
Figure 5.
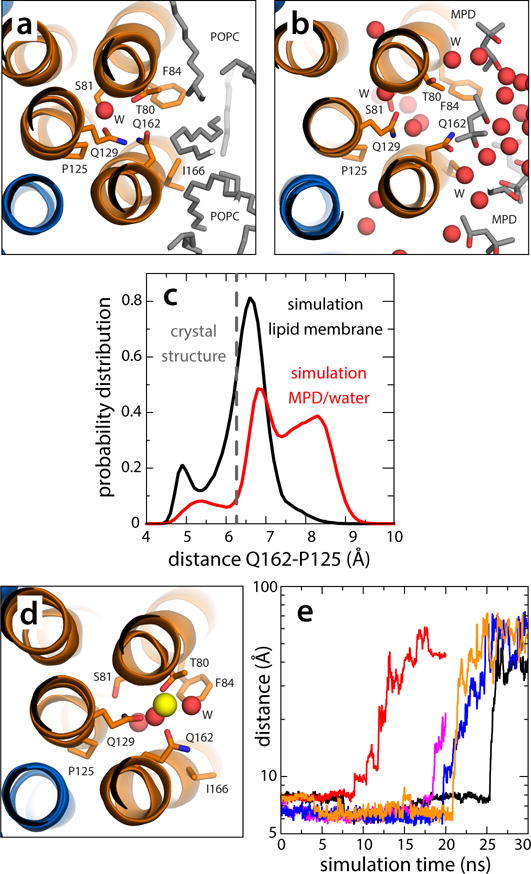
Dynamics of the vacant site in the V-type c1 subunit. (a) Close-up of the c1 site, viewed from the cytoplasm, in a representative snapshot of the simulation of the c-ring in a phospholipid membrane (Supplementary Fig. 7). The configuration closely resembles the locked conformation observed in the crystal structure of the c-ring in detergent. (b) Close-up of the c1 site in a representative snapshot of the simulation of the c-ring in the MPD/water buffer (Supplementary Figs. 7, 8). Polar interactions with the buffer unlock the H-bonding interaction network within the site, allowing Gln162 to project away into the solvent. The c-ring is represented as in Fig. 1. Side-chains and water molecules in the site, as well as neighboring lipid tails or MPD molecules, are highlighted (sticks, spheres). Hydrogen atoms as well as other protein side-chains and lipid head-groups are omitted for clarity. (c) Probability distributions of the distance between the carboxamide group of Gln162 and Pro125, in the neighboring inner helix, in either simulation, demonstrate the propensity of the c1 site to adopt an open conformation in the hydrated environment. (d) Hypothetical configuration of the c1 site with a bound Na+, equilibrated through restrained molecular dynamics simulations. Note the ion is penta-coordinated, as in the actual Na+-binding sites. (e) Rapid, spontaneous dissociation of the Na+ ion modeled in the c1 site in 5 independent simulation trajectories.