

Abstract
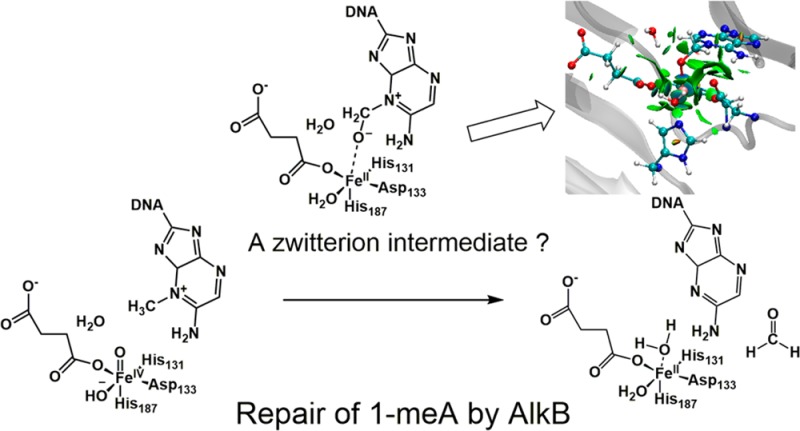
AlkB is the title enzyme of a family of DNA dealkylases that catalyze the direct oxidative dealkylation of nucleobases. The conventional mechanism for the dealkylation of N1-methyl adenine (1-meA) catalyzed by AlkB after the formation of FeIV–oxo is comprised by a reorientation of the oxo moiety, hydrogen abstraction, OH rebound from the Fe atom to the methyl adduct, and the dissociation of the resulting methoxide to obtain the repaired adenine base and formaldehyde. An alternative pathway with hydroxide as a ligand bound to the iron atom is proposed and investigated by QM/MM simulations. The results show OH– has a small impact on the barriers for the hydrogen abstraction and OH rebound steps. The effects of the enzyme and the OH– ligand on the hydrogen abstraction by the FeIV–oxo moiety are discussed in detail. The new OH rebound step is coupled with a proton transfer to the OH– ligand and results in a novel zwitterion intermediate. This zwitterion structure can also be characterized as Fe–O–C complex and facilitates the formation of formaldehyde. In contrast, for the pathway with H2O bound to iron, the hydroxyl product of the OH rebound step first needs to unbind from the metal center before transferring a proton to Glu136 or other residue/substrate. The consistency between our theoretical results and experimental findings is discussed. This study provides new insights into the oxidative repair mechanism of DNA repair by nonheme FeII and α-ketoglutarate (α-KG) dependent dioxygenases and a possible explanation for the substrate preference of AlkB.
1. Introduction
E. coli AlkB is a member of the FeII and α-KG dependent dioxygenase superfamily of enzymes. AlkB can repair alkylated bases such as 1-meA and N3-methyl cytosine (3-meC) via an oxidative dealkylation.1 The proposed mechanism, based on the mechanism of the related enzyme TauD,2 involves a series of steps that can be separated in two parts. The first part is composed of the formation of an FeIV=O (ferryl) intermediate along with the release of CO2 and formation of succinate. After the formation of the iron(IV)–oxo, the oxo moiety undergoes a reorientation from an axial to an equatorial position. The subsequent steps comprise the second part, which involve the oxidation of the methyl moiety on the base as shown in Scheme 1 (see Supporting Information (SI) Scheme S1 for the full mechanism including part 1).
Scheme 1. Proposed Mechanism for the Steps Starting from H Abstraction in the Dealkylation Catalyzed by AlkB Based on the TauD Mechanism.

After the reorientation of the oxo, the FeIV=O moiety abstracts a hydrogen atom from the methyl group of 1-meA, followed by the OH rebound to the carbon radical. Subsequently, the proton on the recently added OH is transferred and the C–N bond breaks, resulting in the formation of formaldehyde. However, the details of the formation of formaldehyde, such as where the proton is transferred and when the C–N bond breaks, are still not clear. Moreover, recent experimental discoveries suggest a possible alternative pathway. The crystal structure of an intermediate in the dealkylation of 3-meC has been recently reported. Based on the crystal data and QM/MM calculations, a zwitterion structure was proposed.3 In addition, time-resolved Raman spectra reveal the possible existence of a metal-coordinated oxygenated intermediate, such as FeII–O–C for TauD, another dioxygenase undergoing a similar mechanism to AlkB.4
In the case of 1-meA as substrate, the zwitterion structure and FeII–O–C complex may form after the deprotonation of the product of the OH rebound process. The crystal structures of AlkB with succinate and different substrates (PDB ID: 2FDG, 2FDJ, 3OIS, 3OIU, 3OIT, 3OIV) show a vacancy between the succinate and the aspartate residue (Asp133) bound to iron (see Figure 1 for 2FDG5). This vacancy is a result of the reorientation of the oxo moiety from an equatorial to an axial position.6,7 This vacancy can be occupied by a water molecule, which results in the traditional pathway as shown in Scheme 2 (H2O pathway). On the other hand, the physiological pH is slightly basic, and the optimal pH for repair of 1-meA is 7.5–8. Thus, this indicates the possibility of hydroxide in the environment.8 Moreover, hydroxide carries a negative charge and is a stronger iron-binding ligand than water. Hence, an alternative pathway (OH– pathway) with the participation of hydroxide is possible (see Scheme 3).
Figure 1.
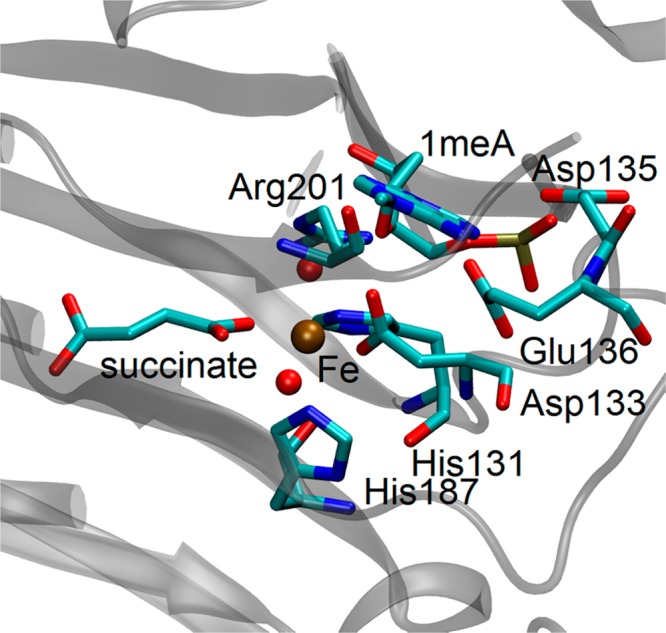
Active site of AlkB with 1-meA in the crystal structure (PDB ID: 2FDG).
Scheme 2. Proposed Detailed H2O Pathway Starting from the Hydrogen Abstraction.

Scheme 3. OH– Pathway: The Newly Proposed Mechanism with OH– Coordinated to the Iron (ROH).

Furthermore, the relative positions of the −CH2OH (or CH2O–) moiety connected to the DNA substrate with respect to the iron atom are not the same for different DNA bases in their corresponding crystal structures (see SI Figure S1). In some crystal structures (3OIU), the substrates are bound to the iron center while for others (3OIS, 3OIV) the oxidized methyls are unbound, and in another case, the moiety is located in an intermediate position (3OIT). In the case when the substrate is unbound from the metal center, the vacancy on the iron may be occupied by a water molecule. This raises the question of whether the unbinding process is always required.
We have previously used ab initio QM/MM to study the rate-limiting H atom abstraction step for the H2O pathway in detail.9 In this contribution, we elucidate the H2O pathway (Scheme 2) after the hydrogen abstraction step, which includes the OH rebound step and the formation of formaldehyde based on the results from QM/MM simulations. In addition, we report results from QM/MM simulations of the new OH– pathway (Scheme 3) and its comparison with the H2O pathway to provide new theoretical insights and their comparison to previous experimental findings. In section 2, we present the details for the setup of the systems including the required structures for the different steps and the computational methods. Subsequently, a detailed analysis of the results for the two different pathways explored, H2O and OH–, is presented.
2. Computational Methods
The computational methods and structure preparation follow our previous study. In brief, after adding hydrogen atoms, water box, and counterions to a crystal structure of AlkB (PDB ID: 2FDG(5)), we carried out Molecular Dynamics (MD) simulations in the NVT (Canonical) ensemble at 300 K using the Amberff99 force field with a 1 fs step size with an 8 Å cutoff for nonbonded interactions and particle mesh Ewald to treat long-range Coulomb interactions.10 The MD simulations were performed with the pmemd program in AMBER11.11 The snapshot with the lowest QM/MM energy among ten selected snapshots was chosen for further optimization on all reactants, intermediates, and products. The QM/MM calculations were performed with an in-house program that links a modified version of Gaussian0912 with a modified version of TINKER.13
All QM/MM optimizations were performed using the iterative method proposed by Zhang et al. using the electrostatic embedding scheme.14 The pseudo-bond approach was used to model the boundary atoms at the QM/MM interface.15 The TSs were optimized using the QST3 method starting with the structure that has the highest energy obtained from the optimized paths calculated with the Quadratic String Method.16 The water coordinated to the iron was replaced by OH– for the OH– pathway without running MD simulations before the QM/MM optimizations. Following the results from our previous simulations, we have employed the ωB97XD17 functional coupled to the 6-31G(d,p) basis set for the QM part. The structures of reactants, intermediates, and products were confirmed to have no imaginary frequency and all transition states (TS) only have one imaginary frequency corresponding to the vibration along the reaction coordinate connecting the two minima for that step.
To understand the interactions between Fe and its surrounding ligands, noncovalent interaction (NCI)18 analysis was performed. NCI analysis plots the reduced density gradient versus the product of the sign of the second eigenvalue (λ2) of the electron-density Hessian matrix and the electron density. In practice, for visualization purposes, a chosen (small) reduced electron gradient is used as the isovalue for the NCI surfaces. The types of the interactions can be distinguished by the sign of λ2 and represented by the different colors. Positive λ2 means repulsion while negative λ2 means attraction. For the color scale (blue/green/red), consistent with the original NCI convention, red represents repulsion, and blue represents attraction. The value of the electron density is represented by the depth of the color. A deeper color means larger electron density, and small electron density is green. Therefore, a red surface indicates relatively strong repulsion; a blue surface represents relatively strong attraction; and a green surface is a sign of relatively weak interaction. This analysis has been proven to be a powerful tool to probe the interactions in small-size molecules19−21 and large-size systems such as enzymes.22 The NCI calculations were performed with the NCIPlot program.23
3. Results and Discussion
In this section, we present the results and discussion for the steps after the formation of FeIV–oxo for the water and hydroxyl pathways. Section 3.1 presents the comparison of the hydrogen abstraction step between the OH– and H2O pathways regarding the energetics and the electronic structure of FeIV–oxo. The differences between these two pathways on the OH rebound step and the unbinding of the DNA base from the metal and the formation of formaldehyde step are discussed in sections 3.2 and 3.3, respectively. Finally, in section 3.4, we present the experimental findings based on the complete energetic picture for these two pathways.
3.1. Hydrogen Abstraction
3.1.1. Energetics of the Critical Structures for the OH– Pathway
Our previous study on the hydrogen abstraction step of the H2O pathway shows there are two substates for each spin state as FeIV–oxo tends to be FeIII–oxyl where one electron is transferred from Fe to oxo. For the total quintet spin state, there are two substates: The first corresponds to HSFeIII–OAF, where s = 5/2 (high spin, HS) Fe is antiferromagnetically (AF) coupled with s = −1/2 O. The second state is ISFeIII–OF where s = 3/2 (intermediate spin, IS) Fe is ferromagnetically coupled with s = 1/2 O. Similarly, for the total triplet spin state, there are also two substates: ISFeIII–OAF where s = 3/2 (intermediate spin, IS) Fe is antiferromagnetically (AF) coupled with s = −1/2 O and LSFeIII–OF where s = 1/2 (low spin, LS) Fe is ferromagnetically coupled with s = 1/2 O (see SI Figure S2 for the electronic configuration diagram).9 Figure 2 depicts the relative energies for the hydrogen abstraction step of the OH– pathway along with the Mülliken spin populations on selected atoms for each of the critical points on the path (see SI Figure S3 for the energy profile for the H2O pathway9).
Figure 2.
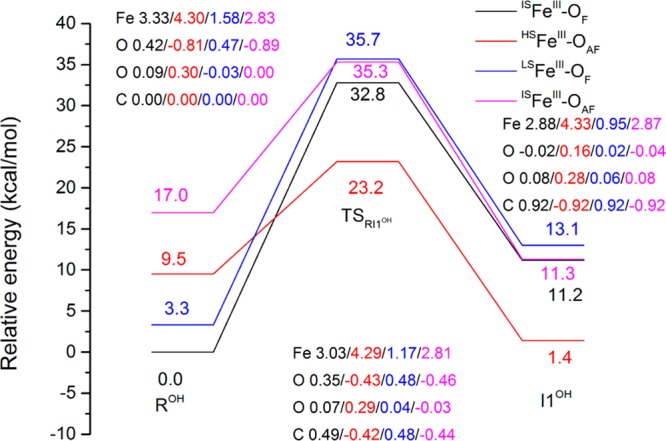
Relative energies (in kcal/mol, with ISFeIII–OF as the reference state) of reactant, TS and I1 and Mülliken spin populations of key atoms (Fe, the first O denotes the oxo, the second O denotes the O of OH– bound to the iron, and C denotes the carbon of methyl group of 1-meA) for the hydrogen abstraction step for OH– pathway in quintet (ISFeIII–OF and HSFeIII–OAF) and triplet states (LSFeIII–OF and ISFeIII–OAF).
Similar to the H2O pathway, the lowest energy state in the hydroxyl pathway for the reactant ROH is the ISFeIII–OF substate. In the same manner, for the transition state (TS) TSRI1OH and intermediate I1OH structures, the HSFeIII–OAF substate has the lowest energy. This indicates an intersystem crossing from ISFeIII–OF ROH to HSFeIII–OAF before the TSRI1OH. As expected for the ISFeIII–OF reactant, the distance between OH– and Fe (1.78 Å) for the OH– pathway is shorter than the distance between H2O and Fe (2.09 Å) for the H2O pathway, and it is a sign of a stronger interaction between OH– and Fe. As mentioned in section 2, we replaced the H2O bound to the iron with OH– without running MD simulations for the OH– pathway. The total electrostatic potential (ESP) fitted charge of the hydroxyl is −0.3466 (sum of the H and O charges) in the optimized reactant, which means part of the negative charge on OH– is delocalized because of its coordination to the iron. It is possible that the OH– may have an impact on its surrounding MM environment due to the change in charge, although this effect might not be significant due to the charge delocalization. We plan on investigating this effect in future studies.
The calculated energy barrier for the OH– pathway (23.2 kcal/mol) is 0.8 kcal/mol higher than the barrier calculated for the H2O pathway (22.4 kcal/mol).9 The slight energy difference indicates that the H2O and OH– pathways are almost equally preferred for the hydrogen abstraction step. The intermediate I1OH is 1.4 kcal/mol higher than the reactant ROH for the OH– pathway, compared to 3.7 kcal/mol lower than R for the H2O pathway (Figure S3). This result suggests OH– stabilizes the reactant more than the intermediate, whereas in the presence of water the opposite is observed. In the present studies, we have not performed free energy calculations based on the minimum energy paths (MEPs), since the potential energy barrier for the rate limiting step for the OH– pathway is very close to the H2O pathway one. Thus, we expect the associated free energies to show a similar trend as observed before.9 Moreover, in our previous study, it was found that the MM environment has only a minor impact on the free energy barrier for the rate limiting step, and it appears its major role for this step is to maintain the geometry around the Fe and its ligands.9
3.1.2. Evolution of Electronic Structure for the H2O Pathway
Despite the slight difference in the barrier, the electronic structure of the FeIV–oxo moiety exhibits different features for the OH– pathway because of the equatorial OH– ligand. For the H2O pathway, the number of unpaired electrons of the iron and oxo O atoms increases during the elongation of the Fe–O distance (d(Fe–Ooxo)) for both ISFeIII–OF and HSFeIII–OAF (see SI Table S1). For the OH– pathway, the trend is the same for HSFeIII–OAF (see Table 1). However, for ISFeIII–OF, the elongation of d(Fe–Ooxo) causes an increase in the number of unpaired electrons on the iron and a decrease on the oxo, which suggests one electron is transferred back to the oxo from the iron. Note that we modified the d(Fe–Ooxo) by only moving the oxo and fixing all other atoms from HSFeIII–OAF reactant instead of relaxing the whole complex with frozen d(Fe–Ooxo)s for both pathways.
Table 1. Mülliken Spin Population for Fe and O (oxo) in HSFeIII–OAF and ISFeIII–OF states with Different Distances between Fe and O (d(Fe–Ooxo)) (Å) for the OH– Pathwaya.
| d(Fe–Ooxo) | spin population (Fe, HSFeIII–OAF) | spin population (Ooxo, HSFeIII–OAF) | spin population (OOH, HSFeIII–OAF) | spin population (Fe, ISFeIII–OF) | spin population (Ooxo, ISFeIII–OF) | spin population (OOH, HSFeIII–OAF) | E(HSFeIII–OAF)–E(ISFeIII–OF) |
|---|---|---|---|---|---|---|---|
| 1.79 | 3.96 | -0.34 | 0.17 | 4.24 | -0.72 | 0.26 | -0.3 |
| 1.75 | 3.80 | –0.13 | 0.15 | 4.22 | 0.68 | 0.25 | 0.6 |
| 1.70 | 3.60 | 0.10 | 0.13 | 4.16 | –0.63 | 0.30 | 7.0 |
| 1.68 | 3.54 | 0.17 | 0.12 | 4.16 | –0.62 | 0.29 | 9.6 |
| 1.65 | 3.47 | 0.25 | 0.11 | 4.13 | –0.59 | 0.28 | 13.7 |
| 1.63 | 3.43 | 0.39 | 0.12 | 4.10 | –0.55 | 0.27 | 16.5 |
The numbers in italics correspond to the structure where the HSFeIII–OAF and ISFeIII–OF states are close in energy (TOH). Except for this point, all other structures are obtained by only moving O with all other atoms fixed starting from the optimized HSFeIII–OAF reactant.
To understand these differences, we turn to the analysis of the canonical molecular orbitals for both pathways. During the H atom abstraction, an electron will be transferred to the oxo O. Figure 3 shows the α-LUMO (lowest unoccupied molecular orbital) and β-LUMO for the reactant structures for the H2O pathway. For the ISFeIII–OF, the α-LUMO (σ* orbital) only changes slightly during the elongation of the d(Fe–oxo) from the reactant (1.60 Å) to the MECP (1.78 Å). In contrast, the percentage of O in β-LUMO (π* orbital) increases and starts to dominate.
Figure 3.

α-LUMO and β-LUMO (canonical orbitals, isovalue = 0.05 au) of the quintet reactants, MECP and TS structures along the H2O pathway. Carbon atoms are colored in gray, hydrogen in white, nitrogen in blue, oxygen in red, iron in purple, and boundary carbon atoms for pseudo-bond in cyan.
According to previous studies24−37 on the reactivity of FeIV–oxo, the path for an α electron being transferred from the substrate to the α-LUMO has been referred to as the σ channel, and the path for a β electron being transferred from the substrate to the β-LUMO has been proposed as the π channel.24−27 For the σ channel, the substrate approaches the FeIV–oxo in a colinear fashion from the top to maximize the overlap between their orbitals. For the π channel, the substrate is supposed to approach the FeIV–oxo horizontally. However, the Pauli repulsion between them will make the angle close to 120° instead of 90°. The two channels were proposed to arise from the same ISFeIII–OF reactant and form a FeIII–oxyl radical on the way to the TS.24−27
As shown in Figure 3, the HSFeIII–OAF and ISFeIII–OF transition state (TS) structures for the H2O pathway correspond to the TSs for the previously proposed σ and π channels respectively originating from the same ISFeIII–OF reactant.24−27 However, the HSFeIII–OAF state, which may become the ground state at a relatively long d(Fe–Ooxo) was not considered in previous studies as it might be in a relatively high energy level for the complexes. The α-LUMOs of both reactant and MECP in HSFeIII–OAF are mostly comprised of oxygen’s p orbital with little covalency with the iron’s d orbital (see Figure 3). Therefore, the moiety in HSFeIII–OAF can be characterized as an oxyl weakly coupled with Fe(III), which is a good α electron acceptor from the substrate. Moreover, the HSFeIII–OAF is more stable than ISFeIII–OF at a relatively long d(Fe–oxo) when approaching the TS. However, one disadvantage for HSFeIII–OAF is that its α-LUMO is perpendicular to the iron–oxo bond. As a result, similar to the π channel mentioned above, the substrate tends to approach the iron–oxo horizontally to maximize the orbital overlap but causes large Pauli repulsion, this interplay yields a bent Fe–O–H angle. For the model complexes used in previous studies, where the substrate can move freely,24−27 both channels can be accessed from the same state. In these studies, the ISFeIII–OF (called Fe(IV)–oxo in these studies) is the lowest energy state. Thus, the σ channel for ISFeIII–OF, which involves less Pauli repulsion, is a better choice in that case than the HSFeIII–OAF state.
Considering all the factors discussed above, the σ channel starting from the HSFeIII–OAF reactant may compete with the σ channel arising from the ISFeIII–OF reactant to accept the α electron from the substrate in the enzyme environment. This competition is likely due to the HSFeIII–OAF state being stabilized by surrounding ligands, or the substrate being oriented in a non-colinear arrangement to Fe–O due to steric constraints. To access the σ channel, the complex needs to transit from ISFeIII–OF to HSFeIII–OAF. For the H2O pathway, the crossing between these two states happens via a minimum energy crossing point (MECP).9 The angles formed by the Fe–O–H atoms for these two quintet states at the TS are around 130°, and the d(Fe–oxo) for the HSFeIII–OAF (1.80 Å) reactant is close to the one for the lowest TS (HSFeIII–OAF, 1.77 Å) and larger than the value for the ISFeIII–OF reactant (1.60 Å).9 It can be seen from Figure 3 that the LUMOs of HSFeIII–OAF TS resemble those of HSFeIII–OAF MECP and the LUMOs of ISFeIII–OF TS resemble those of ISFeIII–OF MECP. The transition from the ISFeIII–OF reactant to a low-lying state at a relatively long d(Fe–O), the description of which matches the features of HSFeIII–OAF, also occurs and results in the non-colinear Fe–O–C in the HSFeIII–OAF TS for some simple model complexes with free substrates and small-size negative charged ligands such as [FeIV(O)(F)5]3–.38
3.1.3. Evolution of Electronic Structure for the OH– Pathway
Regarding the two quintet reactants for the OH– pathway (see Figure 4 for their LUMOs), the α-LUMO of the ISFeIII–OF reactant contains a large component from 1-meA, and the orbital of Fe–oxo is not in a good orientation for overlapping with the orbitals of the methyl group of 1-meA. In contrast, the α-LUMO of HSFeIII–OAF is still mainly comprised of oxygen’s p orbital and should be more likely to accept an α electron from the substrate. It was expected that the elongation of d(Fe–O) would lead to more electrons being transferred from the oxo to the iron for both substates and complete the state transition via a MECP like the H2O pathway. However, the change of Mülliken spin population in Table 1 shows the elongation of d(Fe–oxo) leads to a β electron being transferred from the iron to the oxo for ISFeIII–OF, and smoothly change into HSFeIII–OAF. In practice, the optimization after the structure (TOH, in Table 1d(Fe–oxo) = 1.79 Å) that has a small energy difference for HSFeIII–OAF (and ISFeIII–OF) gives the result that both states converge to HSFeIII–OAF. In terms of d(Fe–O), the value is 1.62 Å in ISFeIII–OF reactant and 1.88 Å in HSFeIII–OAF reactant and TS, which suggests that the HSFeIII–OAF TS resembles the HSFeIII–OAF reactant.
Figure 4.

α-LUMO and β-LUMO (canonical orbitals, isovalue for the surface is 0.05 au) of the quintet reactants, TOH (Table 1) and TS structures along the OH– pathway. Carbon atoms are colored in gray, hydrogen in white, nitrogen in blue, oxygen in red, iron in purple, and boundary carbon atoms for pseudo-bond in cyan.
Figure 4 shows the LUMOs (α and β) of the HSFeIII–OAF TS also resemble the respective LUMOs of the HSFeIII–OAF reactant. In addition, different from the ISFeIII–OF reactant where its β-LUMO is mainly comprised of the orbitals for the substrate, the β-LUMO in ISFeIII–OF TOH is mainly a Fe–O π* orbital, which is more similar to the β-LUMO of the ISFeIII–OF TS. These results underscore the role of the iron–oxo(oxyl) distance, d(Fe–Ooxo), in tuning the reactivity of the FeIV–oxo moiety. In order to obtain the precursor for the ISFeIII–OF TS, the OH– also needs to be considered to investigate the electronic structure change from the ISFeIII–OF reactant to the ISFeIII–OF TS. Since this pathway is on a higher energy level than the MEP (minimum energy pathway), it will be not discussed here. When studying the hydrogen abstraction by FeIV–oxo, the HSFeIII–OAF state should be paid attention, as it may become the ground state on the way to the TS, especially for equatorial electron donating ligands and/or enzymes with constrained substrates.
3.2. OH Rebound
Figure 5 shows the relative energy for this step for both pathways. The lowest energy states for I1 and I1OH are both HSFeIII–OAF, and the two substates for the quintet and triplet merge into one after the OH rebound step at I2 and I2OH. Therefore, only the quintet state is considered for the calculations of this and subsequent steps. The next step for the H2O pathway is a typical OH rebound process. The calculated barrier, TSI1I2, is 12.2 kcal/mol, which is in agreement with a previous QM/MM study.6 For the OH– pathway, the corresponding intermediate I3OH is not stable. During the optimization, the proton is transferred to the iron-bound OH– spontaneously, and the zwitterion structure I2OH forms.
Figure 5.

Relative energies (kcal/mol) of the structures along the minimum energy path (MEP) for the OH rebound step in the quintet state for H2O pathway (a) and OH– pathway (b). The numbers in the parentheses are reaction barriers, which are the energy differences between intermediates and their corresponding TSs. The energy of the corresponding ISFeIII–OF reactant (Figure 2) is taken as zero for each pathway. Carbon atoms are colored in gray, hydrogen in white, nitrogen in blue, oxygen in red, iron in purple, and boundary carbon atoms for pseudo-bond in cyan.
Two possible pathways from I1OH to I2OH can be proposed. One is a concerted pathway in which the OH rebound process is coupled to the proton transfer via TSI1I2OH. The other is a stepwise pathway where the proton is first transferred to form an intermediate I4OH, followed by an oxygen transfer. The calculated TSI1I2OH for the concerted pathway is 11.9 kcal/mol. For the stepwise pathway, the energy for the TSI1I4OH structure between I1OH and I4OH is 24.7 kcal/mol, and the subsequent oxygen transfer is a barrierless downhill process. Based on these barriers, the concerted pathway is favored over the stepwise pathway. Regarding the zwitterion structure I2OH, the distance between Fe and O of −CH2O– (on the adenine base) is short (2.05 Å). As shown in Figure 6, among the ligands coordinated to the iron, the NCI surface between the −CH2O– moiety and the Fe atom has the deepest blue color. This means that this group has the strongest attraction to the Fe. The NCI result suggests that the zwitterion structure also exhibits the characteristic Fe–O–C bond proposed for the TauD mechanism from time-resolved RAMAN studies.4 Similarly, one may expect a concerted pathway to be preferred over a stepwise pathway as proposed for TauD from the radical intermediate to the Fe–O–C intermediate.4
Figure 6.
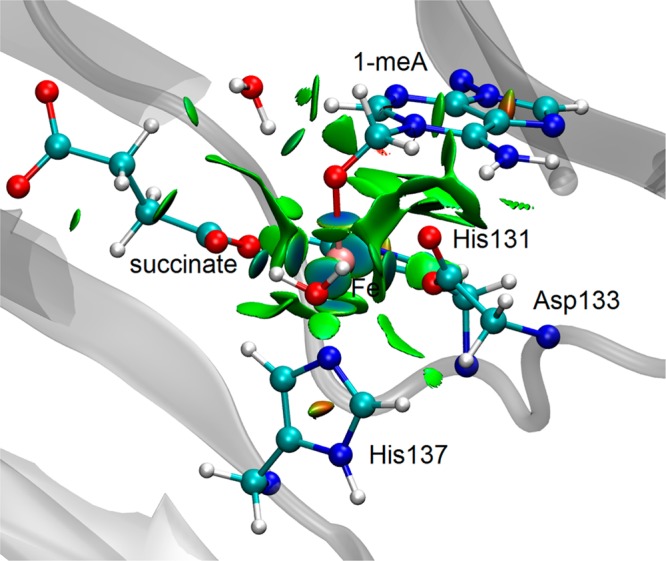
NCI surface (isovalue 0.5 au and a color scale −0.1 < sign(λ2)ρ < 0.1 au) of the zwitterion structure (I2OH) for the OH– pathway.
The lowest barrier for the OH– pathway (TSI1I2OH, 12.0 kcal/mol) is close to the one for the H2O pathway (TSI1I2, 12.2 kcal/mol). The minor difference indicates these pathways are also equally favored for this step. For both pathways, the rebound step leads to an intermediate with much lower energy with respect to the FeIV–oxo structure. To determine whether a zwitterion structure denoted as I5 also exists for the H2O pathway, we carried out the optimization starting from a structure in which the proton is transferred from −CH3OH to Asp133. During the optimization, the proton is spontaneously transferred back to −CH3O–, which suggests that the pKa of the iron-bound Asp133H is larger than the iron-bound −CH3OH when a hydroxyl is coordinated to the Fe atom.
In order to investigate the effect from the positive charge on the nitrogen (N1) of 1-meA, we changed 1-meA to 1-deazameA by replacing N1 with C (see Figure 7a) in the optimized zwitterion I2OH (bound to the iron) and I5OH (unbound from the iron) structures and carried out geometry optimizations. A stable structure for the I2OH analog (Figure 7b) for 1-deazameA was obtained on the PES. Conversely, in the case of the I5OH analog for 1-deazameA, the −CH2O– group abstracts a proton spontaneously from the neighboring water that is coordinated to the iron (Figure 7c). This finding is consistent with previous results for 3-deazameC.3 Additionally, different from 1-meA, we are able to obtain the I3OH analog (Figure 7d) for 1-deazmaeA, although its energy is 8.0 kcal/mol higher than for the I2OH analog. These optimized structures indicate that the proton accepting ability of iron-bound −CH2O– group in 1-deazameA is weaker than when this group is not bound to the iron because of the stabilization effects from the metal. The pKa of iron-unbound −CH2OH in 1-deazameA is larger than the value of H2O coordinated to the iron, and hence larger than 1meA. Since the proton transfer is a necessary step for the repair process and might become the rate-limiting step as discussed in the next section, the pKa difference of −CH2OH may partly explain why 1-meA and 3-meC, which bear a positive charge, are preferred by AlkB over 3-meT and 1-meG, which are charge neutral alkylated DNA bases.3,8
Figure 7.

(a) Chemical structures of 1-meA and 1-deazameA. (b, c, d) 1-DeazameA-related intermediates. Carbon atoms are colored in gray, hydrogen in white, nitrogen in blue, oxygen in red, iron in purple, and boundary carbon atoms for pseudo-bond in cyan.
3.3. Unbinding of the Methoxide Moiety from the Iron Center, Proton Transfer, and the Formation of Formaldehyde
To study whether the unbinding of the methoxide moiety from the metal (Figure 8; from I2 to I3 for the H2O pathway; from I2OH to I5OH for the OH– pathway) is a necessary step, we investigated the formation of the formaldehyde with −CH3OH (for the H2O pathway) or −CH3O– (for the OH– pathway) being coordinated or unbound to the iron. In the case of the H2O pathway with iron-coordinated −CH3OH, the reaction happens in a concerted manner, where the proton transfer to Asp133 and the bond breaking between the C and N1 of 1-meA (C–N bond breaking, I2–P pathway in Scheme 2) leads to the product with the formaldehyde bound to the iron (P). The calculated barrier for this step is 25.9 kcal/mol, which is higher than the hydrogen abstraction step. This result suggests that this step may become the rate-determining step under certain circumstances, such as if no better proton acceptors are available.
Figure 8.

Relative energies (in kcal/mol) for the structures along the minimum energy path (MEP) for the detachment of the DNA base from Fe and the formation of formaldehyde in the quintet state for the H2O pathway (a) and OH– pathway (b). The numbers in the parentheses are reaction barriers, which are the energy differences between intermediates and their corresponding TSs. The ISFeIII–OF reactant (Figure 2) is taken as the reference for each pathway. Carbon atoms are colored in gray, hydrogen in white, nitrogen in blue, oxygen in red, iron in purple, and boundary carbon atoms for pseudo-bond in cyan.
When the −CH3OH moiety is leaving the iron, I3 forms, and its energy is 5.2 kcal/mol higher than the iron-coordinated intermediate I2. The barrier for the unbinding process is 14.3 kcal/mol. The next step is the proton transfer and the C–N bond breaking. If these two processes are stepwise, a zwitterion intermediate (I4) that is unbound to the iron will form. The proton can then be transferred to Asp133 or some other neighboring residue, such as Glu136, in a direct-transfer pattern or via a water bridge. However, in the case of Asp133, the proposed zwitterion structure cannot be obtained, which is partly due to its weaker ability to accept a proton than for iron-unbound Asp133. In other words, the proton transfer to Asp133 has to be coupled with the C–N bonding and form the final product with the formaldehyde unbound to the iron (Pun). The barrier (TSI3P) for this coupled process is 20.6 kcal/mol, and its relative energy is close to the unbound TSI2P.
It is also possible that the proton is transferred to a neighboring residue instead of being transferred to Asp133. As shown in Figure 1, the nearest residue to the active site is Glu136, but its distance from the 1-meA suggests the proton transfer would likely have to occur via a water bridge. Before the proton transfer, the structure rearranges from I3Glu136 to I3′Glu136. (Figure 9). In I3Glu136, the −CH3OH moiety forms a hydrogen bond with Asp133 while −CH3OH forms a hydrogen bond with the bridging water in I3′Glu136. To check the existence of a zwitterion structure, we carried out the optimization of I4Glu136 assuming the proton transfer from −CH3OH to Glu136 with a water molecule as the bridge. However, during the optimization, the structure changes back to I3′Glu136 with the proton being spontaneously transferred back. This indicates that the pKa of −CH3OH is also larger than Glu136H. In other words, if the proton is transferred to Glu136, it has to be coupled with the C–N bond breaking. The calculated barrier for TSI3′Pun-Glu136 for the concerted pathway from I3′Glu136 to PGlu136 is 7.4 kcal/mol, which is much lower than the barrier for the proton being transferred to Asp133. This suggests that Glu136 may be the final proton acceptor when no better acceptor available. The proton acceptor role of Glu136 may partly account for the decreased activity of AlkB in repairing 1-meA when Glu136 is mutated to a leucine.39
Figure 9.
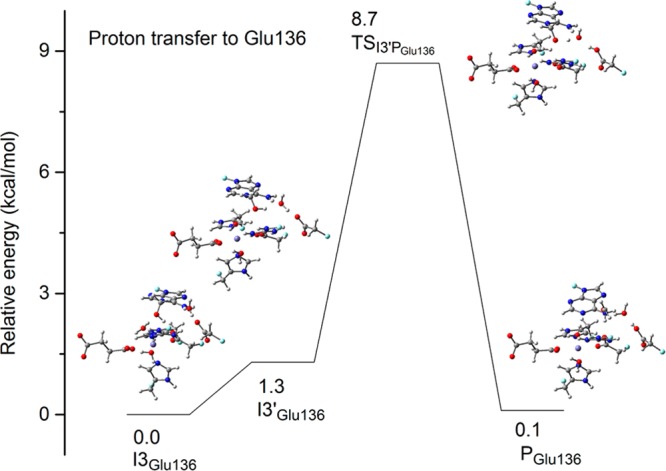
Relative energies (kcal/mol) for the structures along the minimum energy path (MEP) for the proton transferred to Glu136 in the quintet state for the H2O pathway. (Arg210, Glu136, and a bridging water were added to the QM subsystem for these structures). Carbon atoms are colored in gray, hydrogen in white, nitrogen in blue, oxygen in red, iron in purple, and boundary carbon atoms for pseudo-bond in cyan.
The structure of the TSI3′PGlu136 shows a proton is first transferred from H2O to Glu136, followed by the resulting OH– accepting the proton from −CH3OH. It is worth noting that Asp135 could be another possible proton acceptor as well. However, as its relative position to the DNA base in the crystal structure (Figure 1) is not conducive for the proton transfer, the proton transfer process may happen when the DNA base is leaving the active site. If a zwitterion structure (I4) indeed forms, the proton from −CH3OH could be transferred to a hydroxyl molecule in the solvent. In summary, for the H2O pathway, after the hydrogen abstraction and the OH rebound step, the hydroxyl product first unbinds from the iron and loses a proton to Glu136 or Asp135 or solvent with concerted the C–N bond breaking.
For the OH– pathway, as the proton has already been transferred to the iron-bound OH– in the previous rebound step, the final step is only the C–N bond breaking with −CH3O– bound or unbound to the iron. The calculated barrier when the methoxide is bound to the iron (TSI2POH) is 5.9 kcal/mol, compared to the unbound structure (TSI2I5OH) that results in a barrier of 17.3 kcal/mol. This last barrier is 3.0 kcal/mol higher than the barrier for the unbinding process for −CH3OH, which may be due to a stronger attraction between the iron and O of −CH3O– than −CH3OH and the repulsion between the negatively charged O of −CH3O– and one O of Asp133 during the unbinding process. The iron-unbound zwitterion intermediate I5OH is only 0.6 kcal/mol higher than the iron-bound one (I2OH). The barrier for I5OH being finally dissociated into PunOH (TSI5POH) is 1.9 kcal/mol. The imaginary frequency vibrational mode corresponds to the rotation of the formed formaldehyde, which suggests the energy for C–N bond breaking should be lower than 1.9 kcal/mol. Therefore, for the OH– pathway, after the hydrogen abstraction and the OH rebound coupled with a proton transfer to the OH–, the formed zwitterion structure prefers the C–N bond breaking directly over unbinding from the metal center first.
3.4. Comparison between the H2O and OH– Pathways
For both the H2O and OH– MEPs (see SI Figures S4 and S5 for the complete energy profile for these two pathways respectively), the rate-limiting step is the hydrogen abstraction step, and their barriers for this step are close to each other. Once the rate limiting step has been achieved, the mechanisms differ significantly. For the H2O pathway, the last step with the lowest barrier is a proton transfer to Glu136 via a water bridge, and the hydroxyl product has to unbind from the iron center first. Since the water molecule that acts as the bridge and Glu136 can move freely, they may not always be in a perfect arrangement for the proton transfer. In that case, Asp133 may become the best choice. As the barriers for a proton being transferred to Asp133 coupled with the C–N bond breaking are higher than the hydrogen abstraction step, this step may become the rate-limiting step for the H2O pathway. In contrast, the barrier for the last step for the OH– pathway, the C–N bond breaking leading to the formation of formaldehyde is much lower. Taken together, our results suggest that the OH– pathway should be preferred over the H2O pathway, which may partly account for the basic optimal pH for the 1-meA repair catalyzed by AlkB.
The crystal structure for 3-meC (3OIS, SI Figure S1) is proposed to be a zwitterion structure similar to I4 where the base is unbound to the metal based on QM/MM calculations.3 However, those QM/MM calculations cannot rule out the possibility of an alcohol structure. According to our results on 1-meA above, the zwitterion structure generated by just following the OH– pathway is more likely to dissociate with the methoxide moiety bound to the metal, rather than unbinding from the metal. In addition, if the captured crystal structure for 3-meC (3OIS) is indeed a zwitterion, the proton from the alcohol structure has to be transferred to the solvent instead of to Asp133 or Glu136. As a result, another possible pathway may be proposed where the hydrogen abstraction, OH rebound, and unbinding from the iron follow the H2O pathway, and then, the proton is transferred to OH– to form a zwitterion structure. Finally, the zwitterion would dissociate into the repaired DNA base and formaldehyde.
4. Conclusions
In this work, new pathways for the second part of the reaction mechanism, starting from the rate-limiting H atom abstraction, for the dealkylation of 1-meA catalyzed by AlkB have been proposed and investigated by QM/MM simulations based on recent experimental findings. For the hydrogen abstraction and the OH rebound step, the H2O and OH– pathways have close barriers and therefore are equally preferred. For the hydrogen abstraction step, different from most of the previous studies on model systems, the HSFeIII–OAF state, where the iron (s = 5/2) is antiferromagetically coupled with a oxyl (s = −1/2) becomes the ground state when the Fe–oxo distance, d(Fe–Ooxo), is long and similar to the HSFeIII–OAF TS. This finding highlights the electronic structure change of the FeIV–oxo moiety with the binding of the equatorial OH– under the enzymatic environment. Regarding the OH rebound step, a hydroxyl structure forms for the H2O pathway. In contrast, for the OH– pathway, this step is coupled with the proton transfer from −CH3OH to the OH– bound to the iron and forms a zwitterion structure bound to the iron, which can be also characterized as an Fe–O–C complex, which is consistent with experimental findings. Following the OH rebound step, the C–N bond between −CH3O– and the DNA base can easily break to form the final product while the DNA base is bound to Fe. In contrast, for the H2O pathway, the hydroxyl complex needs to unbind from the iron center first and then transfer a proton to the neighboring residue Glu136 via a water bridge or Asp135 or lose it to the solvent. The proton transfer to Glu136 is coupled to the C–N bond breaking. The larger pKa value of −CH3OH in the hydroxyl intermediate for neutral DNA bases may account for its lower repair efficiency compared to positively charged DNA bases. The lower energy barrier for the last step in the OH– pathway compared to that of the H2O pathway when the proton has to be transferred to Asp133 may partly explain the basic optimal pH for the repair of 1-meA by AlkB. Comparison of the energetics for the OH rebound step of 1-meA and 1-deazameA show that the positive charge on the 1N of 1meA is necessary to reduce the barrier, and may help explain AlkB’s substrate preference.
Acknowledgments
This research was supported by Wayne State University. Computing time from Wayne State C&IT is gratefully acknowledged. D.F. also thanks Wayne State University Graduate School for a Rumble Fellowship. This work was partly supported by the National Institutes of Health GM108583-A1 to G.A.C.
Supporting Information Available
Tables and figures as well as Cartesian coordinates of QM part of the structures discussed in this paper. The information is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zheng G.; Fu Y.; He C. Chem. Rev. 2014, 114, 4602–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausinger R. P. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21–68. [DOI] [PubMed] [Google Scholar]
- Yi C.; Jia G.; Hou G.; Dai Q.; Zhang W.; Zheng G.; Jian X.; Yang C.-G.; Cui Q.; He C. Nature 2010, 468, 330–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzyska P. K.; Appelman E. H.; Hausinger R. P.; Proshlyakov D. A. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B.; Edstrom W. C.; Benach J.; Hamuro Y.; Weber P. C.; Gibney B. R.; Hunt J. F. Nature 2006, 439, 879–884. [DOI] [PubMed] [Google Scholar]
- Quesne M. G.; Latifi R.; Gonzalez-Ovalle L. E.; Kumar D.; de Visser S. P. Chem.—Eur. J. 2014, 20, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Llano J.; Gauld J. W. J. Phys. Chem. B 2009, 113, 4887–4898. [DOI] [PubMed] [Google Scholar]
- Mishina Y.; He C. J. Inorg. Biochem. 2006, 100, 670–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D.; Lord R. L.; Cisneros G. A. J. Phys. Chem. B 2013, 117, 6410–6420. [DOI] [PubMed] [Google Scholar]
- Cisneros G. A.; Karttunen M.; Ren P.; Sagui C. Chem. Rev. 2013, 114, 779–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D.; Darden T. A.; Cheatham T. E.; Simmerling C.; Wang J.; Duke R.; Luo R.; Crowley M.; Walker R.; Zhang W.; Merz K. M.; Wang B.; Hayik S.; Roitberg A.; Seabra G.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Wu X.; Brozell S.; Steinbrecher T.; Gohlke H.; Yang L.; Tan C.; Mongan J.; Hornak V.; Cui G.; Mathews D. H.; Seetin M. G.; Sagui C.; Babin V.; Kollman P.. AMBER 11; University of California: San Francisco, 2010. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Ponder J. W.TINKER-Software Tools for Molecular Design; Washington University: Saint Louis, MO, 2008. [Google Scholar]
- Zhang Y.; Liu H.; Yang W. J. Chem. Phys. 2000, 112, 3483–3492. [Google Scholar]
- Parks J. M.; Hu H.; Cohen A. J.; Yang W. J. Chem. Phys. 2008, 129, 154106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger S. K.; Yang W. J. Chem. Phys. 2006, 124, 054109. [DOI] [PubMed] [Google Scholar]
- Chai J.-D.; Head-Gordon M. J. Chem. Phys. 2008, 128, 084106. [DOI] [PubMed] [Google Scholar]
- Johnson E. R.; Keinan S.; Mori-Sánchez P.; Contreras-García J.; Cohen A. J.; Yang W. J. Am. Chem. Soc. 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D.; Piquemal J.-P.; Liu S.; Cisneros G. A. Theor. Chem. Acc. 2014, 133, 1484. [Google Scholar]
- Gillet N.; Chaudret R.; Contreras-García J.; Yang W.; Silvi B.; Piquemal J.-P. J. Chem. Theory Comput. 2012, 8, 3993–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellow J. A.; Fang D.; Kovacevic N.; Martin P. D.; Shearer J.; Cisneros G. A.; Groysman S. Chem.—Eur. J. 2013, 19, 12225–12228. [DOI] [PubMed] [Google Scholar]
- Fang D.; Chaudret R.; Piquemal J.-P.; Cisneros G. A. J. Chem. Theory Comput. 2013, 9, 2156–2160. [DOI] [PubMed] [Google Scholar]
- Contreras-García J.; Johnson E. R.; Keinan S.; Chaudret R.; Piquemal J.-P.; Beratan D. N.; Yang W. J. Chem. Theory Comput. 2011, 7, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik S.; Chen H.; Janardanan D. Nat. Chem. 2011, 3, 19–27. [DOI] [PubMed] [Google Scholar]
- Neidig M. L.; Decker A.; Choroba O. W.; Huang F.; Kavana M.; Moran G. R.; Spencer J. B.; Solomon E. I. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 12966–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C.; Ye S.; Neese F. Angew. Chem. 2010, 122, 5853–5856. [DOI] [PubMed] [Google Scholar]
- Ye S.; Neese F. Proc. Natl. Acad. Sci. U.S.A. 2011, 1228–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao H.; Kumar D.; Thiel W.; Shaik S. J. Am. Chem. Soc. 2005, 127, 13007–13018. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Hirao H.; Que L.; Shaik S. J. Am. Chem. Soc. 2005, 127, 8026–8027. [DOI] [PubMed] [Google Scholar]
- Hirao H.; Kumar D.; Que L.; Shaik S. J. Am. Chem. Soc. 2006, 128, 8590–8606. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Hirao H.; Kumar D. Acc. Chem. Res. 2007, 40, 532–542. [DOI] [PubMed] [Google Scholar]
- Janardanan D.; Wang Y.; Schyman P.; Que L.; Shaik S. Angew. Chem., Int. Ed. 2010, 49, 3342–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser S. P. J. Am. Chem. Soc. 2006, 128, 9813–9824. [DOI] [PubMed] [Google Scholar]
- Johansson A. J.; Blomberg M. R. A.; Siegbahn P. E. M. J. Phys. Chem. C 2007, 111, 12397–12406. [Google Scholar]
- Bernasconi L.; Louwerse M. J.; Baerends E. J. Eur. J. Inorg. Chem. 2007, 2007, 3023–3033. [Google Scholar]
- Wang Y.; Han K. J. Biol. Inorg. Chem. 2010, 15, 351–359. [DOI] [PubMed] [Google Scholar]
- Decker A.; Rohde J.-U.; Klinker E. J.; Wong S. D.; Que L.; Solomon E. I. J. Am. Chem. Soc. 2007, 129, 15983–15996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Geng C.; Huo R.; Ryde U.; Bu Y.; Li J. J. Phys. Chem. B 2014, 118, 1493–1500. [DOI] [PubMed] [Google Scholar]
- Zhu C.; Yi C. Angew. Chem., Int. Ed. 2014, 53, 3659–3662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.