

Background: (2E)-4-hydroxy-2-nonenal (HNE) catabolism and its regulation remain to be fully investigated.
Results: New catabolic pathways of HNE via ω-/ω-1-oxidation are elucidated and up-regulated in a ketogenic diet.
Conclusion: HNE catabolism plays a major role in HNE disposal in certain organs.
Significance: This work will further enhance our understanding of the metabolic fate of HNE and the roles of ω- and ω-1-oxidations in HNE disposal.
Keywords: beta-Oxidation, Coenzyme A (CoA), Cytochrome P450, Diet, Fatty Acid Oxidation, Isotopic Tracer, Lipid Peroxidation, Mass Spectrometry (MS), Metabolism, Metabolomics
Abstract
Oxidative stress triggers the peroxidation of ω-6-polyunsaturated fatty acids to reactive lipid fragments, including (2E)-4-hydroxy-2-nonenal (HNE). We previously reported two parallel catabolic pathways of HNE. In this study, we report a novel metabolite that accumulates in rat liver perfused with HNE or 4-hydroxynonanoic acid (HNA), identified as 3-(5-oxotetrahydro-2-furanyl)propanoyl-CoA. In experiments using a combination of isotopic analysis and metabolomics studies, three catabolic pathways of HNE were delineated following HNE conversion to HNA. (i) HNA is ω-hydroxylated to 4,9-dihydroxynonanoic acid, which is subsequently oxidized to 4-hydroxynonanedioic acid. This is followed by the degradation of 4-hydroxynonanedioic acid via β-oxidation originating from C-9 of HNA breaking down to 4-hydroxynonanedioyl-CoA, 4-hydroxyheptanedioyl-CoA, or its lactone, 2-hydroxyglutaryl-CoA, and 2-ketoglutaric acid entering the citric acid cycle. (ii) ω-1-hydroxylation of HNA leads to 4,8-dihydroxynonanoic acid (4,8-DHNA), which is subsequently catabolized via two parallel pathways we previously reported. In catabolic pathway A, 4,8-DHNA is catabolized to 4-phospho-8-hydroxynonanoyl-CoA, 3,8-dihydroxynonanoyl-CoA, 6-hydroxyheptanoyl-CoA, 4-hydroxypentanoyl-CoA, propionyl-CoA, and acetyl-CoA. (iii) The catabolic pathway B of 4,8-DHNA leads to 2,6-dihydroxyheptanoyl-CoA, 5-hydroxyhexanoyl-CoA, 3-hydroxybutyryl-CoA, and acetyl-CoA. Both in vivo and in vitro experiments showed that HNE can be catabolically disposed via ω- and ω-1-oxidation in rat liver and kidney, with little activity in brain and heart. Dietary experiments showed that ω- and ω-1-hydroxylation of HNA in rat liver were dramatically up-regulated by a ketogenic diet, which lowered HNE basal level. HET0016 inhibition and mRNA expression level suggested that the cytochrome P450 4A are main enzymes responsible for the NADPH-dependent ω- and ω-1-hydroxylation of HNA/HNE.
Introduction
4-Hydroxy acids are a class of compounds that includes both exogenous drugs (C4 and C5) and lipid peroxidation products (C6 and C9) (1, 2). Given the ubiquitous nature of these molecules, their reported toxicities, and their growing importance as pharmaceutical agents (C4), a comprehensive understanding of the mechanisms of detoxifying these compounds in vivo is of great interest.
(2E)-4-Hydroxy-2-nonenal (HNE)3 is one of the most abundant lipid peroxidation products derived from ω-6-polyunsaturated fatty acids. HNE is elevated in oxidative stress-related diseases (3, 4). The etiology of HNE as related to disease states is generally ascribed to increasing HNE-modified proteins, DNA, and phospholipids, which has been documented in many diseases, such as cancer (5), diabetes (6, 7), obesity (8, 9), Alzheimer disease (10, 11), atherosclerosis (12, 13), etc. High concentrations of HNE and its modified proteins, DNA, and lipids could be due to the increased lipid peroxidation or alternatively the down-regulation of detoxification pathways. The detoxification of HNE is generally defined as follows (14–19): (i) conjugation with glutathione; (ii) reduction to 1,4-dihydroxy-2-nonene (DHN); and (iii) oxidation to 4-hydroxynonenoic acid. Our recent liver perfusions with [5,5,6,6,7,7,8,8,9,9,9-2H11]HNE revealed that glutathione conjugation with HNE accounts for only ∼8.7% of total HNE uptake by a perfused rat liver (20), and very little HNE is metabolized to DHN by reduction. Our data indicate that conversion of HNE to 4-hydroxynonanoic acid (HNA) and further catabolism play a dominant role in the detoxification of HNE. HNE catabolism follows two parallel catabolic pathways of 4-hydroxy acids, and leads to the complete degradation of HNE to formic acid, acetyl-CoA, and propionyl-CoA (2). Our data are the first report illustrating how HNE is completely degraded via two parallel catabolic pathways. The significance of the identified catabolic disposal of HNE was demonstrated by our recent dietary and heart perfusion experiments (3, 4). The down-regulated catabolism of HNE in Western diet and ischemic rat heart significantly increases HNE levels.
In addition to the two parallel catabolic pathways of 4-hydroxy acids (2, 21), we recently reported that 4-hydroxybutyric acid (C4, GHB) is catabolized via its own specific pathways, including (i) oxidation to succinic acid and (ii) formation of 3-hydroxypropionic acid from two different processes by removing C-1 or C-4 of GHB (22). Our extensive research on the metabolism 4-hydroxypentanoic acid (C5, GHP) also exhibited its own characteristic metabolism: (i) GHP is oxidized to 4-ketopentanoic acid (levulinic acid) and is further activated to levulinyl-CoA; (ii) levulinyl-CoA is elongated to 3,6-diketoheptanoyl-CoA that yields cyclopentenyl- and cyclopentadienyl-acyl-CoAs via cyclization; and (iii) 3,6-diketoheptanoyl-CoA can pyrrolate free lysine and, presumably, lysine residues from proteins (23). The elucidation of these metabolic pathways sheds light on the mechanism of physiological disposal of these compounds.
Based on findings of the specific catabolic pathways of GHB and GHP, we hypothesize that HNE would also possess its own catabolic pathways in addition to the two common catabolic pathways. The purpose of the present work is to carefully investigate the catabolism of HNE in the perfused rat liver using a combination of metabolomics and isotopomer analysis.4
In the present work, HNA (HNE oxidation/saturation product) is found to be ω- and ω-1-hydroxylated to 4,9-dihydroxynonanoic acid (4,9-DHNA) and 4,8-dihydroxynonanoic acid (4,8-DHNA) in a perfused rat liver. 4,8-DHNA is further catabolized following HNE/HNA catabolic pathways reported previously by us (2). 4,9-DHNA is oxidized to 4-hydroxynonenodioic acid, whose carbon chain length is further shortened by β-oxidation from the C-9 end. In mammals, cytochrome P450 4A and 4F family enzymes (CYP4A and CYP4F) catalyze the ω- and ω-1-hydroxylation of fatty acids. CYP4A1, CYP4A2, CYP4A3, CYP4A4, CYP4F1, CYP4F4, CYP4F5, and CYP4F6 were identified in rat livers according to precedence in the literature (24–26). Given that diet can induce alterations in CYP expression profiles and activities (27), we hypothesized that the ω- and ω-1-hydroxylation activities toward HNE could be modulated by a diet composed of a differential lipid composition. Four different diets (i.e. standard diet, low fat diet, ketogenic (KG) diet (extremely high fat and low carbohydrate), and Western diet (high fat and high carbohydrate)) were used to test the influence of diets with various compositions of fat on HNE ω- and ω-1-hydroxylation. ω- and ω-1-hydroxylation activities in rat liver are significantly stimulated by the KG diet. The enhanced omega- and omega-1-hydroxylation activities toward HNE by the KG diet also explain the lowest basal HNE level found in rat liver from this diet. The CYP family enzymes that are responsible for HNA hydroxylation were characterized using inhibitors and mRNA levels.
EXPERIMENTAL PROCEDURES
Materials
General chemicals, including acyl-coenzyme A, were purchased from Sigma-Aldrich. HNE and [5,5,6,6,7,7,8,8,9,9,9-2H11]HNE were synthesized as described previously (28). [2,2,3,3,4,4-2H6]4-hydroxybutyric acid ([2H6]GHB) was hydrolyzed from the corresponding lactone (Sigma-Aldrich-Isotec) by 10% excess NaOH at 70 °C for 1 h. [3-13C]4-hydroxynonanoic acid (M1 HNA) and [3,4-13C2]4-hydroxynonanoic acid (M2 HNA) were synthesized by us (2). HET0016 was from Cayman Chemical (Ann Arbor, MI). Miconazole was a gift from Dr. Irina Pikuleva (Case Western Reserve University).
Synthesis of 3-([2-2H]5-Oxotetrahydrofuran-2-yl)propanoic Acid (M1 OTHFPA) and 2-OH-[2-2H]Glutaric Acid (M1 2-HG)
M1 OTHFPA was from the reduction of 4-oxoheptanedioic acid (Fisher) by NaBD4. A 0.6 m 4-oxoheptanedioic acid aqueous solution (25 ml) was cooled in ice and mixed with a molar equivalent of NaBD4. After 2 h of incubation at 4 °C, the reaction mixture was adjusted to pH 1–2 with 6 m HCl. M1 OTHFPA was then continuously extracted with diethyl ether for 24 h, and the solvent was removed in vacuo. M1 2-HG was synthesized as above for M1 OTHFPA. The lactone of M1 2-HG was hydrolyzed using 1.1 eq of NaOH at 70 °C for 1 h. The identity and purity of both M1 OTHFPA and M1 2-HG were determined by GC-MS.
Animal Experiments
Male Sprague-Dawley rats (150–300 g) were fed ad libitum for 8–12 days with standard laboratory chow prior to experiments, and rats were studied in the fed (heart perfusion) or overnight fast (liver perfusion) state. All animal procedures were performed on anesthetized animals (2–5% isoflurane). All experiments were performed in accordance with the Institutional Animal Care and Use Committee at Case Western Reserve University.
Liver and Heart Perfusions
Livers from rats were perfused (29) with bicarbonate buffer containing 4 mm glucose and either 4% dialyzed, fatty acid-free, bovine serum albumin (recirculating perfusions) or no albumin (non-recirculating perfusions or when labeled/unlabeled HNE was perfused). After equilibration, a 0–2 mm concentration of various 4-hydroxy acids, OTHFPA and 2-HG, labeled or unlabeled, was added to the perfusate. Perfusates were collected and immediately frozen in dry ice at different perfusion times. Livers were quick-frozen in liquid nitrogen at the end of the experiments. HNA metabolism in rat heart was also carried out by heart perfusion (recirculating perfusion) in Langendorff mode with 1 mm labeled and unlabeled HNA. The details of heart perfusion were described in our previous report (4).
In Vivo Metabolism of HNA
To investigate how HNA is metabolized in various organs in vivo, rats were dosed with HNA (100 mg/kg) by intraperitoneal injection. After 30 min, rats were anesthetized, and a median laparotomy was performed to allow a bolus of heparin (500 units/kg) to be given via the inferior vena cava. Liver, hearts, kidney, and brain were quickly dissected and frozen in liquid nitrogen until used for acyl-CoA analysis.
In Vitro ω- and ω-1-Hydroxylation Assay
100 mg of frozen, powdered tissue was homogenized in 800 μl of phosphate buffer (100 mm, pH 7.4). Tissue homogenate (40 μl) was incubated at 37 °C for 30 min with 1 mm M0 or M2 HNA in reaction buffer (100 mm phosphate, 1 mm NADPH). Reactions were terminated on ice with the addition of 5 volumes of acetonitrile followed by an addition of 4-hydroxydecanoic acid as an internal standard (60 nmol). Samples were centrifuged (2000 × g, 10 min), and supernatants were dried with N2. Samples were derivatized with N,O-bis(trimethylsilyl) trifluoroacetamide and analyzed by GC-MS (1-μl sample injection).
ω- and ω-1-Hydroxylation Inhibition by Miconazole and HET0016
With the assay conditions listed above, two well characterized inhibitors of the CYP enzyme family, miconazole (an inhibitor of most CYP isoforms) and HET0016 (specific for CYP4A and CYP4F isoforms), were tested in liver homogenates from rats fed the KG diet. KG diet liver homogenate was selected in this experiment due to the highest ω-/ω-1-activity. An inhibitory dose-response curve was generated using a range of inhibitor concentrations. Inhibitors were dissolved in dimethyl sulfoxide, which was then used in control experiments.
CYP4A and CYP4F Gene Expression Using Real-time Quantitative PCR
Total RNA was isolated from rat liver tissue samples (30 mg) using the RNeasy Plus Universal minikit (Qiagen, Chatsworth, CA) according to the manufacturer's instructions. RNA was quantified using a NanoDrop spectrophotometer. Reverse transcription was performed using iScript Reverse Transcriptase Supermix (Bio-Rad) with 1 μg of total RNA according to the manufacturer's suggestion to generate cDNA. Primers were designed using an online version of Primer3 via NCBI (30) or referenced from previously published studies (31, 32), and sequences are shown in Table 1. Primers were synthesized by Integrated DNA Technologies (Coralville, IA). Quantitative PCR reactions were run using SYBR Green detection using 300 nm primers in the reaction mix (Quanta Biosciences, Gaithersburg, MD). PCR cycling was set as follows: 6-min denaturation (95 °C), followed by 40 cycles of denaturation (95 °C for 15 s) and extension (60 °C for 45 s). 18 S rRNA expression was used as the endogenous control for target normalization, and the data are presented as relative expression levels (i.e. 2−ΔCt) (33).
TABLE 1.
The sequence of PCR primers for CYP 4A and CYP 4F isoforms
| Primer | Forward/Reverse | Sequence (5′–3′) |
|---|---|---|
| CYP4A1 | Forward | TTCCA GCAGT TCCCA TCACC |
| Reverse | TTGCT TCCCC AGAAC CATCG | |
| CYP4A2 | Forward | CTCGC CATAG CCATG CTTAT C |
| Reverse | CCTTC AGCTC ATTCA TGGCA AAT | |
| CYP4A3 | Forward | CTGCT TCGCT TTGAG TTGCT |
| Reverse | ACAAA TGCAG GTATT GCAGG C | |
| CYP4A8 | Forward | GGAGC AATGT GCGAA TCCAA |
| Reverse | AGTCA ACATT CGTCG GTGCT | |
| CYP4F1 | Forward | GGCCC AAATT CTGAC CCAGA T |
| Reverse | CCCTG CTCTG TAGGA GTGAC | |
| CYP4F4 | Forward | ATCCT TACCA TGGCT GCTTT T |
| Reverse | CCACG AGCTT AGTTA CCTGC T | |
| CYP4F5 | Forward | AGGAT GCCGT GGCTA ACTG |
| Reverse | GGCTC CAAGC AGCAG AAGA | |
| CYP4F6 | Forward | GACTC GTCCA CCCTA ACGTC |
| Reverse | ACTTC TCACC AGCGC TCATC | |
| 18 S | Forward | CGG TAC AGT GAA ACT GCG AAT |
| Reverse | GCT GAC CGG GTT GGT TTT G |
Diet Experiments
To investigate the regulation of ω- and ω-1-hydroxylation activity and its relation with HNE level, rats were fed diets of varying fat content as detailed in our previous work (3). Briefly, the following diets were fed to rats (8 weeks old) for 4 weeks; standard (SD; 27.5% fat, 20.0% protein, 52.5% carbohydrate), low fat (LF; 11.5% fat, 10.4% protein, 78.1% carbohydrate), KG diet (89.5% fat, 10.4% protein, 0.1% carbohydrate), and high fat, high carbohydrate (HF-HC, “Surwit”, Western style diet; 58.1% fat, 16.4% protein, 25.5% carbohydrate). At the termination of the experiment, livers were quickly dissected, freeze-clamped in liquid nitrogen, and kept frozen (−80 °C) until analysis.
Acyl-CoA Profiling by LC-MS/MS
Acyl-CoAs from rat liver were extracted and profiled by LC-MS/MS with our published method (34). Briefly, powdered liver tissue (100 mg) was added to 0.2 nmol of internal standard ([2,2,3,3,4,4,5,5,5-2H9]pentanoyl-CoA) and extracted by 2 ml of methanol/H2O (1:1, v/v) containing 5% acetic acid. The resulting supernatant after homogenization and centrifugation was purified by a 2-(2-pyridyl)ethyl silica gel-packed cartridge. The purified sample was dried by N2 gas and stored at −80 °C until LC-MS/MS analysis (34).
Metabolite Detection by GC-MS
The metabolites of HNE, HNA, OTHFPA, 2-HG, and GHB were characterized and quantified by GC-MS. Samples were deproteinized by acetonitrile, and the dried samples were derivatized by 60 μl of N,O-bis(trimethylsilyl) trifluoroacetamide at 70 °C for 1 h. After derivatization, 1 μl of each sample was analyzed by GC-MS using an Agilent 6890 gas chromatograph and Agilent 5973 mass spectrometer equipped with an Agilent VF-5MS capillary column (53 m × 0.25 mm × 0.25 μm). The injection was in splitless mode. The temperature for both inlet and transfer line was set at 290 and 300 °C, respectively. The ion source and quadrupole temperature were set at 230 and 150 °C. The GC temperature program varied, depending on analytes. All of the masses were measured in electron impact (EI) ionization.
Statistical Analysis
The criterion for significance was set at p < 0.05. Statistical differences were tested using a paired Student's t test (GraphPad Prism software, version 3). The logistic regression fit for inhibitor-dose response and IC50 calculation were performed by Origin version 9.1.
RESULTS
Identification of OTHFPA-CoA
To investigate the metabolism of HNA/HNE, rat livers were perfused with or without labeled and unlabeled HNA/HNE, and acyl-CoAs in the perfused livers were profiled by LC-MS/MS. Results showed that the most abundant acyl-CoA was not 4-hydroxynonanoyl-CoA or acetyl-CoA (the highest acyl-CoA in control rat liver) but an unknown acyl-CoA with m/z at 908 (Fig. 1, A and B), which presents in trace amounts in control rat livers. This unknown acyl-CoA (m/z 908) appeared 16 mass units lower than 4-hydroxynonanoyl-CoA (m/z 924) and eluted earlier than 4-hydroxynonanoyl-CoA in the reversed phase column (C18 column). The early elution time of this unknown acyl-CoA derived from HNA suggests a higher polarity compared with HNA-CoA. Additionally, the m/z of this unknown acyl-CoA shifted to 909 and 910 when perfusions contained [3-13C]HNA and [3,4-13C2]HNA, respectively (Fig. 1, C and D). The presence of 13C from C-3 and C-4 of HNA indicated that this unknown acyl-CoA contains C-3 and C-4 of HNA. Multiple mass isotopomers of this unknown acyl-CoA with m/z from 908 to 913 were found in livers perfused with [5,5,6,6,7,7,8,8,9,9,9-2H11]HNE (Fig. 1E). The loss of 6–11 deuteriums in this unknown acyl-CoA from [5,5,6,6,7,7,8,8,9,9,9-2H11]HNE strongly suggested the loss of carbons from the distal, or ω-, end of HNE (C-9). According to the above observations, two possible chemical structures of this unknown acyl-CoA derived from HNE metabolism were postulated: (i) 4,7-diketoheptanoyl-CoA or (ii) OTHFPA-CoA (see Fig. 1A for structural information). 4,7-Diketoheptanoyl-CoA is very unlikely because (i) diketones are inherently unstable under the given analytical conditions and thus would not be expected to be detectable at an appreciable concentration, and (ii) a relatively small fraction of 4-hydroxynonanoyl-CoA was oxidized to 4-ketononanoyl-CoA under the same redox conditions; this excludes the possibility of accumulating 4,7-diketoheptanoyl-CoA without detecting a significant amount of dihydroxy forms. The identity of OTHFPA-CoA was also supported by the following lines of evidence: (i) fragmentation of m/z 908 by tandem mass spectrometry demonstrates that it is acyl-CoA by the fragmentation patterns of acyl-CoA in mass spectrometry (fragments at m/z 261 and 428 and neutral loss of 507); (ii) the chemical formula of OTHFPA-CoA was confirmed by accurate mass measurement; and (iii) livers perfused with an OTHFPA standard (described below) confirmed OTHFPA-CoA. An unknown metabolite of HNE/HNA was identified as OTHFPA-CoA according to the metabolomics study with the combination of isotopomer analysis.
FIGURE 1.

LC-MS/MS chromatogram and mass spectra of acyl-CoAs in rat livers perfused with control buffer or labeled and unlabeled HNA/HNE. A, overlay of LC-MS/MS chromatogram for acyl-CoA profile from control rat liver and rat liver perfused with 2 mm HNA. B–E, mass spectra of an unknown acyl-CoA from rat livers perfused with unlabeled 2 mm HNA, [3-13C]HNA, [3,4-13C2]HNA, and 1 mm [5,5,6,6,7,7,8,8,9,9,9-2H11]HNE.
Rat Liver Perfused with M0 and M1 OTHFPA
To further confirm the identity of OTHFPA-CoA found in the rat liver perfused with HNA or HNE and its downstream metabolite intermediates, rat livers were perfused with 1 mm unlabeled or M1 OTHFPA. Acyl-CoAs in the perfused rat livers were profiled by LC-MS/MS. In the rat livers perfused with labeled and unlabeled OTHFPA, OTHFPA-CoA was confirmed by m/z 908 as unlabeled OTHFPA and m/z 909 for M1 OTHFPA (data not shown). The retention time of OTHFPA-CoA was consistent with what was found in the HNA/HNE-perfused rat livers. In the perfusion with M1 OTHFPA, 2-HG and GHB in the perfusate were labeled by M1 (Fig. 2, A and B). We postulated that M1 2-HG is the probable metabolite of M1 OTHFPA-CoA after the lactone is hydrolyzed and following one cycle of β-oxidation. The conversion of M1 2-HG to 2-ketoglutaric acid (2-KG) is coupled to deuterium transfer from M1 2-HG to succinic semialdehyde (SSA), which is reduced to M1 GHB (Fig. 2C). GHB was accumulated in the perfusates from rat livers perfused with both HNA and OTHFPA. GHB was not labeled from the perfusion with [3,4-13C2]HNA, although 2-HG was M2-labeled in the perfusion with [3,4-13C2]HNA. Clearly, GHB production is coupled with the oxidation of 2-HG to 2-KG (35). In this experiment, OTHFPA-CoA catabolized from HNA/HNE was confirmed by perfusing rat livers with labeled and unlabeled OTHFPA. OTHFPA-CoA is further metabolized to 2-HG via β-oxidation and to GHB.
FIGURE 2.

The metabolism of OTHFPA to 2-HG and GHB. A and B, labeling of 2-HG and GHB from the metabolism of M1 2-HG and M1 OTHFPA. C, schematic diagram of metabolism of OTHFPA to M1 2-HG and M1 GHB. D, labeling of 2-HG from the metabolism of [2H6]GHB. E, coupled reaction of 2-KG to 2-HG with GHB to SSA by hydrogen transfer. Error bars, S.D. of measurements (n = 5).
Rat Liver Perfused with M0 and M1 2-HG
To verify the hydrogen transfer from 2-HG to GHB after OTHFPA is degraded to 2-HG, rat livers were perfused with labeled and unlabeled 2-HG. Indeed, M1 GHB was enriched in the perfusate of M1 2-HG (Fig. 2B). This is direct evidence that hydrogen is transferred from 2-HG to SSA to form GHB (Fig. 2C).
Rat Liver Perfused with [2H6]GHB
To investigate the reversibility of the coupled reactions between GHB/SSA and 2-HG/2-KG, rat livers were perfused with 2 mm [2H6]GHB, and the enrichment of M1 2-HG in the perfusate was 90% (see Fig. 2D). The coupled reactions between GHB/SSA and 2-HG/2-KG were reversible and were outlined in Fig. 2, C and E.
HNA ω-Oxidation in the Perfused Heart
ω-Oxidation of HNA was also investigated in the perfused rat heart. As was expected, HNA was activated to HNA-CoA, and metabolites of two parallel catabolic pathways of HNA were also observed in the perfused hearts, such as 4-phosphononanoyl-CoA and 2-hydroxyheptanoyl-CoA. However, no ω-oxidation product of HNA was found in the perfused rat hearts.
ω-Oxidation of HNA following Acute Exposure in the Whole Animal
Because acyl-CoAs are intracellular metabolites, the assay of acyl-CoAs from different organs would reflect organ-specific metabolic status. To investigate HNA metabolism in different organs, rats received 100 mg/kg HNA by intraperitoneal injection. After 30 min, rats were anesthetized, and brain, kidney, liver, and heart were quickly dissected and frozen in liquid nitrogen. Acyl-CoAs in rat brain, kidney, liver, and heart were profiled by LC-MS/MS. The results are shown in Fig. 3. HNA-CoA, 4-phosphononanoyl-CoA, and propionyl-CoA were found in all organs assayed (Fig. 3, B–D). However, the ω-oxidation metabolite, OTHFPA-CoA (Fig. 3A), was undetectable in brain and heart. In vivo experiments again confirmed that rat heart and brain display little ω-oxidation activity toward HNA (Fig. 3A).
FIGURE 3.
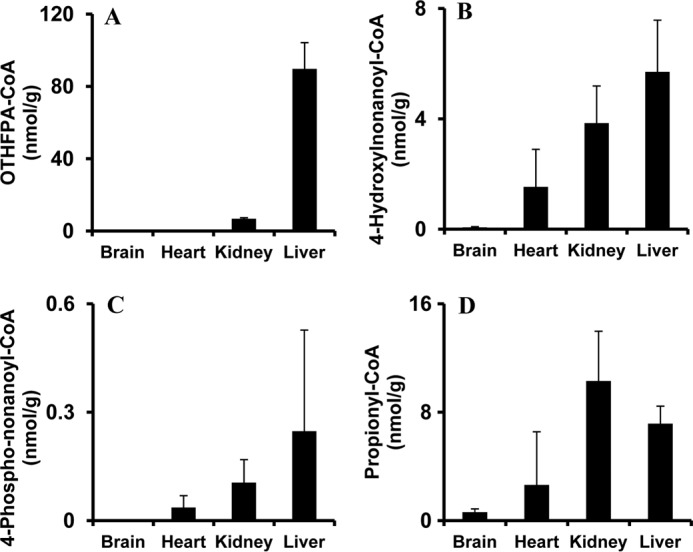
Acyl-CoAs assayed in brains, hearts, kidneys, and livers of rats treated with HNA. Five rats received an intraperitoneal injection of 100 mg/kg HNA. Rats were terminated after 30 min, and organs (brains, hearts, kidneys, and livers) were quickly dissected and snap-frozen in liquid nitrogen for acyl-CoA analysis. A–D, OTHFPA-CoA, 4-hydroxynonanoyl-CoA, 4-phosphononanoyl-CoA, and propionyl-CoA detected in rat brain, heart, kidney, and liver. Error bars, S.D. of measurements (n = 5).
ω- and ω-1-Hydroxylation Metabolites of HNA in Rat Liver Homogenate
To test the ω-hydroxylation activity toward HNA/HNE in rat liver, in vitro incubation of HNA with liver homogenate was conducted. Five milligrams of rat liver homogenate was incubated with 2 mm HNA or [3,4-13C2]HNA and NADH or NADPH in 100 mm phosphate buffer for 90 min. Samples taken at 0 and 90 min were subsequently assayed by GC-MS. As shown in Fig. 4A, two new peaks (at 25.4 and 27 min, respectively) appeared in the 90-min sample with the addition of NADPH (NADH did not facilitate the production of these metabolites) (data not shown). EI mass spectra of both peaks demonstrated the same molecular weight and very similar fragmentation patterns. The m/z 391 is the derivative of hydroxylated HNA with 3 trimethylsilyl (TMS) groups minus 15 atomic mass units (accounted for by the loss of one CH3). When [3,4-13C2]HNA was included in the incubation, m/z of both metabolites had the same mass shift, suggesting that both metabolites are the hydroxylation products of HNA (Fig. 4, B–E). Based on GC-MS data and literature (36), the metabolite eluting at 27 min is 4,9-DHNA (ω-hydroxylation product), and the metabolite at 25.4 min is 4,8-DHNA (ω-1-hydroxylation product). The identification of both metabolites was based on the following lines of evidence: (i) 4,8-DHNA is predicted to have a shorter retention time than 4,9-DHNA because carboxylic acids with the hydroxyl group further from the C-1 carboxylic group have a longer retention time in GC-MS based on our previous work (37); (ii) m/z 103 is the typical fragment of TMS derivative of an ω-hydroxylated fatty acid that would not be observed with a ω-1-hydroxylated fatty acid (Fig. 4, D and E) (37); (iii) 4,8-DHNA, which has two chiral carbons (C-4 and C-8), shows unresolved double peaks in the extended GC-MS chromatogram (Fig. 4A, inset). Only one peak was observed for 4,9-DHNA because enantiomers with only one chiral center are not separated on non-chiral capillary GC columns. An in vitro experiment confirmed the NADPH-dependent ω-/ω-1-hydroxylation of HNA and their corresponding metabolites in rat liver.
FIGURE 4.

Metabolites profiled by GC-MS before and after rat liver homogenates were incubated with labeled and unlabeled HNA. A, overlay of GC-MS chromatograms from liver homogenates incubated with 1 mm HNA in the presence of 1 mm NADPH at 0 and 90 min. The inset is an extended GC-MS chromatogram (long running method with better separation) of peak 1. B and C, EI mass spectra of peak I (4,8-DHNA, ω-1-hydroxylation product) when liver homogenates were incubated with unlabeled HNA and [3,4-13C2]HNA. D and E, EI mass spectra of peak II (4,9-DHNA, ω-hydroxylation product) when liver homogenates were incubated with unlabeled HNA and [3,4-13C2]HNA.
Additional Metabolites of HNA ω- and ω-1-Oxidation
All of the above experimental data clearly suggested further catabolic pathways of 4,9-DHNA: (i) 4,9-DHNA is oxidized and activated to 6-hydroxynonenodioyl-CoA; (ii) 6-hydroxynonenodioyl-CoA is catabolized to 4-hydroxyheptanedioyl-CoA that can be quickly equilibrated to lactone form (OTHFPA-CoA); (iii) the further catabolism of 4-hydroxyheptanedioyl-CoA is to generate 2-hydroxyglutaryl-CoA, which is hydrolyzed to form 2-HG; (iv) 2-HG enters the citric acid cycle via the oxidation to 2-KG, and this oxidation is coupled by the reduction of SSA to GHB.
Two parallel catabolic pathways of 4,8-DHNA were identified in the perfused rat liver from the acyl-CoA profile by LC-MS/MS. Catabolic pathway A of 4,8-DHNA is the following route: 4,8-DHNA-CoA → 4-phospho-8-hydroxynonanoyl-CoA → 3,8-dihydroxynonanoyl-CoA → 6-hydroxyheptanoyl-CoA → 4-hydroxypentanoyl-CoA → acetyl-CoA and propionyl-CoA that enter the citric acid cycle. Catabolic pathway B of 4,8-DHNA follows the following steps: 4,8-DHNA-CoA → 2,6-dihydroxyheptanoyl-CoA → 5-hydroxyhexanoyl-CoA → 3-hydroxybutyryl-CoA → acetyl-CoA that enters the citric acid cycle. The current proposed catabolic pathways of HNA/HNE are depicted in a diagram in Fig. 5. The metabolites involved in various pathways were assayed in rat livers perfused with M2 HNA, and the concentrations and labeling of metabolites from ω- and ω-1-oxidation were not detectable in the perfused hearts with M2 HNA (Table 2).
FIGURE 5.

Identified catabolic pathways of HNE/HNA via ω- and ω-1-oxidations in the perfused rat livers. Compounds 1–13 are as follows: HNE (1), HNA (2), 4,8-DHNA (3), 4,9-DHNA (4), 6-hydroxynonenodioyl-CoA (5), 4-hydroxyheptanedioyl-CoA (6), 2-HG (7), 4-phospho-8-hydroxynonanoyl-CoA (8), 3,8-dihydroxynonanoyl-CoA (9), 6-hydroxyheptanoyl-CoA (10), 4-hydroxypentanoyl-CoA (11), 2,6-dihydroxyheptanoyl-CoA (12), 5-hydroxyhexanoyl-CoA (13).
TABLE 2.
Concentrations of acyl-CoAs catabolized from HNA via ω- and ω-1-oxidation from rat livers and hearts perfused with [3,4-13C2]HNA
| Acyl-CoAs | m/z | Liver | Heart | ω-/ω-1-Metabolites |
|---|---|---|---|---|
| nmol/g | nmol/g | |||
| HNA-CoA | 926 (M2) | 15.0 ± 5.3 | 7.6 ± 2.4 | |
| 4,9-Dihydroxynonanoyl-CoA | 942 (M2) | 0.5 ± 0.2 | ω | |
| 4,8-Dihydroxynonanoyl-CoA | 942 (M2) | 0.1 ± 0.05 | ω-1 | |
| 8-OH-4-phospho-nonanoyl-CoA | 1022 (M2) | 0.2 ± 0.05 | ω-1 | |
| 4-OH-nonenodioyl-CoA (lactone) | 938 (M2) | 0.5 ± 0.1 | ω | |
| OTHFPA-CoA | 910 (M2) | 118.0 ± 25.8 | 0.09 ± 0.02 | ω |
| 5-OH-hexanoyl-CoA | 883 (M1) | 0.5 ± 0.1 | ω-1 | |
| 6-OH-heptanoyl-CoA | 898 (M2) | 0.4 ± 0.2 | ω-1 | |
| 4-OH-pentanoyl-CoA | 868 (M0) | 1.8 ± 0.49 | ω-1 | |
| 4-Phosphopentanoyl-CoA | 948 (M0) | 0.5 ± 0.2 | ω-1 |
HNA ω- and ω-1-Oxidation Activities in Other Organs
The discovery of ω-oxidation metabolites in liver but not heart using whole organ perfusion models coupled with the identification of ω- and ω-1-hydroxylated HNA in the rat liver homogenate compelled us to investigate the ω- and ω-1-hydroxylation activities of HNA in various tissue homogenates (brain, heart, kidney, and liver; see Fig. 6). Comparable with the results of ω- oxidation metabolites found in heart and liver perfusions, heart homogenates possessed little ω- and ω-1-hydroxylation of HNA, with similar trends in the brain. Kidney and liver homogenates had similar activities of ω-oxidation (3.7–4 nmol/(g·min)) and ω-1-oxidation (1.2–1.5 nmol/(g·min)) (Fig. 6, A and B). The total production of both 4,8-DHNA and 4,9-DHNA was shown in Fig. 6C. The ratio of ω- hydroxylation/ω-1-hydroxylation was 2.3 ± 0.4 in rat liver homogenate and 3.2 ± 0.8 in kidney homogenate (Fig. 6D).
FIGURE 6.
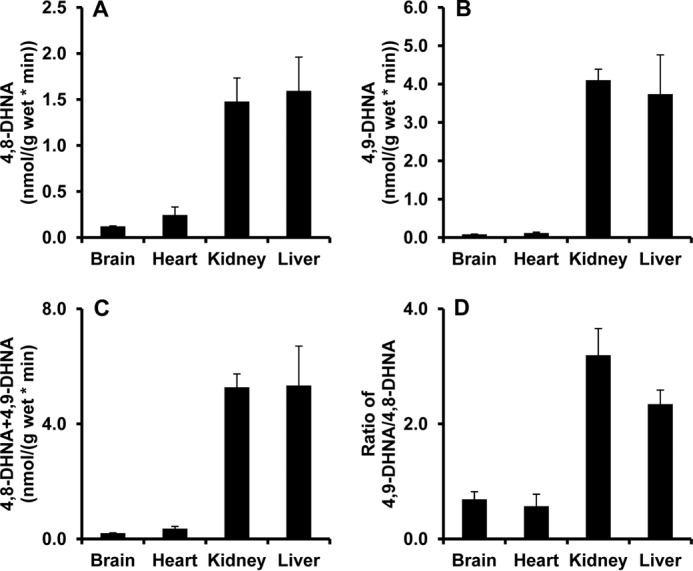
4,8-DHNA and 4,9-DHNA production in rat brain, heart, kidney, and liver homogenates incubated with HNA via ω-1- and ω-hydroxylation. A and B, 4,8-DHNA and 4,9-DHNA productions from HNA incubated in rat brain, heart, kidney, and liver homogenates. C, total amount of 4,8-DHNA and 4,9-DHNA production. D, ratio of 4,9-DHNA/4,8-DHNA. Rat organ homogenates were incubated with 1 mm HNA, 1 mm NADPH, and 100 mm phosphate buffer at 37 °C for 30 min. ω- and ω-1-hydroxylation products were assayed by GC-MS. Error bar, S.D. of measurements (n = 5).
The Impact of Diet on Hepatic ω- and ω-1-Hydroxylation Activity
Our previous work investigated the HNE level in livers from rats fed diets with varying fat content (SD, LF, KG, and HF-HC diets). Livers taken from KG rats had the lowest level of HNE compared with all other diet treatments. In light of our newly identified ω- and ω-1-oxidation pathways of HNE, it was of great interest to define how KG alters the catabolism of HNE. HNA at 1 mm was incubated with 40 μl of liver homogenate containing 5 mg of wet liver tissue with the presence of 100 mm phosphate buffer (pH 7.4) and 1 mm NADPH. The results are shown in Fig. 7, A and B. ω- and ω-1-Hydroxylation activities in KG liver samples were increased by 7- and 2-fold compared with SD, respectively. Neither LF nor HF-HC had a significant influence on the activity of ω- and ω-1-hydroxylation compared with SD.
FIGURE 7.
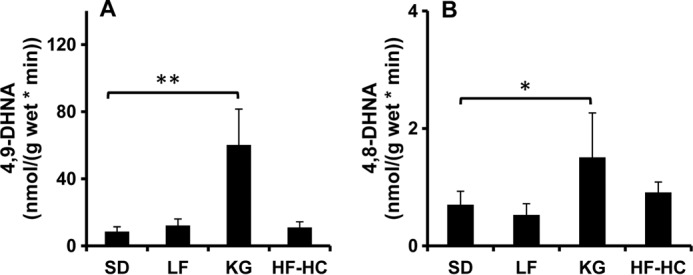
Hepatic ω- and ω-1-hydroxylation activities of rats fed diets with varying lipid and carbohydrate content. Rats were fed with four different diets: SD, LF, KG, and HF-HC. Liver homogenate was incubated with 1 mm HNA, 1 mm NADPH, and 100 mm phosphate buffer at 37 °C for 30 min. ω- and ω-1-hydroxylation products were assayed by GC-MS. A and B, ω- and ω-1-hydroxylation activity in rat livers. **, p < 0.001; *, 0.01 < p < 0.05. Error bars, S.D. of measurements (n = 5).
Inhibition by Miconazole and HET0016
Miconazole is the known inhibitor of many CYP enzymes. And HET0016 is a specific inhibitor of CYP4A and CYP4F enzymes. To elucidate which enzymes have hydroxylation activities toward HNA, various doses of miconazole and HET0016 were tested for the inhibition of HNA ω-hydroxylation. Miconazole does not inhibit the HNA ω-hydroxylation, even in the millimolar range (data not shown). HET0016 efficiently inhibits HNA ω-hydroxylation at low concentration. The dose response of inhibition is shown in Fig. 8. The half-maximal inhibitory concentration (IC50) of HET0016 is 214 nm. Inhibition experiments suggested that CYP4A or CYP4F is the enzyme catalyzing the ω-/ω-1-hydroxylation of HNA/HNE.
FIGURE 8.
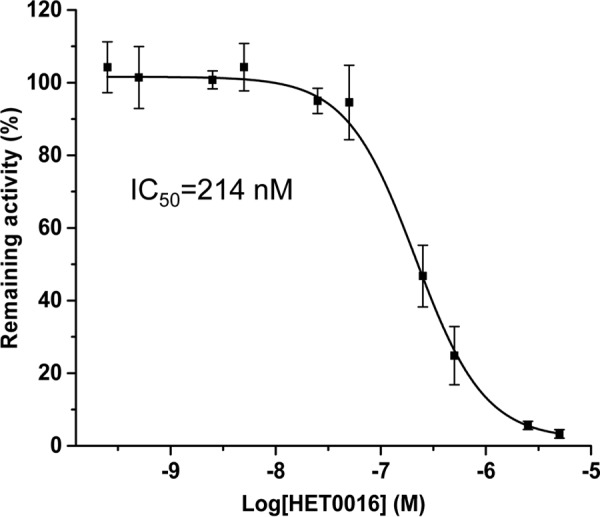
The inhibition of HNA ω-hydroxylation by the CYP inhibitor, HET0016. Liver homogenates from rats fed 4 weeks of a KG diet were incubated with 1 mm HNA, 1 mm NADPH, and 100 mm phosphate buffer at 37 °C for 30 min, and various amounts of HET0016 (n = 5 at each concentration of HET0016) were added in the incubation mixtures. Error bars, S.D. of measurements (n = 5).
The Impact of Diet on CYP4A and CYP4F Gene Expression
To further characterize CYP, which catalyzes the ω-/ω-1-hydroxylation of HNE/HNA and is modulated by diets, the expression of CYP4A and CYP4F mRNA in livers from rat fed with various diets was quantified by quantitative PCR. The results are shown in Figs. 9 and 10. The expressions of Cyp4a1, Cyp4a2, and Cyp4a3 were significantly increased, although that of Cyp4a8 was significantly down-regulated (Fig. 9). No influence of diet was determined for the expression of Cyp4f1, Cyp4f4, and Cyp4f5. KG trended toward a modest effect on expression of Cyp4f6, and its relative expression was low in liver tissue. The data of mRNA expression clearly showed that CYP4A but not CYP4F involved the ω-/ω-1-hydroxylation of HNE/HNA.
FIGURE 9.
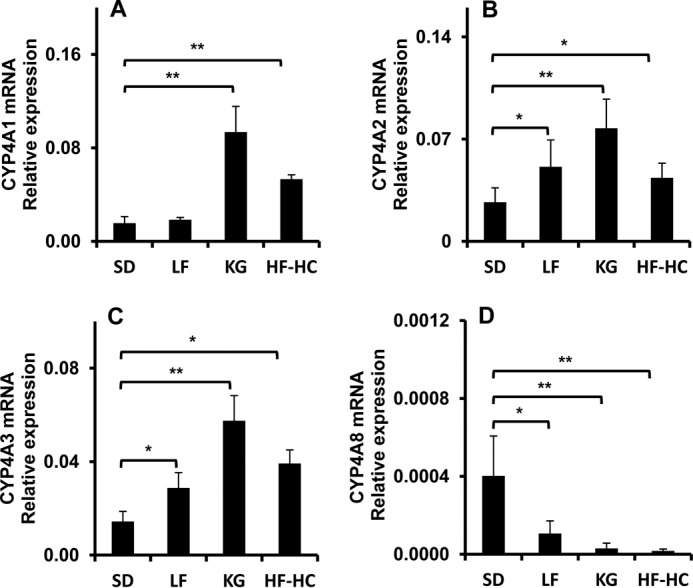
CYP4A mRNA expression in livers from rats fed with various diets. A–D, mRNA expression levels of CYP4A1, CYP4A2, CYP4A3, and CYP4A8 in livers from rat fed with SD, LF, KG, and HF-HC diets. **, p < 0.001; *, 0.01 < p < 0.05. Error bars, S.D. of measurements (n = 5).
FIGURE 10.
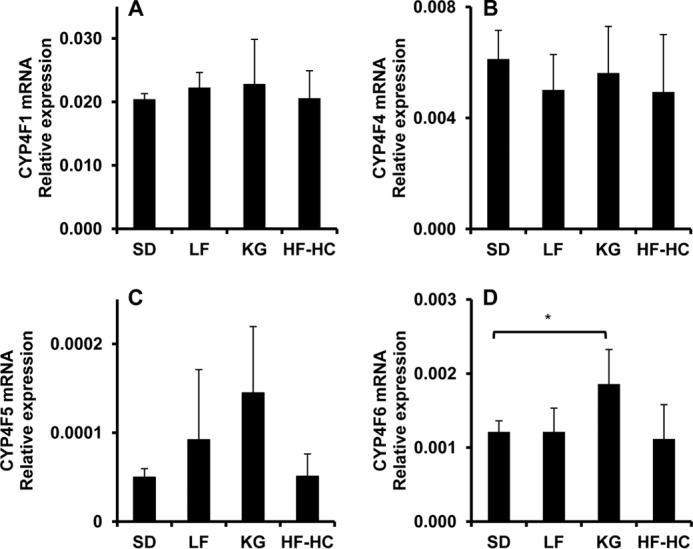
CYP4F mRNA expression in livers from rat fed with various diets. A–D, mRNA expression levels of CYP4F1, CYP4F4, CYP4F5, and CYP4F6 in livers from rats fed with SD, LF, KG, and HF-HC diets. **, p < 0.001; *, 0.01 < p < 0.05. Error bar, S.D. of measurements (n = 5).
DISCUSSION
HNE is the major lipid peroxidation product of ω-6-polyunsaturated fatty acids. The biological properties of protein, DNA, and lipid are altered by HNE modification, which exemplifies the essential role of HNE metabolism in its disposal. Our recent work has shown that conjugation with glutathione, previously believed to be the major route of HNE disposal, constitutes only 8.7% of total [5,5,6,6,7,7,8,8,9,9,9-2H11]HNE taken up by perfused rat liver (20). In addition, very little DHN was detected from [5,5,6,6,7,7,8,8,9,9,9-2H11]HNE reduction in liver perfusion experiments. These results imply the presence of other metabolic pathways that contribute to HNE disposal. We have thoroughly investigated the catabolic pathways of HNE using labeled and unlabeled HNE and HNA. In the HNA perfused rat liver, the highest abundance acyl-CoA is OTHFPA-CoA, which was identified and confirmed by isotope labeling, fragmentation, and retention time. Experiments in which livers were perfused with OTHFPA also confirm the identity of OTHFPA-CoA. Based on the weight of evidence, we concluded that OTHFPA-CoA is the metabolite of HNE/HNA ω- oxidation.
The first step of ω-oxidation is ω-hydroxylation presumably catalyzed by CYP family enzymes. The ω-hydroxylation of HNA was confirmed by incubating labeled and unlabeled HNA with rat liver homogenate. In addition to 4,9-DHNA derived from ω-hydroxylation, 4,8-DHNA from ω-1-hydroxylation was also identified (see Fig. 4). The identities of 4,9-DHNA and 4,8-DHNA as products of ω- and ω-1-hydroxylation of HNA were collectively deduced from the following evidence. (i) No 4,9-DHNA or 4,8-DHNA was found at the baseline control (0 min). (ii) Both incubation metabolites from M0 HNA display an m/z 391 fragment, which is shifted to m/z 393 when M2 HNA is added in place of M0 HNA. (iii) Sanders et al. (36) have reported the fragmentation patterns of the ω- and ω-1-hydroxylated C26:0 fatty acid-TMS derivative, and the ω-hydroxylated C26:0-TMS derivative has the typical fragment at m/z 103 ((CH3)3Si-O-CH2). Also, the ω-1-hydroxylated C26:0-TMS derivative has a highly abundant fragment at m/z 117 ((CH3)3Si-O-CH-CH3). In the mass spectra of two peaks found in our HNA incubation (Fig. 4), peak 1 is an ω-1-hydroxylation product due to the abundant m/z 117, and peak 2 is an ω-hydroxylation product as evidenced by the presence of the m/z 103 fragment. (iv) Another piece of evidence for the identities of two peaks is from the chirality of the two dihydroxynonanoic acids. Racemic compounds with one chiral center cannot be separated by a non-chiral GC column; however, a non-chiral GC column is able to separate racemic compounds with two chiral centers under proper separation conditions. 4,9-DHNA only has one chiral carbon at C-4, whereas 4,8-DHNA has two chiral centers at C-4 and C-8, respectively. When a GC method with a longer temperature gradient is applied, 4,8-DHNA (peak 1) containing two peaks starts partially separated (see Fig. 4A, inset).
4,8-DHNA and 4,9-DHNA were further catabolized in their specific pathways. 4,9-DHNA is obviously oxidized and activated to 6-hydroxynonenodioyl-CoA (C-9 of HNA is oxidized to carboxylic acid and activated to CoA) and degraded to OTHFPA-CoA after one cycle of β-oxidation. M1 OTHFPA and M1 2-HG experiments showed that OTHFPA is degraded to 2-HG that is converted to 2-KG by d-2-hydroxyglutarate transhydrogenase (see Fig. 2). Hydrogen transfer between 2-HG/2-KG and GHB/SSA was confirmed by rat liver perfusion with [2H6]GHB, and M1 2-HG was labeled by 90% from [2H6]GHB. Hydrogen transfer between 2-HG and GHB by transhydrogenase was also reported by Struys et al. (35).
The catabolism of 4,8-DHNA follows two parallel catabolic pathways of 4-hydroxy acids reported by us previously (2). One route of 4,8-DHNA catabolism involves isomerization to 3,8-DHNA-CoA that is subjected to two cycles of β-oxidation and conversion to 4-hydroxypentanoyl-CoA. Further metabolism of 4-hydroxypentanoyl-CoA to acetyl-CoA and propionyl-CoA was characterized in our previous reports (1, 23). GHP is known as a drug of abuse and metabolite from levulinic acid that is used as a food additive and calcium carrier. This work is the first to report an endogenous source of GHP. In the second route of 4,8-DHNA catabolism, 4,8-DHNA-CoA is degraded to 2,6-dihydroxyheptanoyl-CoA via one cycle of β-oxidation. 2,6-Dihydroxyheptanoyl-CoA loses one carbon by α-oxidation to form 5-hydroxyhexanoyl-CoA that is completely oxidized to acetyl-CoA by two cycles of β-oxidation.
The formation of 4,9-DHNA and 4,8-DHNA via ω- and ω-1-hydroxylation is the first step of catabolic pathways characterized in the present work. The characterizations of ω- and ω-1-hydroxylation in rat organs and enzymes were attempted. ω- and ω-1-hydroxylation activities in rat organs (brain, heart, kidney, and liver) were compared (Fig. 6). Kidney and liver possess higher ω- and ω-1-hydroxylation activities toward HNA compared with brain and heart. Different ω- and ω-1-hydroxylation activities in various organs were also confirmed by in vivo experiments. The result in Fig. 3 matched the finding of in vitro incubation experiments on HNA hydroxylation activity, and confirmed that the brain and heart have little ω- or ω-1-hydroxylation activity toward HNA. Lack of ω- or ω-1-oxidation activities in the brain and heart might leave these organs vulnerable to HNE accumulation and damage, particularly for the brain because it has low β-oxidation activity that is important to HNE catabolism (2). Indeed, studies from Alzheimer disease patients reported the accumulation of HNE adducts as a component of disease etiology (38).
In our previous report (2, 21), we identified two parallel catabolic pathways of HNE in the perfused rat livers. These degradation pathways overlap in that a majority of the reactions involve β-oxidation. Therefore, modulation of β-oxidation could regulate HNE catabolic disposal (3). Our work with altered dietary lipid supports this possibility because the decreased β-oxidation activity and increased lipid synthesis induced with HF-HC (Western diet) is accompanied by a decrease in efficiency of HNE disposal and results in hepatic HNE accumulation. The converse is true with KG rat liver wherein basal HNE levels were the lowest. The finding of catabolic disposal of HNE through ω- and ω-1-oxidation drove us to investigate how diets regulate ω- and ω-1-hydroxylation activity. Fig. 7 shows that the KG diet, but not other diets, significantly induces ω- and ω-1-hydroxylation activity in liver. KG is well known to induce the expression of β-oxidation genes, but the additional findings of this study broaden the network of reaction pathways related to fatty acid breakdown that may contribute to the catabolism of HNE to a recycled lipid chain re-entering energy metabolism. The dramatic increase of ω- and ω-1-hydroxylation of HNE in KG liver correlates well with observations of lower [HNE] compared with the other diets. HF-HC diet (Western diet) induces obesity and diabetes (39). KG diet with extremely high fat composition has been used worldwide for refractory childhood epilepsy (40). The KG diet recently has also been reported to improve diabetes and malignant glioma (41, 42), although the long term compliance of the KG diet is problematic. The present work provides another mechanism to explain these different impacts of Western diet and KG diet on healthy outcomes based upon their converse contribution to HNE catabolism.
Enzymes that catalyze ω- and ω-1-hydroxylation of HNE/HNA are part of the CYP family, and isoforms CYP4A and CYP4F appear to be most directly involved based on drug inhibition and mRNA assay experiments. Miconazole is a general inhibitor of CYP enzymes but not CYP4A or CYP4F enzymes. Miconazole did not show any inhibition of ω- or ω-1-hydroxylation of HNA at various concentrations (data not shown). CYP4A or CYP4F is known to hydrolyze fatty acid at ω- and ω-1-carbons. HET0016 is a specific inhibitor of CYP4A or CYP4F enzymes. Both ω- and ω-1-hydroxylations of HNA were inhibited by HET0016. The reported IC50 of HET0016 is ∼10–40 nm using arachidonic acid as a substrate. The high IC50 value found in this work is probably due to substrate affinity (HNA versus arachidonic acid) and liver homogenate used in the present work. The inhibition experiments using these two inhibitors implicate CYP4A or CYP4F in the catalysis of ω- and ω-1-hydroxylation of HNA. The mRNA expression in livers from rats fed with various diets demonstrated an induction of Cyp4a but not Cyp4f, providing additional evidence for the role of these enzymes in the ω- and ω-1-hydroxylation of HNA. This is the first report to show that CYP4A but not CYP4A8 is induced by KG. The up-regulated CYP4A1, CYP4A2, and CYP4A3 are probably due, in part, to PPAR activation by fatty acids in the KG diet. The differential expression of CYP4A and CYP4F by the KG diet remains to be investigated further. ω- or ω-1-oxidation of HNA or fatty acids is one of the mechanisms to degrade the excess amount of HNE or fatty acids. Induction of CYP4A probably increases ω-hydroxylation of arachidonic acid to generate 20-hydroxy-(5Z,8Z,11Z,14Z)-eicosatetraenoic acid that is proinflammatory and might contribute to oxidative stress. 20-Hydroxy-(5Z,8Z,11Z,14Z)-eicosatetraenoic acid production influenced by diets is beyond the scope of this work. The effect of decreased CYP4A8 expression by KG is an interesting observation, but the underlying mechanism requires further investigation.
Compelling evidence shows that ω-/omega-1-hydroxylation of fatty acids is in microsomes (43–46); probably, it is the same for the ω-/ω-1-hydroxylation of HNE/HNA. The further degradation via β-oxidation and α-oxidation may proceed in peroxisome and mitochondria, as we observed in our previous study (2).
In summary, three catabolic pathways of HNE/HNA via ω- and ω-1-oxidation were characterized with a combination of metabolomics and isotopic analysis in the present work. The newly identified catabolism of HNE/HNA via ω-1- and ω-oxidation is active in rat liver and kidney but not in the brain or heart. NADPH-dependent ω- and ω-1-hydroxylation is probably catalyzed by CYP4A enzymes (probably not CYP4A8) that are highly up-regulated by the KG diet in rat liver.
This work was supported, in whole or in part, by National Institutes of Health (NIH) Roadmap Grants R33DK070291 and R01ES013925 (to Henri Brunengraber) and by NIH, NCI, Grant CA157735 (to G. P. T.). This work was also supported by Case Western Reserve University/Cleveland Clinic CTSA Grant UL1 RR024989 (to G. F. Z.) from the National Center for Research Resources (NCRR), a component of the NIH and NIH Roadmap for Medical Research. This work was also supported by National Science Foundation Grant MCB-084480 (to G. P. T.), American Heart Association Grant 12GRNT12050453 (to G. F. Z.), and a grant from the Cleveland Mt. Sinai Health Care Foundation.
Mass isotopomers are designated as M0, M1, M2, … Mn, where n is the number of heavy atoms in the molecule.
- HNE
- (2E)-4-hydroxy-2-nonenal
- SD
- standard diet
- LF
- low fat
- KG
- ketogenic
- HF-HC
- high fat and high carbohydrate
- DHN
- 1,4-dihydroxy-2-nonene
- HNA
- 4-hydroxynonanoic acid
- GHB
- 4-hydroxybutyric acid
- [2H6]GHB
- [2,2,3,3,4,4-2H6]4-hydroxybutyric acid
- GHP
- 4-hydroxypentanoic acid
- 4,9-DHNA
- 4,9-dihydroxynonanoic acid
- 4,8-DHNA
- 4,8-dihydroxynonanoic acid
- EI
- electron impact
- 2-KG
- 2-ketoglutaric acid
- 2-HG
- 2-hydroxyglutaric acid
- OTHFPA
- 3-(5-oxotetrahydrofuran-2-yl)propanoic acid
- SSA
- succinic semialdehyde
- TMS
- trimethylsilyl.
REFERENCES
- 1. Harris S. R., Zhang G. F., Sadhukhan S., Murphy A. M., Tomcik K. A., Vazquez E. J., Anderson V. E., Tochtrop G. P., Brunengraber H. (2011) Metabolism of levulinate in perfused rat livers and live rats: conversion to the drug of abuse 4-hydroxypentanoate. J. Biol. Chem. 286, 5895–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang G. F., Kombu R. S., Kasumov T., Han Y., Sadhukhan S., Zhang J., Sayre L. M., Ray D., Gibson K. M., Anderson V. A., Tochtrop G. P., Brunengraber H. (2009) Catabolism of 4-hydroxyacids and 4-hydroxynonenal via 4-hydroxy-4-phosphoacyl-CoAs. J. Biol. Chem. 284, 33521–33534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li Q., Tomcik K., Zhang S., Puchowicz M. A., Zhang G. F. (2012) Dietary regulation of catabolic disposal of 4-hydroxynonenal analogs in rat liver. Free Radic. Biol. Med. 52, 1043–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Q., Sadhukhan S., Berthiaume J. M., Ibarra R. A., Tang H., Deng S., Hamilton E., Nagy L. E., Tochtrop G. P., Zhang G. F. (2013) 4-Hydroxy-2(E)-nonenal (HNE) catabolism and formation of HNE adducts are modulated by β oxidation of fatty acids in the isolated rat heart. Free Radic. Biol. Med. 58, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okamoto K., Toyokuni S., Uchida K., Ogawa O., Takenewa J., Kakehi Y., Kinoshita H., Hattori-Nakakuki Y., Hiai H., Yoshida O. (1994) Formation of 8-hydroxy-2′-deoxyguanosine and 4-hydroxy-2-nonenal-modified proteins in human renal-cell carcinoma. Int. J. Cancer 58, 825–829 [DOI] [PubMed] [Google Scholar]
- 6. Akude E., Zherebitskaya E., Roy Chowdhury S. K., Girling K., Fernyhough P. (2010) 4-Hydroxy-2-nonenal induces mitochondrial dysfunction and aberrant axonal outgrowth in adult sensory neurons that mimics features of diabetic neuropathy. Neurotox. Res. 17, 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mali V. R., Ning R., Chen J., Yang X. P., Xu J., Palaniyandi S. S. (2014) Impairment of aldehyde dehydrogenase-2 by 4-hydroxy-2-nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high-fat diet and injected with low-dose streptozotocin. Exp. Biol. Med. 239, 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mattson M. P. (2009) Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 44, 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang X., Wang Z., Li J., Gu D., Li S., Shen C., Song Z. (2013) Increased 4-hydroxynonenal formation contributes to obesity-related lipolytic activation in adipocytes. PLoS One 8, e70663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calingasan N. Y., Uchida K., Gibson G. E. (1999) Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer's disease. J. Neurochem. 72, 751–756 [DOI] [PubMed] [Google Scholar]
- 11. Ando Y., Brännström T., Uchida K., Nyhlin N., Näsman B., Suhr O., Yamashita T., Olsson T., El Salhy M., Uchino M., Ando M. (1998) Histochemical detection of 4-hydroxynonenal protein in Alzheimer amyloid. J. Neurol. Sci. 156, 172–176 [DOI] [PubMed] [Google Scholar]
- 12. Brown M. S., Goldstein J. L. (1983) Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu. Rev. Biochem. 52, 223–261 [DOI] [PubMed] [Google Scholar]
- 13. Steinberg D., Parthasarathy S., Carew T. E., Khoo J. C., Witztum J. L. (1989) Beyond cholesterol: modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 320, 915–924 [DOI] [PubMed] [Google Scholar]
- 14. Choudhary S., Srivastava S., Xiao T., Andley U. P., Srivastava S. K., Ansari N. H. (2003) Metabolism of lipid derived aldehyde, 4-hydroxynonenal in human lens epithelial cells and rat lens. Invest. Ophthalmol. Vis. Sci. 44, 2675–2682 [DOI] [PubMed] [Google Scholar]
- 15. Enoiu M., Herber R., Wennig R., Marson C., Bodaud H., Leroy P., Mitrea N., Siest G., Wellman M. (2002) γ-Glutamyltranspeptidase-dependent metabolism of 4-hydroxynonenal-glutathione conjugate. Arch. Biochem. Biophys. 397, 18–27 [DOI] [PubMed] [Google Scholar]
- 16. Falletti O., Cadet J., Favier A., Douki T. (2007) Trapping of 4-hydroxynonenal by glutathione efficiently prevents formation of DNA adducts in human cells. Free Radic. Biol. Med. 42, 1258–1269 [DOI] [PubMed] [Google Scholar]
- 17. Laurent A., Perdu-Durand E., Alary J., Debrauwer L., Cravedi J. P. (2000) Metabolism of 4-hydroxynonenal, a cytotoxic product of lipid peroxidation, in rat precision-cut liver slices. Toxicol. Lett. 114, 203–214 [DOI] [PubMed] [Google Scholar]
- 18. Ramana K. V., Bhatnagar A., Srivastava S., Yadav U. C., Awasthi S., Awasthi Y. C., Srivastava S. K. (2006) Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J. Biol. Chem. 281, 17652–17660 [DOI] [PubMed] [Google Scholar]
- 19. Siems W., Grune T. (2003) Intracellular metabolism of 4-hydroxynonenal. Mol. Aspects Med. 24, 167–175 [DOI] [PubMed] [Google Scholar]
- 20. Sadhukhan S., Han Y., Jin Z., Tochtrop G. P., Zhang G. F. (2014) Glutathionylated 4-hydroxy-2-(E)-alkenal enantiomers in rat organs and their contributions toward the disposal of 4-hydroxy-2-(E)-nonenal in rat liver. Free Radic. Biol. Med. 70, 78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sadhukhan S., Han Y., Zhang G. F., Brunengraber H., Tochtrop G. P. (2010) Using isotopic tools to dissect and quantitate parallel metabolic pathways. J. Am. Chem. Soc. 132, 6309–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang G. F., Sadhukhan S., Ibarra R. A., Lauden S. M., Chuang C. Y., Sushailo S., Chatterjee P., Anderson V. E., Tochtrop G. P., Brunengraber H. (2012) Metabolism of γ-hydroxybutyrate in perfused rat livers. Biochem. J. 444, 333–341 [DOI] [PubMed] [Google Scholar]
- 23. Harris S. R., Zhang G. F., Sadhukhan S., Wang H., Shi C., Puchowicz M. A., Anderson V. E., Salomon R. G., Tochtrop G. P., Brunengraber H. (2013) Metabolomics and mass isotopomer analysis as a strategy for pathway discovery: pyrrolyl and cyclopentenyl derivatives of the pro-drug of abuse, levulinate. Chem. Res. Toxicol. 26, 213–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnston J. B., Ouellet H., Podust L. M., Ortiz de Montellano P. R. (2011) Structural control of cytochrome P450-catalyzed ω-hydroxylation. Arch. Biochem. Biophys. 507, 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalsotra A., Cui X., Anakk S., Hinojos C. A., Doris P. A., Strobel H. W. (2005) Renal localization, expression, and developmental regulation of P450 4F cytochromes in three substrains of spontaneously hypertensive rats. Biochem. Biophys. Res. Commun. 338, 423–431 [DOI] [PubMed] [Google Scholar]
- 26. Simpson A. E. (1997) The cytochrome P450 4 (CYP4) family. Gen. Pharmacol. 28, 351–359 [DOI] [PubMed] [Google Scholar]
- 27. Theken K. N., Deng Y., Schuck R. N., Oni-Orisan A., Miller T. M., Kannon M. A., Poloyac S. M., Lee C. R. (2012) Enalapril reverses high-fat diet-induced alterations in cytochrome P450-mediated eicosanoid metabolism. Am. J. Physiol. Endocrinol. Metab. 302, E500–E509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gioacchini A. M., Calonghi N., Boga C., Cappadone C., Masotti L., Roda A., Traldi P. (1999) Determination of 4-hydroxy-2-nonenal at cellular levels by means of electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 13, 1573–1579 [DOI] [PubMed] [Google Scholar]
- 29. Brunengraber H., Boutry M., Lowenstein J. M. (1973) Fatty acid and 3-β-hydroxysterol synthesis in the perfused rat liver: including measurements on the production of lactate, pyruvate, β-hydroxy-butyrate, and acetoacetate by the fed liver. J. Biol. Chem. 248, 2656–2669 [PubMed] [Google Scholar]
- 30. Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T. L. (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bleicher K. B., Pippert T. R., Glaab W. E., Skopek T. R., Sina J. F., Umbenhauer D. R. (2001) Use of real-time gene-specific polymerase chain reaction to measure RNA expression of three family members of rat cytochrome P450 4A. J. Biochem. Mol. Toxicol. 15, 133–142 [DOI] [PubMed] [Google Scholar]
- 32. Kalsotra A., Anakk S., Boehme C. L., Strobel H. W. (2002) Sexual dimorphism and tissue specificity in the expression of CYP4F forms in Sprague-Dawley rats. Drug Metab. Dispos. 30, 1022–1028 [DOI] [PubMed] [Google Scholar]
- 33. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 34. Li Q., Zhang S., Berthiaume J. M., Simons B., Zhang G. F. (2014) Novel approach in LC-MS/MS using MRM to generate a full profile of acyl-CoAs: discovery of acyl-dephospho-CoAs. J. Lipid Res. 55, 592–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Struys E. A., Verhoeven N. M., Jansen E. E., Ten Brink H. J., Gupta M., Burlingame T. G., Quang L. S., Maher T., Rinaldo P., Snead O. C., Goodwin A. K., Weerts E. M., Brown P. R., Murphy T. C., Picklo M. J., Jakobs C., Gibson K. M. (2006) Metabolism of γ-hydroxybutyrate to d-2-hydroxyglutarate in mammals: further evidence for d-2-hydroxyglutarate transhydrogenase. Metabolism 55, 353–358 [DOI] [PubMed] [Google Scholar]
- 36. Sanders R. J., Ofman R., Duran M., Kemp S., Wanders R. J. (2006) ω-Oxidation of very long-chain fatty acids in human liver microsomes. Implications for X-linked adrenoleukodystrophy. J. Biol. Chem. 281, 13180–13187 [DOI] [PubMed] [Google Scholar]
- 37. Li Q., Zhang G. F. (2012) Identification of n-hydroxy acid metabolites in electron impact ionization mass spectrometry. Rapid Commun. Mass Spectrom. 26, 1355–1362 [DOI] [PubMed] [Google Scholar]
- 38. Butterfield D. A., Reed T., Sultana R. (2011) Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer's disease. Free Radic. Res. 45, 59–72 [DOI] [PubMed] [Google Scholar]
- 39. Manrique C., DeMarco V. G., Aroor A. R., Mugerfeld I., Garro M., Habibi J., Hayden M. R., Sowers J. R. (2013) Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a Western diet. Endocrinology 154, 3632–3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharma S., Tripathi M. (2013) Ketogenic diet in epileptic encephalopathies. Epilepsy Res. Treat. 2013, 652052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mobbs C. V., Mastaitis J., Isoda F., Poplawski M. (2013) Treatment of diabetes and diabetic complications with a ketogenic diet. J. Child Neurol. 28, 1009–1014 [DOI] [PubMed] [Google Scholar]
- 42. Woolf E. C., Scheck A. C. (2014) The ketogenic diet for the treatment of malignant glioma. J. Lipid Res. 10.1194/jlr.R046797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hayashi S., Yasui H., Sakurai H. (2005) Essential role of singlet oxygen species in cytochrome P450-dependent substrate oxygenation by rat liver microsomes. Drug Metab. Pharmacokinet. 20, 14–23 [DOI] [PubMed] [Google Scholar]
- 44. Okita R. T., Jakobsson S. W., Prough R. A., Masters B. S. (1979) Lauric acid hydroxylation in human liver and kidney cortex microsomes. Biochem. Pharmacol. 28, 3385–3390 [DOI] [PubMed] [Google Scholar]
- 45. Okita R. T., Masters B. S. (1980) Effect of phenobarbital treatment and cytochrome P-450 inhibitors on the laurate ω- and (ω-1)-hydroxylase activities of rat liver microsomes. Drug Metab. Dispos. 8, 147–151 [PubMed] [Google Scholar]
- 46. Tanaka S., Imaoka S., Kusunose E., Kusunose M., Maekawa M., Funae Y. (1990) ω- and (ω-1)-hydroxylation of arachidonic acid, lauric acid and prostaglandin A1 by multiple forms of cytochrome P-450 purified from rat hepatic microsomes. Biochim. Biophys. Acta 1043, 177–181 [DOI] [PubMed] [Google Scholar]