b;93:3318–3324. doi: 10.1210/jc.2008-0255. [DOI] [PubMed] [Google Scholar]
- Rojas-Garcia PP, Recabarren MP, Sarabia L, Schon J, Gabler C, Einspanier R, Maliqueo M, Sir-Petermann T, Rey R, Recabarren SE. Prenatal testosterone excess alters Sertoli and germ cell number and testicular FSH receptor expression in rams. Am J Physiol Endocrinol Metab. 2010;299:E998–E1005. doi: 10.1152/ajpendo.00032.2010. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL, Barnes RB, Ehrmann DA. Studies of the nature of 17-hydroxyprogesterone hyperresponsiveness to gonadotropin-releasing-hormone agonist challenge in functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1994;79:1686–1692. doi: 10.1210/jcem.79.6.7989476. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL, Perovic N, Ehrmann DA, Barnes RB. Acute hormonal responses to the gonadotropin releasing hormone agonist leuprolide: dose-response studies and comparison to nafarelin—a clinical research center study. J Clin Endocrinol Metab. 1996;81:3408–3411. doi: 10.1210/jcem.81.9.8784105. [DOI] [PubMed] [Google Scholar]
- Sam S, Legro RS, Essah PA, Apridonidze T, Dunaif A. Evidence for metabolic and reproductive phenotypes in mothers of women with polycystic ovary syndrome. Proc Natl Acad Sci USA. 2006;103:7030–7035. doi: 10.1073/pnas.0602025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam S, Coviello AD, Sung YA, Legro RS, Dunaif A. Metabolic phenotype in the brothers of women with polycystic ovary syndrome. Diabetes Care. 2008a;31:1237–1241. doi: 10.2337/dc07-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam S, Sung YA, Legro RS, Dunaif A. Evidence for pancreatic beta-cell dysfunction in brothers of women with polycystic ovary syndrome. Metabolism. 2008b;57:84–89. doi: 10.1016/j.metabol.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Zhao H, Shi Y, Cao Y, Yang D, Li Z, Zhang B, Liang X, Li T, Chen J, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet. 2012;44:1020–1025. doi: 10.1038/ng.2384. [DOI] [PubMed] [Google Scholar]
- Sir-Petermann T, Angel B, Maliqueo M, Carvajal F, Santos JL, Perez-Bravo F. Prevalence of Type II diabetes mellitus and insulin resistance in parents of women with polycystic ovary syndrome. Diabetologia. 2002;45:959–964. doi: 10.1007/s00125-002-0836-3. [DOI] [PubMed] [Google Scholar]
- Sir-Petermann T, Cartes A, Maliqueo M, Vantman D, Gutierrez C, Toloza H, Echiburu B, Recabarren SE. Patterns of hormonal response to the GnRH agonist leuprolide in brothers of women with polycystic ovary syndrome: a pilot study. Hum Reprod. 2004;19:2742–2747. doi: 10.1093/humrep/deh512. [DOI] [PubMed] [Google Scholar]
- Spratt DI, Finkelstein JS, Butler JP, Badger TM, Crowley WF., Jr Effects of increasing the frequency of low doses of gonadotropin-releasing hormone (GnRH) on gonadotropin secretion in GnRH-deficient men. J Clin Endocrinol Metab. 1987;64:1179–1186. doi: 10.1210/jcem-64-6-1179. [DOI] [PubMed] [Google Scholar]
- Tee MK, Miller WL. Phosphorylation of human cytochrome P450c17 by p38alpha selectively increases 17,20 lyase activity and androgen biosynthesis. J Biol Chem. 2013;288:23903–23913. doi: 10.1074/jbc.M113.460048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toscano V, Balducci R, Bianchi P, Mangiantini A, Sciarra F. Ovarian 17-ketosteroid reductase deficiency as a possible cause of polycystic ovarian disease. J Clin Endocrinol Metab. 1990;71:288–292. doi: 10.1210/jcem-71-2-288. [DOI] [PubMed] [Google Scholar]
- Urbanek M, Sam S, Legro RS, Dunaif A. Identification of a polycystic ovary syndrome susceptibility variant in fibrillin-3 and association with a metabolic phenotype. J Clin Endocrinol Metab. 2007;92:4191–4198. doi: 10.1210/jc.2007-0761. [DOI] [PubMed] [Google Scholar]
- Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab. 2006;91:2100–2104. doi: 10.1210/jc.2005-1494. [DOI] [PubMed] [Google Scholar]
- Yildiz BO, Yarali H, Oguz H, Bayraktar M. Glucose intolerance, insulin resistance, and hyperandrogenemia in first degree relatives of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:2031–2036. doi: 10.1210/jc.2002-021499. [DOI] [PubMed] [Google Scholar]
- Zhou R, Bird IM, Dumesic DA, Abbott DH. Adrenal hyperandrogenism is induced by fetal androgen excess in a rhesus monkey model of polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90:6630–6637. doi: 10.1210/jc.2005-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]