

Abstract
Advances in light microscopy have enabled the visualization of DNA in the interphase nucleus with more detail than is visible with conventional light microscopy. The nuclear architecture is assumed to be different in cancer cells compared to normal cells. In this paper we have studied, for the first time, the organization of nuclear DNA and that of DNA-free space in control lymphocytes, Hodgkin cells and Reed–Sternberg cells using 3D structured illumination microscopy (SIM). We have observed detail in these SIM images that was not observed in conventional widefield images. We have measured the size distribution of the DNA structure using granulometry and noted a significant, progressive increase in the amount of sub-micron structures from control lymphocytes to Hodgkin cells to Reed–Sternberg cells. The DNA-free space changes as well; “holes” in the DNA distribution start to appear in the malignant cells. We have studied whether these “holes” are nucleoli by staining for upstream binding factor (UBF), a protein associated with the nucleolus. We have found that the relative UBF content progressively and significantly decreases—or is absent—in the DNA-free space when measured as either the Pearson correlation coefficient with the DNA-free space or as the number of “holes” that contain UBF. Similar differences exist within the population of Reed–Sternberg cells between binucleated and multinucleated cells with four or more subnuclei. To our knowledge, this is the first study that investigates the changes of the nuclear DNA structure in any disease with superresolution light microscopy. J. Cell. Biochem. 115: 1441–1448, 2014. © 2014 The Authors. Journal of Cellular Biochemistry published by Wiley Periodicals, Inc. This is an open access article under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs License, which permits use and distribution in any medium, provided the original work is properly cited, the use is non-commercial and no modifications or adaptations are made.
Keywords: STRUCTURED ILLUMINATION MICROSCOPY, NUCLEAR ARCHITECTURE, QUANTITATIVE MICROSCOPY, HODGKIN’S LYMPHOMA
The nuclear architecture and its cancer-related changes have been studied since Boveri first postulated that the nuclear architecture differs between normal and cancer cells [Boveri, 1914, 2008]. Over the course of the last century the structure of DNA has been unraveled at various length scales. The structure by itself does not, however, reveal its spatial organization within the nucleus. Many current models about the nuclear architecture are studied in animals and human cell lines. For clinical applications such models also need to be validated in primary human tumor cells.
The existence of individual chromosomes in dividing nuclei was first observed in mitotic cells [Flemming, 1882]. Chromosomes occupy distinct regions in the interphase nucleus, designated as chromosome territories (CTs) [Cremer and Cremer, 2006a,b]. The position of each human CT inside the nucleus is determined by its size and gene density [Tanabe et al., 2002]. As the spatial distribution of DNA is non-random, it is important to assess the spatial DNA structure. This would include measurements at length scales larger than the typical sizes of the quaternary nucleic acid structure.
Microscopic analyses of the DNA structure in cell nuclei have been performed since the wide-scale availability of digital image processing. Automatic estimation of the number of low- and high-density DNA regions within a white blood cell has been performed since the 1980s [Bins et al., 1981]. Several additional features, including the granularity of the spatial DNA distribution, were also measured during that time [Young et al., 1986]. It has been noted that chromatin is structurally organized on various length scales that can be made visible using light microscopy [Einstein et al., 1998]. Differences in the microscopic DNA structure have been described using various names, including chromatin condensation, chromatin structure, and chromosome packaging, in a variety of diseases, including cancer [Hannen et al., 1998; Vergani et al., 1999; Natarajan et al., 2012].
3D structured illumination microscopy (SIM) is a superresolution imaging modality that has only recently found its way to the biology laboratory. This methodology offers a higher image resolution than conventional epifluorescence widefield microscopy through the use of heterodyne detection of a fluorescent sample illuminated by a periodic pattern [Heintzmann and Cremer, 1999; Cragg and So, 2000; Frohn et al., 2000; Gustafsson, 2000]. It has been shown that 3D-SIM images of DNA, stained with DAPI, reveals structural information that had not been seen with conventional microscopy methods [Schermelleh et al., 2008]. Investigation of the nuclear architecture using fluorescent in situ hybridization (FISH) showed that, during FISH experiments, key characteristics of the ultrastructure are preserved [Markaki et al., 2012]. This suggests that the nuclear architecture, as observed by 3D-SIM, remains stable for different sample preparation techniques.
The DNA structure inside the interphase nucleus can be visualized with 3D-SIM at microscopic length scales. Visual inspection of 3D-SIM images of different cell types shows qualitative differences in the DNA structure between cell types. In order to measure these differences objectively, a method to explore and quantify the structure is needed. In particular, the granularity of the DAPI-stained DNA structure can then be assessed using 3D-SIM.
We choose to study the DNA structure in Hodgkin’s lymphoma (HL) cells, because of the unique nature of this form of lymphoid cancer. Malignant cells in HL are mononucleated Hodgkin cells (H) and bi- or multinucleated Reed–Sternberg cells (RS). The RS cell is the diagnostic cell for this malignancy. A variety of cellular functions are affected in these cells in comparison to the normal B lymphocytes from which they originate [Kuppers et al., 2012]. A multitude of translocations have been identified in RS cells [MacLeod et al., 2000] and their nuclear architecture becomes progressively more disorganized as the number of subnuclei increases [Knecht et al., 2009; Guffei et al., 2010]. This includes an increase in the number of centrosomes and aberrant multi-polar mitotic spindles [Martin-Subero et al., 2003; Knecht et al., 2009]. The nuclear architecture of and the difference between H and RS cells has also been linked to the clinical outcome of the disease [Knecht et al., 2012].
In this article, we first aimed to quantitatively describe the size distribution and assess the differences of the DNA structure and the DNA-free space(s) in lymphocytes, H cells, and RS cells. When we refer to DNA size in this paper, we refer to the physical “cluster” sizes visible in the 3D-SIM images, rather than the number of base pairs of these DNA clusters. When we refer to density, we discuss the relative local intensity in images and not the absolute concentration of DNA. Second, we have investigated the spatial relation between the nucleolus-related protein UBF and the DNA-free space, which is part of the complement (or negative) of the DNA structure. We have found a significant and progressive difference in DNA structure and DNA-free space between normal, Hodgkin, and Reed–Sternberg cells.
MATERIALS AND METHODS
CELL PREPARATION
Normal blood was treated with Ficoll (GE Healthcare, Uppsala, Sweden) to obtain the control lymphocytes. The removed buffy coat was washed in a PBS solution and the cell pellet collected. The cells were subsequently placed onto slides. The HDLM-2 cell line [Drexler et al., 1986] was grown in RPMI-1640 medium, supplemented with 20% fetal bovine serum (FBS), 1% l-glutamine, and 1% penicillin–streptomycin (reagents from Invitrogen/Gibco, Burlington, ON). Cells were incubated at 37°C with 5% CO2 in a humidified atmosphere. After 2 days, 1–2 ml of fresh media was added. The following day, half of the cells were split into a fresh plate; the other half were used to prepare slides. The slides, both the control and HDLM-2 slides, were incubated in 3.7% formaldehyde (Sigma-Aldrich, Oakville, ON) for 10 min. The slides were dehydrated using a standard ethanol series, air dried and stored at −20°C until needed. The slides were later rehydrated using a reverse ethanol series and permeabilized with 0.2% Triton X-100. Primary UBF anti-body (H-300, sc-9131, Santa Cruz, Dallas) was used at a concentration of 1:60 and incubated for 45 min at room temperature (RT). Slides were washed in 1× PBS/50 mM MgCl2 and UBF was visualized with goat-anti-rabbit Alexa 488 (Molecular Probes, Eugene, OR) at a concentration of 1:500 and incubated for 30 min at RT. Slides were then washed in 1× PBS/50 mM MgCl2 and 50 μl of 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole) was added and incubated for 5 min. Excess DAPI was drained, 1 drop of Vectashield (Vector Labs, Burlingame, CA) was added to the slide and a coverslip (No. 1½, Schott, Mainz, Germany) was placed and sealed with nail polish. Slides were stored at 4°C until imaging.
MICROSCOPY
The cells were recorded with a Zeiss Elyra PS1 SIM equipped with a Plan-Apochromat 63×/1.40 Oil immersion objective using an Andor EM-CCD iXon 885 camera and a 1.6× tube lens at room temperature. The DAPI channel was obtained with 405 nm laser excitation, 23 μm diffraction grating and filter cube SR Cube 07; the UBF channel with 488 nm laser excitation, 28 μm diffraction grating and filter cube SR cube 11. The lateral pixel size, Δx and Δy, was 79 nm in the recorded images and 40 nm in the reconstructed image, the step between z-planes, Δz, was 91 nm. The 3D-SIM and widefield images were reconstructed with ZEN 2012 black edition (Carl Zeiss, Jena, Germany) with the standard settings except for the regularization parameter, which was set to 10−3, and clipping, which was turned off. The regularization parameter was empirically determined based on visual inspection of image quality. Clipping the image in the reconstruction stage artificially sets the background to zero (black), but hides actual image information and was, therefore, not done.
IMAGE ANALYSIS
The image processing and measurement steps were performed in Matlab (MathWorks, Natick, MA) with the toolbox DIPimage [Luengo Hendriks et al., 1999]. A central z-plane was manually selected for processing (see Supplement for justification). The cell was automatically detected by isodata thresholding [Ridler and Calvard, 1978] the widefield DAPI image and filling the holes in the binary image. The grayscale DAPI images were error-function clipped between the 10th and 90th percentile of the intensity over the detected cell [Verbeek and van Vliet, 1993]. The granulometry of the DNA structure and DNA-free space was subsequently measured with a morphological sieve applied to the error-function clipped images [Luengo Hendriks et al., 2007]. The coefficient of variation (the standard deviation divided by the mean) and skewness of the intensity histogram over the detected region was computed as well. To assess the significance of the measured difference, we used two-sided, two-sample Kolmogorov–Smirnov (KS) tests, in which the sample size was determined by the relative area over the median structure size (see Supplement for details). A linear classification line based on the Fisher linear discriminant assuming equal priors was performed for the histogram features using the PRTools toolbox for Matlab [Duin et al., 2007]. The significance of differences in the classification error was assessed with the McNemar test. For each apparent hole in the SIM DAPI image (Fig. 1) it was determined by visual inspection whether or not it was filled with UBF. Pearson’s correlation coefficient, R, was calculated over the nucleus between the DNA-free space, the negative of the DNA image, and both the original unclipped SIM and widefield UBF images. The significance of the differences between the UBF-based measures was assessed with the two-sample Student t-test with unequal variances.
Figure 1.
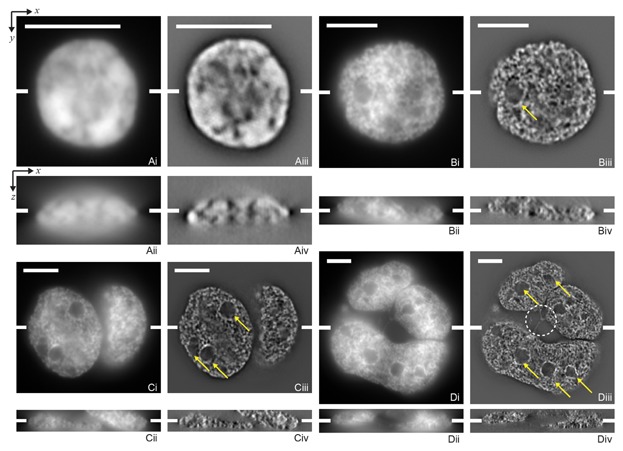
Slices from DAPI stained cell nuclei of a control lymphocyte (A), a mononucleated Hodgkin cell (B), a binucleated Reed–Sternberg cell (C), and a multinucleated Reed–Sternberg cell (D). (See Fig. S1 for tri- and tetranucleated cells.) Widefield images are shown in images (i,ii) and unclipped SIM images in (iii,iv). The top row (i,iii) shows a lateral (x,y)-slice and the bottom row (ii,iv) an axial (x,z)-slice. The scale bars are 5 μm in each lateral slice—note the increasing image size from A to D—the tick marks in the middle of each panel indicate the positions of the corresponding orthogonal planes. The arrows denote “holes” in the DNA distribution of these cells. Note that not all apparent holes are indicated to maintain clarity. The multinucleated cell (D) contains a tri-directional anaphase bridge between several of the subnuclei (dashed circle in Diii). The intensities of all images are linearly stretched between their minimum and maximum value. Note that the DNA structure becomes visible by comparing the SIM images to the widefield images. Also note the difference in the structure between the different images.
RESULTS
DNA STRUCTURE AND STRUCTURE OF DNA-FREE SPACE
In order to investigate the DNA structure, lymphocytes were isolated and imaged using 3D-SIM, see Figure 1A for the recorded DAPI channel of a typical lymphocyte (L). The widefield image does not reveal details of the DNA distribution within the nucleus. Structure in the DNA distribution within the nucleus becomes visible in the 3D-SIM image, although it is still mostly a relative uniform distribution for normal lymphocytes, with some intensity variation in the middle of the nucleus.
To assess the DNA distribution within the nucleus and to determine whether there are differences between normal and cancer cells, we studied Hodgkin lymphoma (HL) cells, where mono-nucleated Hodgkin (H) cells give rise to bi- or multinucleated Reed Sternberg (RS) cells. The nuclear organization in RS cells becomes progressively more disturbed with increasing multinuclearity [Guffei et al., 2010]. We recorded both H and RS cells from the HL cell line HDLM-2 for which we display several representative images in Figures 1 and S1. The full z-stacks for these cells are shown in Movies 1–6. Although some structure variations are visible in the widefield images, all 3D-SIM images reveal more of the internal DNA structure than the respective widefield images of the same cells. Several qualitative observations can be made. The DNA structure inside the nuclei shows some granularity, that is, it is non-constant and shows structure at smaller length scales than the open spaces. There are “holes” in the DAPI stained nuclei. These “holes” are areas within the nucleus that have a low DNA density—or contain no DNA at all—as exemplified by the arrows in Figure 1. Note that such large scale open areas are rare in lymphocytes. Finally, it seems that both the DNA structure and the structure of the DNA-free space differ between lymphocytes, H cells, and RS cells.
We have used granulometry to quantify the distribution of length scales present in the DNA structure as well as the DNA-free space, the dark regions in the figures. By “length” we refer to equivalent diameters of the granule-like regions in the DNA and DNA-free spaces. The resulting cumulative distributions of the typical granule sizes in these cells are plotted in Figure 2A,B for the three cell types. The granule size distribution of the DNA distribution is smallest for the control lymphocytes. Both HL cell types contain relatively more DNA structures at both the low end of the size distribution scale, 200–700 nm, representing the actual intranuclear DNA structure, and the high end of the size distribution in these images, 1–3 μm, representing structure with length scales close to the size of the cells. We evaluated the significance of the measured differences with the two-sided, two-sample Kolmogorov–Smirnov test (KS-test) for triplicate experiments and found the differences to be significant at the 5% level. We then combined the triplicate experiments for a total cell count of 137 lymphocytes, 129 H cells, and 97 RS cells. The KS-test determined that all three groups were significantly different, P = 10−18 for L versus H cells and L versus RS cells, P = 10−11 for H cells versus RS cells. The length scales measured by the granulometries are mostly larger than the traditional microscopic diffraction limit. The structure itself is hard to detect visually in the widefield images (Figs. 1 and S1) and is not picked up by the granulometry when applied to these images (Fig. S4). In the widefield case the granulometry measures highlight differences in the global size of the cell (3–15 μm) rather than the intranuclear DNA structure. The differences are, however, also significant for the widefield images: P = 10−12 for L versus H cells, P = 10−37 for L versus RS cells, and P = 10−7 for H versus RS cells. But again, the differences occur at lengths scales on the order of the size of the nuclei, rather than the size of the intranuclear structure for the widefield images.
Figure 2.
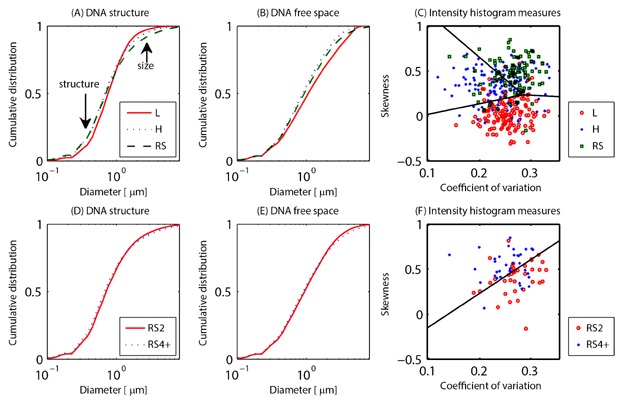
Measurements on SIM images of DAPI-stained nuclei of the size distribution of the DNA structure and DNA-free space, as well as the intensity histogram. These visible structures are depicted in Figure 1. The top row (A–C) shows these measures for lymphocytes (L, red solid lines and circles), Hodgkin cells (H, blue dotted lines and crosses), and Reed–Sternberg cells (RS, green dash lines and squares). The bottom row (D–F) illustrates binucleated RS cells (RS2, red solid lines and circles) and RS cells with 4 or more subnuclei (RS4+, blue dotted lines and crosses). A,D: Cumulative size distribution of the DNA structure, (B,E) the cumulative size distribution of the DNA-free space. All these size distributions are measured with a granulometry. The difference in the sub-micron size range is caused by differences in the DNA structure size. Differences in size in the order of several μm are due to the difference in cell size. Both regimes are indicated with an arrow in the top left plot. C,F: Coefficient of variation and skewness of the intensity histogram of these cells. The black lines are the Fisher linear discriminants. See Figure S4 for measurements on the widefield images of the same cells. All differences between the granulometries are significant at the 5% significance level, except RS2 versus RS4+ for the DNA-free space.
Next we characterized the DNA-free space to investigate whether there are changes between normal and cancer cells. The control lymphocytes contain DNA-free space at larger length scales; this is also visible in the cell images (Fig. 1). Both the H and RS cells display smaller open areas/holes than lymphocytes. The largest difference between the distributions occurs at the length scales of 0.6–2.0 μm. These are the typical sizes of the DNA-free space as well as the “holes” visible in the DAPI-stained images (Fig. 1). Neither the DNA structure nor the DNA-free space show apparent differences in the widefield image. The difference in the DNA-free space is, however, significant for the 3D-SIM image; the KS-test yields P = 10−20 for L versus H cells, P = 10−10 for L versus RS cells, and P = 10−7 for H versus RS cells. For the widefield images these measurements (Fig. S4) are: P = 10−34 for L versus H cells, P = 10−86 for L versus RS cells, and P = 10−23 for H versus RS cells. Again, the differences occur at length scales on the order of the size of the nuclei, rather than the size of the intranuclear structure for the widefield images.
We also noted visually that there was a difference in the DAPI intensity over the nucleus. The intensity histogram itself has different properties for the different cell groups (3D-SIM, Fig. 2C,F; widefield, Fig. S4C,F). When the coefficient of variation (c.o.v.) and skewness are plotted for each cell there is no correlation between these measures for the 3D-SIM image (R = 0.065). These measures are, however, somewhat correlated in the widefield image (R = 0.500). Since the three cell groups seem to occupy somewhat different regions of the 2D space spanned by c.o.v. on one axis and skewness on the other axis, we linearly divided the 2D space between the cell groups. If there were no difference between the cell types, the resulting discrimination functions would not be meaningful and 2/3 = 67% would be classified erroneously. Classifying the cells based on their histogram, the error rate was 30% for the 3D-SIM images and 38% for the widefield images. Most of these errors occur close to the boundary lines between the regions. This is expected, because we see a transition from H to RS cells. We did not aim to optimize this as a classification tool in this study. Note, however, that the error rate of the classification decreases significantly for the SIM images compared to the widefield images (P = 0.021). For the 3D-SIM image the skewness of both the H and RS cells is higher than for the lymphocytes. This indicates that the former have a relatively higher number of high-intensity pixels. The c.o.v. in RS cells is higher than in H cells. This indicates there is a larger spread of intensity values in the multinucleated RS cells.
UBF CONTENT IN THE DNA-FREE SPACE
For the HL cells, both H and RS, some “open spaces” (Fig. 1) are clearly visible. These open spaces do not appear in the control lymphocytes. Nucleoli display the same morphology and could also be associated with the absence of DNA. To investigate whether the DNA-free space might be associated with nucleoli, we stained the upstream binding factor (UBF). UBF is a protein that is present in the nucleolus [Hein et al., 2013]. The spatial position of UBF within the nucleus is depicted for its widefield image in Figure 3, Figure S5 shows the SIM UBF image. Areas with higher concentrations of UBF do occur in some of these holes, but not in all of them. For the HL cell line we counted how many of these holes are filled with UBF and how many are not. We found that 85% of the holes in the H cells were filled with UBF, compared to 50% of the RS cells (Fig. 3G). This difference is significant (P = 10−12). To assess this objectively we also computed Pearson’s correlation coefficient of the DNA-free space (the negative of the DAPI image) and the UBF image. We first computed the correlation coefficient between the SIM UBF image and SIM DNA-free space (Fig. 3E). The coefficient was relatively low (between 0.05 and 0.14), because the UBF signals in the 3D-SIM images appear very spiky and not as homogeneous as in the widefield images. The correlation between both channels, however, monotonically decreases from the control lymphocytes to H cells to RS cells. This indicates an increase in DNA-free space that is not occupied by this transcription factor. This is also exemplified by the correlation coefficient between the SIM DNA-free space and the widefield UBF image (Fig. 3F), in which the UBF signal appears spatially homogeneous. The correlation coefficient again decreases, from 0.38 for the lymphocytes through 0.27 for the H cells to 0.18 for the RS cells. The difference between the lymphocytes and the H and RS cells are significant in all cases (L vs. H: P = 10−5 for DAPI-SIM and UBF-SIM, P = 10−8 for DAPI-SIM and UBF-widefield; L vs. RS: P = 10−7 for DAPI-SIM and UBF-SIM, P = 10−14 for DAPI-SIM and UBF-widefield). The difference between H and RS cells is also significant for the correlation between the SIM DNA-free space and the SIM UBF image (P = 0.029) as well as for correlation between the SIM DNA-free space and widefield UBF image (P = 10−6).
Figure 3.

DAPI SIM image in grayscale and UBF widefield image in green for the same cells as Figure 1 with a lymphocyte (A), an H cell (B), a binucleated RS cell (C), and a multinucleated RS cell (D). The top row (i) shows a lateral (x,y)-slice and the bottom row (ii) a (x,z)-slice from the same cell. The scale bars are 5 μm in each lateral slice. The tick marks in the middle of each panel indicate the positions of the corresponding orthogonal planes. The DAPI signal intensities are linearly stretched between the minimum and maximum value, the widefield UBF signal intensities are linearly scaled between the 5th and 100th percentile over the nucleus. Values below the 5th percentile were clipped. Note that clusters of UBF, a transcription factory, are located in DNA-free space, including some of the holes identified by arrows in Figure 1. Not all holes contain UBF, as is most apparent in panels (D). The bottom row (E–G) shows measurements relating to the spatial UBF distribution with regard to the nucleus for the five groups of cells. The bars indicate the sample mean and the error bars the standard error of the mean. The bottom left (E) indicates the correlation coefficient between the DNA-free space SIM image and SIM UBF image. The bottom middle (F) displays the correlation coefficient between the DNA-free space SIM image and widefield UBF image. The bottom right (G) indicates the manually counted fill rate for the “holes” in the DNA structure (arrows in Fig. 1). Because there are no clearly visible holes in the lymphocytes, the H and RS numbers are only shown. All differences in correlation coefficients and filled-hole-rates on either side of the dashed lines are significant at the 5% level, except RS2 versus RS4+ in panel (E).
PROGRESSIVE TREND WITH THE POPULATION OF REED–STERNBERG CELLS
Because RS cells can consist of different numbers of subnuclei, which form progressively during tumor development, we compared binucleated Reed–Sternberg (RS2) cells with RS cells that contain four or more subnuclei (RS4+). Both groups consisted of 36 cells in our study; the remaining 25 cells were trinucleated RS cells. We performed the same measures on this two-group system. The DNA structure is different between the two (KS-test P = 10−5), in particular the RS4+ cells had a larger spread in the size of their structure than the RS2 cells. The differences for the DNA-free space were, however, not significant (KS-test, P = 0.09). For the widefield images this led to P = 0.0016 for the DNA structure and P = 10−9 for the DNA-free space. The 2D c.o.v.-skewness space classification leads to a 25% error rate for the SIM images and a 35% error rate for the widefield images. As before, the error rate decreased for the SIM images compared to the widefield images; although not significantly (P = 0.18). Note that this is a two-group system in which complete overlap would mean a classification error of 50%. The differences between RS2 and RS4+ cells display the same trend as the differences between H and RS cells. The UBF-based measures revealed differences as well, although not significant in all cases. The relative number of “holes” that is filled with UBF is 69% for the RS2 cells and 41% for the RS4+ cells, a significant difference (P = 10−5). The difference in the mean correlation coefficient between the two groups was significant when comparing the DNA-free space in SIM with the widefield UBF image (P = 0.021). The difference between RS2 and RS4+ cells was, however, not significant when the correlation coefficient was calculated between the UBF and DAPI SIM images (P = 0.22). We did not further subdivide the RS4+ cell into subgroups of tetranucleated cells and cells with eight or more subnuclei. We do, however, hypothesize that the described trends continue toward cells with increasing numbers of subnuclei.
DISCUSSION
We have described, to the best of our knowledge for the first time, the intranuclear DNA structure of normal and cancer cells using a superresolution microscopy method. We have been able to quantify the DNA structure revealed by high resolution light microscopy successfully. In particular, we have been able to measure structures at the 200–700 nm size range. We have observed that many more of these sub-micron structures are present and that they are smaller in size in HL cells than in control lymphocytes. When we measured the properties of the SIM DAPI intensity histograms, we noted an increased skewness for the HL cells. This means that these malignant cells have a more asymmetric DNA distribution than lymphocytes. This can be attributed to the apparent higher degree of clustering in these cells. The RS cells have a larger spread in pixel intensities than H cells, as evidenced by their increased c.o.v. This means that the DNA density in these multinucleated cells is more varied than for the mononucleated H cells.
These structures might appear due to changes in the condensation of the DNA. As H and RS cells are larger, the DNA might also be spread out over a larger volume. If the spreading is uneven, this could lead to local “patches” of DNA. It might also be linked to a difference in chromatin organization, possibly measured with chromosome conformation capture techniques [Nagano et al., 2013], between these cells and healthy lymphocytes. We have previously shown that, in the HD cell-line U-HO1-PTPN1, PTPN1 induced down-regulation of STAT5A was associated with multi-nuclearity and high apoptotic index compared to the U-HO1 cell-line [Knecht et al., 2010]. The DNA content varies between different subnuclei in RS cells, some subnuclei are more DNA-rich than others [Guffei et al., 2010].
We measured the DNA-free space in these cells as well. We observed an increase in the DNA-free space in HL compared to lymphocytes, as well as the formation of “holes” in the nucleus. To check whether the DNA-free space or the holes represented nucleoli, we stained for UBF. We found during HL progression from H cells to RS cells with increased multinuclearity, that both the portion of DNA-free space filled with UBF and the rate of visible holes filled with UBF decreased significantly. Nucleoli can be disrupted in cancer [Boulon et al., 2010], which would explain why the UBF signal is not confined to one nucleolus per subnucleus in the malignant cells. The reduction of relative UBF content in the DNA-free space for H and RS cells poses the question whether something else takes its place. This space might now be filled by other nucleolar proteins, different nuclear bodies [Fong et al., 2013], transcription factories or could simply be devoid of stable sub-nuclear organelles.
We do not claim that this is the only microscope modality that could visualize these structures. Other superresolution methods, or microscopes that perform optical sectioning, could lead to similar images as in Figures 1 and S1. Such images should then lead to similar granulometry results. The structures with frequency content that is within the pass-band of the objective lens might be recovered from widefield microscopy images using post-processing methods.
Previous studies measuring aspects of the nuclear architecture within Hodgkin and Reed–Sternberg cells have detected a progressive disruption of the nuclear organization [Martin-Subero et al., 2003; Knecht et al., 2009; Guffei et al., 2010]. Our measurements quantitatively revealed the progressive disruption of nuclear DNA organization in HL. We have, for the first time, shown a progressive trend in the organization of DNA using superresolution microscopy. This trend starts at the control lymphocytes, moves towards Hodgkin cells, and then progresses to Reed–Sternberg cells. Within the population of Reed–Sternberg cells we found the same trend with increasing multinuclearity.
We have performed our study on an HL cell line. The measurements described in this article can be extended in two ways. First, it would be desirable to perform the same measurements in primary tumor cells of clinical HL samples. Care should be taken in choosing an embedding medium with proper refractive index. As we currently perform the measurements in 2D slices, it should be possible to do this in tissue, in particular lymph node biopsies. Second, it should be possible to investigate other cancers to determine whether our observations are specific for HL or whether they would apply more generally to other cancers as well. If these changes to the nucleus are common to cancer cell nuclei, then the question arises if the same changes would be detected for different cancers or whether they would be cancer specific. The initial candidates for similar studies should be other hematological malignancies.
The features of the nuclear architecture we measure follow a progressive trend with progressive cell conditions in HL. More aggressive cases of HL can, in some cases, be identified based on the nuclear organization of the H and RS cells in those tumors [Knecht et al., 2012]. The nuclear DNA structure might, therefore, also be related to the aggressiveness of HL. Whether the measures presented here are correlated with clinical outcome, could be investigated by comparing HL cases of patients who respond to treatment versus those who recur.
The measures of the DNA structure and DNA-free space were performed over the entire nucleus. There might be a difference in these features between chromosomes, for instance the length, gene density, activity, and/or radial position of each chromosome might play a role in the DNA organization. To test this, CTs would need to be stained individually. It should then be possible to check whether there are differences between the DNA organization between CT’s, or for the same CT between a healthy and disease state. It might also be possible to determine the location of the DNA-free space in relation to the interface between CT’s. This could ultimately determine which boundary model—either the interchromatin domain model or the interchromosomal network [Branco and Pombo, 2006]—is correct; it could lead also to a different model altogether.
Cancer is a disease of DNA organization [Pienta et al., 1989] and for the first time we can address the changes in the organization in detail. We have shown a difference for both the DNA structure and DNA-free space in the nucleus and found both nuclear and nucleolar remodeling. This study can be seen as the tip of an iceberg, where we have measured a selected set of structural changes in one cell line. There are a wide variety of other features and other systems in which this can be studied. This is, however, the first superresolution view of these nuclear alterations. The DNA architecture in cancer might be a hallmark of the underlying genomic instability and cellular reprogramming that promotes cell proliferation, evasion from apoptosis and immunosurveilance.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
supporting information Fig. S1. Slices from a trinucleated Reed-Sternberg cell (A) and a tetranucleated Reed-Sternberg cell (B).
supporting information Fig. S2. Several images illustrating the granulometry tests discussed in the supplementary materials and methods.
supporting information Fig. S3. Granulometry input images for a lymphocyte (A), an H cell (B), a binucleated RS cell (C), a trinucleated RS cell (D), a tetranucleated RS cell (E), and a multinucleated RS cell (F).
supporting information Fig. S4. Granulometries and histogram-based cell properties measured from widefield images.
supporting information Fig. S5. SIM image of the UBF channel for a lymphocyte (A), an H cell (B), a binucleated RS cell (C), a trinucleated RS cell (D), a tetranucleated RS cell (E), and a multinucleated RS cell (F).
supporting information Movie S1. All z-positions for the cell shown in Figure 1A.
supporting information Movie S2. All z-positions for the cell shown in Figure 1B.
supporting information Movie S3. All z-positions for the cell shown in Figure 1C.
supporting information Movie S4. All z-positions for the cell shown in Figure S1A.
supporting information Movie S5. All z-positions for the cell shown in Figure S1B.
supporting information Movie S6. All z-positions for the cell shown in Figure 1D.
REFERENCES
- Bins M, Landeweerd GH, Gelsema ES, van Montfort LH, Halie MR. Texture of white blood cells expressed by the counting densitogram. Cytometry. 1981;1:321–324. doi: 10.1002/cyto.990010504. [DOI] [PubMed] [Google Scholar]
- Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T. Zur Frage der Entstehung maligner Tumoren. Jena: Fischer; 1914. [Google Scholar]
- Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121:1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- Branco MR, Pombo A. Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol. 2006;4:e138. doi: 10.1371/journal.pbio.0040138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg GE, So PT. Lateral resolution enhancement with standing evanescent waves. Opt Lett. 2000;25:46–48. doi: 10.1364/ol.25.000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Cremer C. Rise, fall and resurrection of chromosome territories: A historical perspective. Part I. The rise of chromosome territories. Eur J Histochem. 2006a;50:161–176. [PubMed] [Google Scholar]
- Cremer T, Cremer C. Rise, fall and resurrection of chromosome territories: A historical perspective. Part II. Fall and resurrection of chromosome territories during the 1950s to 1980s. Part III. Chromosome territories and the functional nuclear architecture: Experiments and models from the 1990s to the present. Eur J Histochem. 2006b;50:223–272. [PubMed] [Google Scholar]
- Drexler HG, Gaedicke G, Lok MS, Diehl V, Minowada J. Hodgkin’s disease derived cell lines HDLM-2 and L-428: Comparison of morphology, immunological and isoenzyme profiles. Leuk Res. 1986;10:487–500. doi: 10.1016/0145-2126(86)90084-6. [DOI] [PubMed] [Google Scholar]
- Duin RPW, Juszczak P, Paclik P, Pekalska E, de Ridder D, Tax DMJ, Verzakov S. PRTools, a Matlab toolbox for pattern recognition. Delft, The Netherlands: Delft University of Technology; 2007. [Google Scholar]
- Einstein AJ, Wu H-S, Sanchez M, Gil J. Fractal characterization of chromatin appearance for diagnosis in breast cytology. J Pathol. 1998;185:366–381. doi: 10.1002/(SICI)1096-9896(199808)185:4<366::AID-PATH122>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Flemming W. Zellsubstanz, Kern und Zelltheilung. Leipzig: F.C.W. Vogel; 1882. [Google Scholar]
- Fong KW, Li Y, Wang W, Ma W, Li K, Qi RZ, Liu D, Songyang Z, Chen J. Whole-genome screening identifies proteins localized to distinct nuclear bodies. J Cell Biol. 2013;203:149–164. doi: 10.1083/jcb.201303145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohn JT, Knapp HF, Stemmer A. True optical resolution beyond the Rayleigh limit achieved by standing wave illumination. Proc Natl Acad Sci USA. 2000;97:7232–7236. doi: 10.1073/pnas.130181797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guffei A, Sarkar R, Klewes L, Righolt C, Knecht H, Mai S. Dynamic chromosomal rearrangements in Hodgkin’s lymphoma are due to ongoing three-dimensional nuclear remodeling and breakage-bridge-fusion cycles. Haematologica. 2010;95:2038–2046. doi: 10.3324/haematol.2010.030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MG. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Hannen EJM, Van Der Laak JAWM, Manni JJ, Pahlplatz MMM, Freihofer HPM, Slootweg PJ, Koole R, De Wilde PCM. An image analysis study on nuclear morphology in metastasized and non-metastasized squamous cell carcinomas of the tongue. J Pathol. 1998;185:175–183. doi: 10.1002/(SICI)1096-9896(199806)185:2<175::AID-PATH69>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Hein N, Hannan KM, George AJ, Sanij E, Hannan RD. The nucleolus: An emerging target for cancer therapy. Trends Mol Med. 2013;19:643–654. doi: 10.1016/j.molmed.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Heintzmann R, Cremer CG. Laterally modulated excitation microscopy: improvement of resolution by using a diffraction grating. Proc SPIE. 1999;3568:185–196. [Google Scholar]
- Knecht H, Bruderlein S, Wegener S, Lichtensztejn D, Lichtensztejn Z, Lemieux B, Moller P, Mai S. 3D nuclear organization of telomeres in the Hodgkin cell lines U-HO1 and U-HO1-PTPN1: PTPN1 expression prevents the formation of very short telomeres including “t-stumps”. BMC Cell Biol. 2010;11:99. doi: 10.1186/1471-2121-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knecht H, Kongruttanachok N, Sawan B, Brossard J, Prevost S, Turcotte E, Lichtensztejn Z, Lichtensztejn D, Mai S. Three-dimensional telomere signatures of Hodgkin- and Reed-Sternberg cells at diagnosis identify patients with poor response to conventional chemotherapy. Transl Oncol. 2012;5:269–277. doi: 10.1593/tlo.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knecht H, Sawan B, Lichtensztejn D, Lemieux B, Wellinger RJ, Mai S. The 3D nuclear organization of telomeres marks the transition from Hodgkin to Reed-Sternberg cells. Leukemia. 2009;23:565–573. doi: 10.1038/leu.2008.314. [DOI] [PubMed] [Google Scholar]
- Kuppers R, Engert A, Hansmann ML. Hodgkin lymphoma. J Clin Invest. 2012;122:3439–3447. doi: 10.1172/JCI61245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo Hendriks CL, Rieger B, van Ginkel M, van Kempen GMP, van Vliet LJ. DIPimage: A scientific image processing toolbox for MATLAB. Delft, The Netherlands: Delft University of Technology; 1999. [Google Scholar]
- Luengo Hendriks CL, van Kempen GMP, van Vliet LJ. Improving the accuracy of isotropic granulometries. Pattern Recognit Lett. 2007;28:865–872. [Google Scholar]
- MacLeod RA, Spitzer D, Bar-Am I, Sylvester JE, Kaufmann M, Wernich A, Drexler HG. Karyotypic dissection of Hodgkin’s disease cell lines reveals ectopic subtelomeres and ribosomal DNA at sites of multiple jumping translocations and genomic amplification. Leukemia. 2000;14:1803–1814. doi: 10.1038/sj.leu.2401894. [DOI] [PubMed] [Google Scholar]
- Markaki Y, Smeets D, Fiedler S, Schmid VJ, Schermelleh L, Cremer T, Cremer M. The potential of 3D-FISH and super-resolution structured illumination microscopy for studies of 3D nuclear architecture. BioEssays. 2012;34:412–426. doi: 10.1002/bies.201100176. [DOI] [PubMed] [Google Scholar]
- Martin-Subero JI, Knippschild U, Harder L, Barth TF, Riemke J, Grohmann S, Gesk S, Hoppner J, Moller P, Parwaresch RM, Siebert R. Segmental chromosomal aberrations and centrosome amplifications: Pathogenetic mechanisms in Hodgkin and Reed-Sternberg cells of classical Hodgkin’s lymphoma? Leukemia. 2003;17:2214–2219. doi: 10.1038/sj.leu.2403129. [DOI] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan S, Juneja M, Pallam NK, Boaz K, Mohindra A, Lewis A. A novel technique to assess chromatin texture using pixel optical densitometry in oral squamous cell carcinoma. Microsc Res Tech. 2012;75:1119–1123. doi: 10.1002/jemt.22039. [DOI] [PubMed] [Google Scholar]
- Pienta KJ, Partin AW, Coffey DS. Cancer as a disease of DNA organization and dynamic cell structure. Cancer Res. 1989;49:2525–2532. [PubMed] [Google Scholar]
- Ridler TW, Calvard S. Picture thresholding using an iterative selection method. IEEE Trans Syst Man Cybernet. 1978;8:630–632. [Google Scholar]
- Schermelleh L, Carlton PM, Haase S, Shao L, Winoto L, Kner P, Burke B, Cardoso MC, Agard DA, Gustafsson MG, Leonhardt H, Sedat JW. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 2008;320:1332–1336. doi: 10.1126/science.1156947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe H, Muller S, Neusser M, von Hase J, Calcagno E, Cremer M, Solovei I, Cremer C, Cremer T. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc Natl Acad Sci USA. 2002;99:4424–4429. doi: 10.1073/pnas.072618599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek PW, van Vliet LJ. Estimators of 2D edge length and position, 3D surface area and position in sampled grey-valued images. Bioimaging. 1993;1:47–61. [Google Scholar]
- Vergani L, Fugazza G, Chessa L, Nicolini C. Changes of chromatin condensation in one patient with ataxia telangiectasia disorder: A structural study. J Cell Biochem. 1999;75:578–586. doi: 10.1002/(sici)1097-4644(19991215)75:4<578::aid-jcb4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Young IT, Verbeek PW, Mayall BH. Characterization of chromatin distribution in cell nuclei. Cytometry. 1986;7:467–474. doi: 10.1002/cyto.990070513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supporting information Fig. S1. Slices from a trinucleated Reed-Sternberg cell (A) and a tetranucleated Reed-Sternberg cell (B).
supporting information Fig. S2. Several images illustrating the granulometry tests discussed in the supplementary materials and methods.
supporting information Fig. S3. Granulometry input images for a lymphocyte (A), an H cell (B), a binucleated RS cell (C), a trinucleated RS cell (D), a tetranucleated RS cell (E), and a multinucleated RS cell (F).
supporting information Fig. S4. Granulometries and histogram-based cell properties measured from widefield images.
supporting information Fig. S5. SIM image of the UBF channel for a lymphocyte (A), an H cell (B), a binucleated RS cell (C), a trinucleated RS cell (D), a tetranucleated RS cell (E), and a multinucleated RS cell (F).
supporting information Movie S1. All z-positions for the cell shown in Figure 1A.
supporting information Movie S2. All z-positions for the cell shown in Figure 1B.
supporting information Movie S3. All z-positions for the cell shown in Figure 1C.
supporting information Movie S4. All z-positions for the cell shown in Figure S1A.
supporting information Movie S5. All z-positions for the cell shown in Figure S1B.
supporting information Movie S6. All z-positions for the cell shown in Figure 1D.