


Abstract
Vibrio parahaemolyticus is the leading worldwide cause of seafood-associated gastroenteritis, yet little is known regarding its intraintestinal gene expression or physiology. To date, in vivo analyses have focused on identification and characterization of virulence factors—e.g. a crucial Type III secretion system (T3SS2)—rather than genome-wide analyses of in vivo biology. Here, we used RNA-Seq to profile V. parahaemolyticus gene expression in infected infant rabbits, which mimic human infection. Comparative transcriptomic analysis of V. parahaemolyticus isolated from rabbit intestines and from several laboratory conditions enabled identification of mRNAs and sRNAs induced during infection and of regulatory factors that likely control them. More than 12% of annotated V. parahaemolyticus genes are differentially expressed in the intestine, including the genes of T3SS2, which are likely induced by bile-mediated activation of the transcription factor VtrB. Our analyses also suggest that V. parahaemolyticus has access to glucose or other preferred carbon sources in vivo, but that iron is inconsistently available. The V. parahaemolyticus transcriptional response to in vivo growth is far more widespread than and largely distinct from that of V. cholerae, likely due to the distinct ways in which these diarrheal pathogens interact with and modulate the environment in the small intestine.
INTRODUCTION
Vibrio parahaemolyticus is a gram-negative enteric pathogen and the leading worldwide cause of seafood-associated gastroenteritis. Intestinal colonization by V. parahaemolyticus results in marked disruption of the intestinal epithelium, inflammation and diarrhea, both in infected humans (1) and in infant rabbits, which were recently developed as a model host for study of V. parahaemolyticus-linked pathogenesis (2). Analyses using infant rabbits, as well as using ligated intestinal loops in adult rabbits, have enabled identification of several factors that are critical for virulence. In particular, one of V. parahaemolyticus’ two type 3 secretion systems (T3SS) has been found to be essential for intestinal colonization, and a subset of its secreted effectors play prominent roles in pathogenesis (2–5). However, in vivo analyses to date have primarily investigated the clinical significance of candidate virulence loci identified through comparisons of pathogenic and non-pathogenic isolates and based on knowledge of virulence genes in other organisms. Studies have not addressed the overall physiology of V. parahaemolyticus within the host; consequently, transcriptomic (RNA-Seq) analyses of gene expression in vivo can illuminate a wealth of previously unexplored pathways and processes. RNA-Seq analyses provide a genome-wide overview of factors that may contribute to in vivo adaptation and growth; additionally, comparison of in vivo gene expression profiles to regulons dissected in vitro can provide insight into the environment that V. parahemolyticus encounters and generates within a mammalian host.
Comparative genomic analyses revealed that pathogenic isolates of V. parahemolyticus encode two T3SS, while environmental isolates commonly encode only a single system (6). The ubiquitous secretion system, T3SS1, contributes to V. parahemolyticus’ cytotoxicity toward cultured cells (reviewed in (7)), but does not appear to play a significant role in intestinal colonization or induction of intestinal pathology (2,3,8). It may instead influence interactions between V. parahaemolyticus and other hosts, such as shellfish. In contrast, T3SS2, which along with one or more hemolysins is encoded in a genomic island associated with pathogenic isolates (VP-PAI) (9), is essential for intestinal colonization and consequently all downstream features of disease in infant rabbits (2). It is also required for disruption of the intestinal epithelium and villus structure and for the bacteria's enterotoxicity in ligated loops, in which colonization factors can be dispensible (3). In particular, the T3SS2 effectors VopZ and VopV have been found to be critical virulence factors in these model systems (4,5). Expression of T3SS2 structural genes and effectors is controlled in vitro by two transcription factors, VtrA and VtrB, and can be induced by bile, which is presumed to induce its expression in vivo as well (10,11). Expression of T3SS1 is governed by the transcription activator ExsA and the associated factors ExsCDE, as well as by calcium and iron (12,13); however, no analyses regarding T3SS1 expression in vivo have been reported.
V. parahaemolyticus also has the capacity to produce two flagellar systems, which function in different environments associated with distinct morphological states. In liquid cultures, the organism is relatively small (2–3 μm), and produces a single polar flagellum. In contrast, growth on a surface or in a viscous environment, which impedes rotation of the polar flagellum, can induce formation of longer (up to 30 μm) swarmer cells with multiple lateral flagella as well as the polar flagellum (14). Induction of lateral flagella, as well as of many genes associated with the swarmer state/surface sensing, is controlled by the sigma 54-dependent transcription regulator LafK (15,16). It has been hypothesized that induction of the swarmer state might occur during infection (2,16); however, previous analyses have not assessed the prevalence of such transitions, and the importance in vivo of lateral flagella or other LafK-regulated factors have not been reported.
A newly recognized feature of V. parahaemolyticus is production of Type VI secretion systems (T6SS), which have been found to mediate interbacterial competition for several other bacterial species (17,18). As with its T3SS, pathogenic V. parahaemolyticus encodes two T6SS, one of which is predominantly found in clinical, rather than environmental, isolates (19). Precise roles and mechanisms of action have not been identified for either system; however, it has been reported that V. parahaemolyticus’ T6SS1 and T6SS2 are active under distinct conditions, and thus that they may promote bacterial survival within different environments. In particular, the pathogen-associated T6SS1 is activated by surface sensing and high salinity and negatively regulated by quorum sensing (i.e. by high cell density), whereas the uniformly present T6SS2 is inhibited by surface sensing and activated by quorum sensing and low salinity (20,21). Thus, the relative expression of these systems during infection can provide some insight into the environmental conditions encountered by V. parahaemolyticus within the host.
The infant rabbit model of infection used for investigation of V. parahaemolyticus pathogenicity has also been used to characterize infections caused by a related enteric pathogen, Vibrio cholerae, which causes the severely dehydrating diarrheal disease, cholera (22). Like V. parahaemolyticus, V. cholerae primarily colonizes the small intestine; thus, the two pathogens are expected to encounter similar environmental stimuli during the infection process. However, in contrast to V. parahaemolyticus, V. cholerae does not closely associate with or damage enterocytes or elicit a marked inflammatory response; instead, cholera toxin, a secreted AB5 type toxin underlies its pathogenicity (23). We previously developed the tools to carry out a comparative transcriptome analysis on V. cholerae isolated from in vitro cultures and from the fluid that accumulates within the ceca of infected infant rabbits (24). This study allowed identification of numerous genes whose expression is altered by the infection process, including a high number of known virulence genes that exhibited increased expression in vivo. Genome-wide analyses of V. parahaemolyticus gene expression in vivo have not previously been performed; however, microarrays have been used to investigate changes in V. parahaemolyticus gene expression induced by bile and by the absence of VtrB, and have been used to define several additional regulons (e.g. genes regulated by ExsA, LafK and the quorum-sensing regulator, OpaR) (10,11,13,16,21,25).
Here, we compare the transcriptomes of V. parahaemolyticus cultured under standard laboratory conditions, in the presence of bile or VtrB overexpression, and isolated from the ceca of infected infant rabbits. Our analyses reveal that expression of the genes encoding T3SS2 and its effectors is markedly upregulated in vivo, and that these genes constitute the majority of those upregulated in common (relative to growth in Luria Bertani Broth (LB) alone) by all three experimental conditions tested. However, despite the significant overlap between these regulons, our analyses indicate that there is also a large set of genes (n = 277) whose expression is only altered in vivo, suggesting that V. parahaemolyticus gene expression in vivo is subject to significant regulatory influences in addition to bile/VtrB induction. Notably, even though V. parahaemolyticus and V. cholerae both colonize the small intestine, relatively few genes shared by these two pathogens were found to be differentially regulated in vivo in both organisms. Thus, our observations suggest that the distinct modes with which these two small bowel pathogens interact with the host profoundly affect the environment perceived by the bacteria, and thereby shape their disparate transcriptional responses.
MATERIALS AND METHODS
Strains and culture conditions
All experiments were performed with the sequenced V. parahaemolyticus pandemic isolate RMID2210633 (serotype O3:K6) or derivatives of this strain. The vscN1 and vscN2 mutants have been previously described (8). Cultures were grown in LB (Miller) at 37°C, and supplemented as indicated with bile (0.04%) or 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG), which was used to induce expression of plasmid-encoded vtrB from a derivative of pMMB207 (26). The spf mutants were constructed by homologous recombination using a suicide vector, pDM4, as previously described (12). Complementation of the spf mutation was performed by cloning wild-type spf into pMMB207. For analyses of VtrB expression, the HA tag was introduced at the C terminus of the chromosomal vtrB locus using pDM4.
Rabbit infections
Infant rabbits were infected basically as described previously (2). Briefly, cimetidine-treated rabbits were orogastrically inoculated with ∼109 cfu of V. parahaemolyticus, and bacterial colonization (cfu/g intestinal tissue) and fluid accumulation ratio were assessed 38 h post-infection.
All animal experiments were performed in accordance with a protocol approved by the Harvard IACUC.
RNA isolation
In vitro grown bacteria were harvested 2 h after addition of supplements (if used), at an OD600 ∼1.0, and rinsed once with phosphate buffered saline (PBS). For the in vivo RNA samples, infected animals were euthanized ∼38 h post-infection, and cecal fluid was collected. RNA was isolated from cell pellets using Trizol according to the manufacturer's instructions. Samples were treated with DNAse, then rRNA was removed from 1 to 5 μg of total RNA using the RiboZero kit for Gram-negative bacteria (Epicentre). rRNA-depleted samples were concentrated by EtOH precipitation.
cDNA library construction and sequencing
First strand cDNA synthesis from rRNA-depleted samples was primed using the Elute, Prime, Fragment Mix of TruSeq RNA sample Prep (Illumina), bypassing the kit's mRNA enrichment protocol. TruSeq first strand cDNA synthesis was also performed, with a 50 min incubation at 50°C rather than 42°C. cDNA was purified using the RNA Clean and Concentrator-25 kit (Zymo Research), and eluted with 64 μl H20. For second strand synthesis, first strand cDNA was supplemented with 5× FSS (4 μl), 0.1 M DTT (2 μl), 10 mM dNTP (dTTP replaced with dUTP; 4 μl), second strand buffer (20 μl), Escherichia coli DNA ligase (1 μl), E. coli DNA polymerase I (4 μl), and E. coli RNAse H (1 μl), and incubated at 16°C for 2 h. Reactions were cleaned up with AMPure XP beads as instructed, followed by end repair, adenylation of 3′ ends and ligation of adapters. Reactions were cleaned 2× with AMPure XP beads, then treated with Uracil-N-glycosylase (AmpErase kit, Applied Biosystems). Finally, polymerase chain reaction was used (10 cycles) to amplify the library and enrich for fragments ligated to adapters. Reactions were cleaned with AMPure XP beads and eluted in 32.5 ul of resuspension buffer. Libraries were sequenced on the HiSeq 2000 platform to yield 76 base paired-end reads.
RNA-Seq analysis
Reads were aligned to the chromosomes I and II (RefSeq NC_004603 and 004605, respectively) using BWA (27) version 5.9. Gene annotations were obtained from RefSeq and Rfam (28). The overall fragment coverage of genomic regions corresponding to features, such as open reading frames (ORFs) and rRNAs, was conducted as described (24). Differential expression analysis was conducted using DESeq (29). Random sampling of reads from RNA-Seq data sets to control for differences in total read depth was conducted as described (30). Gene ontology (GO) groups for V. parahamolyticus and V. cholerae genes were designated using Blast2GO (31).
Visualization of read coverage on genome maps was conducted using Artemis (32).
Western analysis
For western analysis of VtrB expression, a strain expressing chromosomally encoded VtrB-HA was used. V. parahaemolyticus cultures were grown in LB with 0.04% bile to OD600 ∼1.0, then bacteria and culture supernatants were harvested. Western analyses were performed as described in (33). Blots were probed with mouse anti-HA, rabbit anti-VopD2 and mouse anti-RNAP.
Effector translocation
Translocation of VopV-CyaA into differentiated Caco-2 cells was assessed as described (4).
Bacterial adhesion
Differentiated Caco-2 cells were infected with V. parahaemolyticus for 1.5 h. Infected Caco-2 monolayers were washed with PBS for five times to remove non-adherent bacteria and subsequently the number of adherent bacterial cfu was enumerated via plating. For each experiment, the frequency of adhesion by the ΔvscN1 strain was set to 1, and the frequencies of mutant and complement strains were calculated relative to this control strain.
RESULTS AND DISCUSSION
RNA-Seq profiling of the V. parahaemolyticus transcriptome
To begin to profile V. parahaemolyticus physiology during the infection process, we performed RNA-Seq analysis on RNA isolated from mid-exponential phase LB cultures (n = 4) and from the cecal fluid of orally infected rabbits (CF; n = 3). Greater than 7 million fragments were obtained for all RNA samples but one (CF1) (Supplementary Figure S1A). Due to targeted depletion of rRNAs, the vast majority (greater than 97%) of these reads corresponded to the sense strands of annotated ORFs and non-coding RNAs (ncRNAs) or to unannotated intergenic regions (Supplementary Figure S1B), yielding a sequencing depth previously shown sufficient to reliably quantify and compare transcript abundance across a wide dynamic range (30). The correlation (R2) of normalized expression per gene between biological replicates ranged from 0.72 to 0.94 and 0.66 to 0.88 for LB and CF samples, respectively (Supplementary Figure S2).
Comparative analyses of V. parahaemolyticus gene expression in culture and during infection
To identify genes that are differentially expressed in LB cultures and in vivo, we compared the RNA-Seq data for these conditions using DESeq, a differential expression analysis package for RNAseq data that presumes read abundance can be modeled by a negative binomial distribution (29). When data from all three rabbits and all four LB cultures were compared, 416 (9%) and 181 (4%) of the 4868 annotated V. parahaemolyticus ORFs and ncRNAs were found to be significantly up- and down-regulated (>3-fold, Padj < 1 × 10−5), respectively, in cecal fluid compared to LB (Supplementary Tables S1 and S2). Of note, the majority of differentially expressed genes (57%) came from chromosome II, despite the fact that this replicon encodes only 36% of annotated V. parahaemolyticus genes. Forty-four of the genes induced in vivo (11%) lie within the region of chrII that encodes T3SS2 (VPA1321-VPA1370) (Figure 1), consistent with its known role in pathogenesis and with previous reports that it is expressed at low levels under non-inducing conditions in vitro. Nearly all of the genes associated with T3SS1 (VP1656-VP1697) were also found to be highly induced in vivo, as were two unlinked T3SS1 effectors (VPA0450/1; (34)) and a subset of genes associated with T6SS1 (particularly VP1408-1414). In contrast, all 27 annotated ORFs associated with T6SS2 were among the genes whose expression was reduced in CF. Our data and previous analyses, which reported that T6SS2 secretion (though not expression) is limited at 37°C, suggest that T6SS2 does not play a significant role in vivo, and this conclusion is supported by experimental analysis of colonization by a T6SS2-deficient strain (VPA1039; Supplementary Figure S3) (20). Over one-third of all the genes identified as differentially expressed in vivo are annotated as hypothetical proteins; additional investigation of their functions may reveal new factors that enable the pathogen to survive and multiple in vivo.
Figure 1.
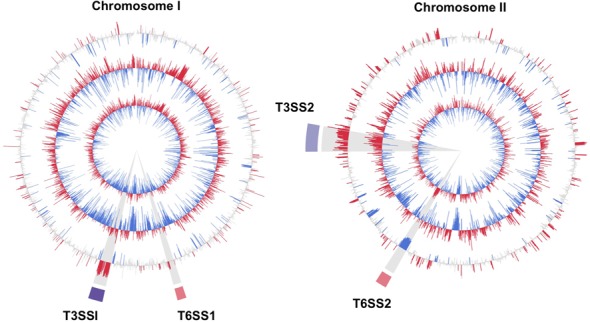
Comparison of V. parahaemolyticus gene expression in vitro (in LB) and in vivo (in CF). The two inner circles correspond to log2 FPKMO (fragments per kilobasebair per million ORF fragments) in LB and CF, respectively, and are averages from 4 (LB) and 3 (CF) experimental replicates. In these plots, ORFs whose abundance is above and below the mean are shown in red and blue, respectively. The third circle shows the log2 of differential abundance in CF versus LB with ORFs whose fold expression is significantly higher or lower (fold > 3, Padj < 1 × 10−5) in vivo shown in red and blue, respectively. Regions highlighted in purple and pink correspond to the T3SS and T6SS, respectively.
To gain additional insight into V. parahaemolyticus physiology in vivo, we grouped the differentially expressed genes based on their GO annotations. This analysis revealed that several GO categories are significantly enriched (>2-fold, P < 1 × 10−3) among V. parahaemolyticus genes down-regulated in CF compared to LB, including those involved in branched-chain aliphatic amino acid transport, histidine catabolism and the trichloroacetic acid cycle (Figure 2A; Supplementary Table S3). We did not detect differential expression of these pathways in our previous RNA-Seq based investigation of the V. cholerae transcriptome during infection of infant rabbits ((24), Figure 2 and see below), suggesting that the metabolic requirements for in vivo growth may differ between these pathogens. Among upregulated genes, overrepresented GOs include protein secretion, transport activity, maltose and phosphate transport and pathogenesis (Figure 2; Supplementary Table S3). It should be noted though that assignments to the ‘pathogenesis’ category are based primarily on homology, rather than experimental evidence in V. parahaemolyticus; in fact, T3SS1 genes are classified within pathogenesis, whereas T3SS2 genes are not associated with any GO group. Many V. cholerae virulence genes (e.g. the components of the critical colonization factor TCP) are also not associated with the GO pathogenesis category. However, classification of genes associated with more conserved functions is likely more reliable, and thus supports the idea that V. parahaemolyticus and V. cholerae rely on distinct metabolic processes to grow in the small intestine.
Figure 2.
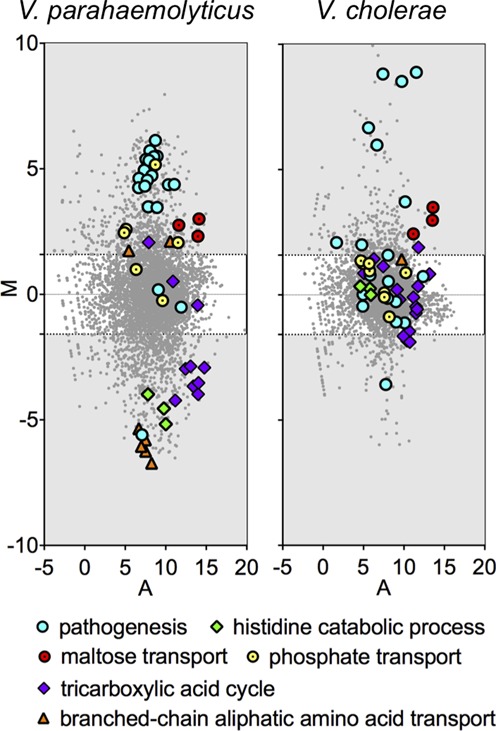
MA plots of relative transcript abundance in cecal fluid versus LB. The log2 of the ratio of abundances of each transcript between the two conditions (M) is plotted against the average log2 of abundance of that transcript in both conditions (A). Members of GO groups that are significantly overrepresented among up- and down-regulated V. parahaemolyticus genes are highlighted. Transcriptome data is plotted for V. parahaemolyticus (left) and V. cholerae (right).
Variability in the samples from infected rabbits
Results from animal models typically are less consistent than those from broth cultures, and, as expected, there was more variability in V. parahaemolyticus gene expression profiles among CF samples than among independent LB cultures (Figure 3 and Supplementary Figure S2). In agreement with their essential role in colonization, a vast majority of T3SS2 genes were induced in all three rabbits. In contrast, genes of T3SS1 were significantly induced in only two samples (CF1 and CF2), and the correlation (R2) between CF3 and the other samples for T3SS1 was only ∼0.5, whereas it was 0.97 for CF1 and CF2 (Figure 3). This finding supports our previous report that T3SS1 is dispensable for pathogenesis (2), since infection occurred despite low expression of T3SS1, and suggests that activation of T3SS1 activity may be a response to (as yet undefined) host/microbiome factor(s) that are not invariably present. We also observed significant variability in induction of several other gene clusters, including those encoding iron transport systems and nitrate/nitrite reductases (Supplementary Figure S4), further suggesting that the metabolic environment encountered by V. parahaemolyticus during infection is not consistent even among genetically identical animals grown under controlled laboratory conditions.
Figure 3.
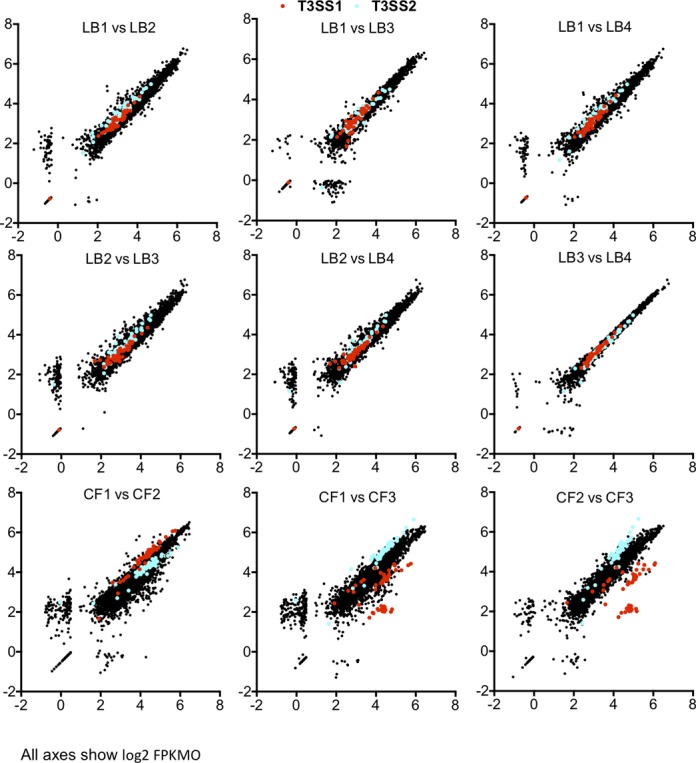
Scatter plots of log2 FPKMO values per ORF in biological replicates. ORFs with no reads were assigned a value of 0.5 fragments. Genes of T3SS1 are depicted in red, and genes of T3SS2 are depicted in blue.
Exposure to bile and induction of VtrB likely account for induction of many genes in vivo
Previous analyses have demonstrated that bile and the transcription factor VtrB both induce expression of T3SS2 in vitro, and it is hypothesized that these factors are related to changes in V. parahaemolyticus gene expression during infection (10,11). To gain insight into the extent to which bile and VtrB account for in vivo gene expression profiles, we compared the RNA-Seq data from V. parahaemolyticus isolated from CF to those derived from mid-exponential phase V. parahaemolyticus cultures supplemented with bile (n = 3) or overexpressing the transcription factor VtrB (n = 2). We observed significant overlap between induced genes (>3-fold, Padj < 1 × 10−5) under the three conditions (Figure 4). Most bile-induced genes (53/69) were also up-regulated in vivo (including VtrB), and 101 of the in vivo-induced genes were also induced by exogenous expression of VtrB (Figure 4A). As expected, both bile and VtrB induced expression of T3SS2 genes, which comprise the majority of genes in the three-way overlap (Figure 4B, also Supplementary Table S4), while neither appears to account for induction of T3SS1 in vivo. There was also some overlap among genes down-regulated under the three conditions, although this was less marked. Both in vivo growth and VtrB overexpression were associated with reduced expression of T6SS2; however, since this did not also occur in response to bile (despite its induction of VtrB), it is unclear whether elevated VtrB in vivo accounts for changes in T6SS2 expression. It should be noted that transcripts from exogenously expressed VtrB were more abundant than in vivo- or bile-induced transcripts, raising the possibility that some changes observed in response to pVtrB are not physiologically meaningful.
Figure 4.
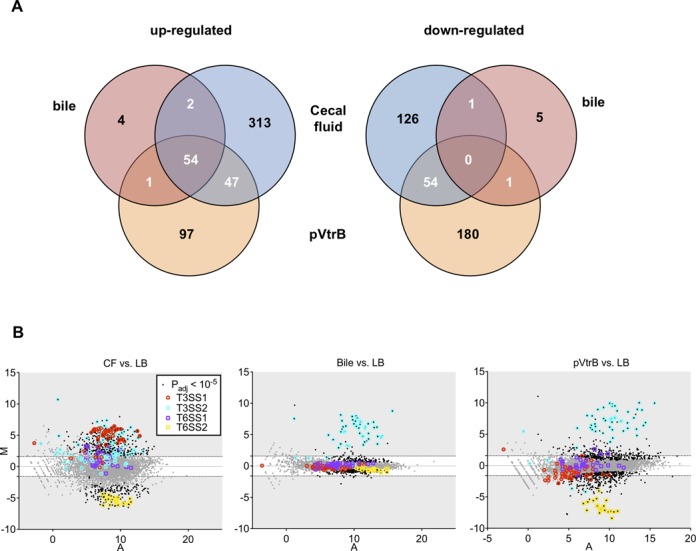
Comparative analyses of the V. parahaemolyticus transcriptional response to intraintestinal growth, bile and VtrB overexpression. (A) Venn diagrams showing overlaps among genes with increased (left) or decreased (right) transcript abundance in response to the three conditions, relative to growth in LB. (B) MA plots depicting changes in gene expression for each condition, with genes of T3SS and T6SS highlighted.
In vivo-regulated genes are related to the previously defined ExsA and OpaR regulons
Despite the extensive overlap in the three data sets compared above, we also identified numerous in vivo-regulated genes that do not appear to respond to bile and/or VtrB. Therefore, to better define the regulatory networks that govern V. parahaemolyticus adaptation to the intestinal environment, we compared the set of genes showing altered expression in vivo to previously defined V. parahaemolyticus regulons. In particular, we explored the potential roles of the T3SS1 regulator ExsA, the surface sensing/swarming regulators LafK and ScrABC and the quorum sensing regulator, OpaR (10,11,16,21). It should be remembered, however, that the regulons controlled by these factors were often ascertained under different experimental conditions than we have used (e.g. in plate-grown bacteria), complicating interpretation of these comparisons.
Our data indicate that ExsA is up-regulated in vivo (Supplementary Tables S1 and S2), and comparative analyses suggest that increased ExsA is responsible for up-regulation of numerous additional genes in vivo. The set of in vivo-induced genes includes a majority (50 of 70) genes previously reported to be induced by ExsA, including nine genes that are not in the T3SS1 gene cluster (Figure 5A, also Supplementary Table S5). Furthermore, although levels of ExsA transcripts are elevated in CF from all three rabbits, they are markedly less increased in CF3, in which T3SS1 up-regulation was not apparent. This is consistent with ExsA levels needing to reach a threshold to mediate downstream events; however, it is also possible that ExsA activity is dependent upon an additional (undefined) factor absent in bacteria in CF3. Levels of ExsA are thought to be responsive to increased calcium and to reduced iron levels (13), raising the possibility that intraintestinal variability in these ions accounts for at least some of the transcriptional heterogeneity we observed among the cecal fluid samples.
Figure 5.
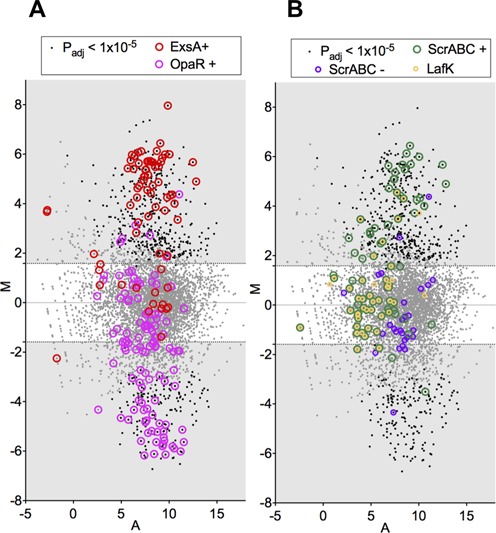
MA plots for V. parahaemolyticus gene expression in CF versus in LB, highlighting genes regulated by known transcription factors. (A) Genes down-regulated in an OpaR deletion mutant (21) or up-regulated in a strain overexpressing ExsA (13) are highlighted. (B) Genes up- and down-regulated in ScrABC deletion mutant (ScrABC+ and ScrABC-, respectively) (25) or up-regulated during growth on a surface only in a LafK+ strain (16) are highlighted.
V. parahaemolyticus genes linked to quorum sensing also may account for some of the differences between gene expression in vitro and in vivo. Expression of AphA (VP2762), a recently described repressor of the quorum sensing regulator, OpaR, was elevated in vivo, consistent with growth of cells under relatively low density conditions (35). Additionally, the altered expression of a number of genes regulated by OpaR (which is repressed by AphA) also might indicate that OpaR was less active in vivo. Approximately 40% of the OpaR regulon showed altered expression in vivo that is consistent with reduced activity of this regulator, including changes in expression of T6SS1 and T6SS2 (Figure 5A, also Supplementary Table S5). Many of the affected loci (e.g. T6SS2) also showed altered expression in response to exogenous production of VtrB, and it is possible that the OpaR and VtrB pathways are interrelated; however, the precise nature of their relationship remains to be determined.
Intraintestinal growth does not induce a swarmer gene expression profile
In contrast to the ExsA and OpaR regulons, we saw relatively little overlap between in vivo-regulated genes and genes controlled by surface sensing, LafK or ScrABC, all of which govern V. parahaemolyticus’ production of lateral flagella and transition to a swarmer state (25,36). Although a few genes associated with lateral flagella (VPA1538-VPA1543) as well as ExsA and members of its regulon were induced in cecal fluid, the majority of genes in the LafK and ScrABC regulons are not differentially expressed in CF, clearly suggesting that induction of the swarmer state does not predominate in vivo (Figure 5B, also Supplementary Table S5). Additional analyses indicate that induction is not imperative for even a subset of cells, or for intestinal sites other than the cecum; we observed that deletion of numerous lateral flagella genes involved in the transition from the swimmer to the swarmer state (VPA0264-VPA0275) does not impair colonization of the small intestine by V. parahaemolyticus (Supplementary Figure S3).
Intraintestinal growth alters the abundance of regulatory RNAs
Our transcriptome analyses also revealed several known or putative V. parahaemolyticus regulatory RNAs whose abundance is significantly altered during infection compared to growth in culture (Supplementary Tables S1 and S2). One of these is Spot 42, a trans-acting sRNA implicated as a regulator of catabolite repression, central metabolism and motility in E. coli and Aliivibrio salmonicida (37,38), whose expression was elevated more than 35-fold in vivo. Notably, we also detected reduced transcript abundance for homologs of genes down-regulated by Spot 42 in E. coli (e.g. citrate synthase (VP0842 and VP1647), malate dehydrogenase (VP0325) and pyridine nucleotide transhydrogenase (VP2942)), suggesting that V. parahaemolyticus Spot 42 likely regulates similar processes as the E. coli sRNA. Additionally, this result suggests that preferred carbon sources, such as glucose, are readily available to intestinal V. parahaemolyticus, which is perhaps surprising given our observation that maltose transport genes are also up-regulated. One of the five V. parahaemolyticus qrr sRNAs was also significantly up-regulated in CF from all three rabbits. Qrrs are key regulators of quorum sensing that are expressed at low cell density and up-regulate expression of AphA (35); thus, this observation is consistent with our analyses of AphA, OpaR, and downstream genes, which suggested cells in vivo do not perceive the presence of a bacterial ‘quorum’. Also consistent with our earlier analyses, we observed that transcript abundance for RyhB, an sRNA that is expressed when cellular iron concentrations are low (39), varied considerably among animals; this data provides further evidence that iron is not uniformly available within the intestines of infected animals. Finally, we found that the abundance of a region encoding two tandem GEMM domains on chromosome I was significantly up-regulated in vivo, and two other tandem GEMM domains on chromosome II met slightly less stringent criteria for in vivo change (>3-fold, Padj < 1 × 10−4). These domains typically mediate cyclic di-guanosine monophosphate (c-di-GMP) controlled regulation of downstream sequences, and we observed that expression of downstream genes, including TfoX (VP1028), a putative regulator of bacterial competence, was also increased in vivo. Thus, these data suggest the possibility that levels of c-di-GMP differ between in vivo and in vitro grown cells; however, as GEMM domain binding can have either positive or negative effects (40), the direction of change cannot be inferred from our data.
Spot 42 modulates T3SS2 production but is not required for V. parahaemolyticus virulence
Given the marked up-regulation of Spot 42 in vivo, we wondered whether this sRNA might be important for V. parahaemolyticus adaptation to and colonization of the intestinal environment. Therefore, we examined the effect of deleting spf (which encodes Spot 42) on virulence-related phenotypes in vitro and on V. parahaemolyticus pathogenicity. Unexpectedly, we observed that the absence of Spot 42 reduced levels of VtrB in V. parahaemolyticus, and also reduced (but did not eliminate) production and secretion of T3SS2 translocon and effector proteins (VopD2 and VPA1350, respectively) that are regulated by VtrB (Figure 6A and B and data not shown). Consistent with these findings, the spf mutant displayed reduced adhesion to cultured epithelial cells and had a reduced capacity to translocate effector proteins into these cells (Figure 6C and D). The mutant's deficiencies could be complemented by exogenous expression of Spot 42 or VtrB, providing further evidence that Spot 42 influences production of VtrB. However, despite the reduced activity of T3SS2 in vitro in the spf mutant, the mutant retained full virulence in the rabbit model of infection, indicating that the residual levels of T3SS2 expression and activity in the mutant are sufficient to mediate intestinal colonization and induction of diarrhea (Figure 6E–G). Thus, Spot 42's effects on T3SS2 and metabolic functions do not appear to be critical for pathogenesis.
Figure 6.
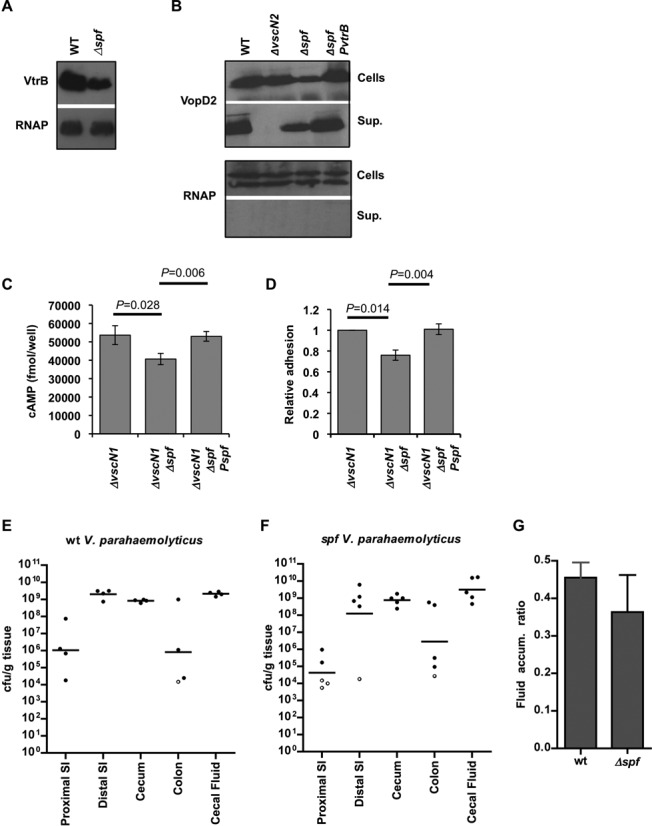
The influence of spf deletion upon T3SS2-related phenotypes. Western blots show the effect of spf deletion on expression of VtrB (A), and expression and secretion of the T3SS2 translocon protein VopD2 (B). Antisera against VopD2 and HA (for chromosome-encoded VtrB-HA) was used. As a loading control, blots were probed with antisera to RNAP. (C) The effect of spf deletion and complementation on translocation of the T3SS2 effector VopV into differentiated Caco-2 cells was measured using a VopV-CyaA fusion protein. (D) The effect of spf deletion and complementation on V. parahaemolyticus adhesion to Caco-2 cells is shown relative to adhesion of the vscN1 strain. The T3SS1 gene vscN1 was deleted for adhesion and translocation assays (C and D) in order to avoid the cytotoxicity associated with T3SS1. Mean and SEM are shown. (E and F) Colonization (cfu/g tissue) of the intestinal tracts of infant rabbits by wt (E) and spf (F) V. parahaemolyticus. Bars mark geometric means. Open circles represent the limit of detection, based on isolation of a single cfu, when no cfu were detected. (G) Fluid accumulation ratio in the ceca of infant rabbits infected with wt and spf V. parahaemoloyticus. Mean and SEM are shown.
The host intestine elicits distinct transcriptional responses in V. parahaemolyticus and V. cholerae
Since we had previously profiled the transcriptome of V. cholerae in both LB and in the infant rabbit model using the same RNA-Seq approach used in this study (24), we compared the transcriptional responses of these two pathogenic vibrios to the host environment. Notably, DESeq analysis employing the same cutoffs used here yielded only 91 (2%) of all annotated V. cholerae genes, a much smaller response than we observed in V. parahaemolyticus, where >12% of genes were differentially regulated. This difference does not appear to be due to technical differences in read depth or number of replicates tested in these two studies, as similar results were seen with randomly sampled subsets of each of these data sets containing an equal number of total non-rRNA reads. We speculate that the more severe host response to V. parahaemolyticus (e.g. inflammation, epithelial disruption) may in turn elicit a greater transcriptional response from this pathogen, which relies on T3SS2 to efface the intestinal microvilli and establish a more intimate connection with host epithelium than V. cholerae (41). It is possible that the longer period of colonization by V. parahaemolyticus (38 h versus 22 h for V. cholerae) also provides greater opportunity for host responses that influence pathogen gene expression; however, signs of V. parahaemolyticus infection are minimal at 22 h p.i. (the peak of V. cholerae infection), and thus the disparate timepoints probably provide a more meaningful comparison of the pathogens’ transcriptional responses to the infection process.
To further compare the transcriptional responses of V. parahaemolyticus and V. cholerae to the host environment, we examined the expression patterns of V. parahaemolyticus genes whose homologs were differentially expressed in V. cholerae during infection. There was little correlation between the relative expression of these homologous genes in V. parahaemolyticus and V. cholerae in vivo compared to LB cultures (Figure 7), and, as noted above, enriched GO terms were discordant for the two organisms (Figure 2B). Other than ‘pathogenesis’ (which depends upon disparate mechanisms in the two species), the only shared response appears to be induction of maltose transport; transcripts related to maltose transport are extremely abundant in vivo for both pathogens. V. cholerae in vivo-induced genes are also enriched for factors involved in sugar transport (GOs 0005351 and 0009401), and while several genes involved in sugar transport were also induced in vivo in V. parahaemolyticus, neither of these GOs was significantly enriched among V. parahaemolyticus genes induced in vivo. Thus, despite their genetic similarity and their shared capacity to colonize the small intestine, V. parahaemolyticus and V. cholerae have distinct transcriptional responses to the host environment during infection.
Figure 7.
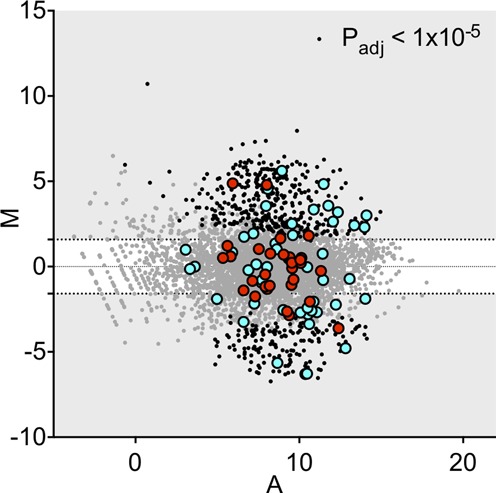
MA plots of V. parahaemolyticus gene abundance in cecal fluid versus LB. The log2 of the ratio of abundances of each ORF between these two conditions (M) is plotted against the average log2 of abundance of that ORF in both conditions (A). M and A values are based on data from all biological replicates of each growth condition. Homologs of genes up- and down-regulated in V. cholerae are highlighted in blue and red, respectively.
In conclusion, our RNA-Seq analyses provided a wealth of information regarding V. parahaemolyticus gene expression during infection, yielding clues about the environment it encounters within the host and the bacterial regulators that govern the organism's response to this environment. Our analyses provide further evidence that one of V. parahaemolyticus’ T3SS (T3SS1) is dispensable for pathogenesis, and confirm the prior supposition that expression of T3SS2 is induced in vivo, probably due to the presence of bile and induction of VtrB. Differentiation of bacteria into the swarmer state and expression of surface-induced genes is not apparent in vivo; additionally, despite accumulation of 109 bacteria per gram tissue or milliliter fluid in vivo, V. parahaemolyticus colonizing the rabbit small intestine and cecum do not perceive the presence of an abundant bacterial community, and genes activated by quorum sensing are not expressed. Although iron limitation can sometimes restrict bacterial growth in vivo, our results indicate that V. parahaemolyticus does not routinely encounter an environment in which iron is scarce. Similarly, iron is also not thought to be limiting during V. cholerae infections of infant rabbits (24). However, we observed relatively little concordance overall between the sets of genes from V. parahaemolyticus and V. cholerae that are differentially regulated in vivo. Moreover, a far greater fraction of the V. parahaemolyticus genome is differentially expressed in vivo compared with V. cholerae (12% versus 2%). This marked difference likely reflects V. parahaemolyticus' and V. cholerae's fundamentally distinct modes of interacting with the host. Unlike V. cholerae, which does not intimately interact with and disrupt the host epithelium, V. parahaemolyticus depends on its TTSS to attach to and efface the brush border of the intestinal epithelium (2,41). Furthermore, and perhaps related to its disruption of host tissue, V. parahaemolyticus, unlike V. cholerae, stimulates considerable inflammation. Thus, V. parahaemolyticus' more extensive transcriptional changes likely reflect its adaptation to the changes (tissue disruption and inflammation) that it has elicited in the host. Finally, we anticipate that future RNA-seq-based analyses will also allow examination of the host transcriptome during infection, enabling even greater understanding of the interplay between the host and these intestinal pathogens.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
Present address: Xiaohui Zhou, Department of Pathobiology and Veterinary Science, University of Connecticut, Storrs, CT, USA.
FUNDING
National Institutes of Health [R37 AI042347 to M.K.W.]; Howard Hughes Medical Institute [M.K.W.]. Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services [HHSN272200900018C]. Funding for open access charge: HHMI.
Conflict of interest statement. None declared.
REFERENCES
- 1.Qadri F., Alam M.S., Nishibuchi M., Rahman T., Alam N.H., Chisti J., Kondo S., Sugiyama J., Bhuiyan N.A., Mathan M.M., et al. Adaptive and inflammatory immune responses in patients infected with strains of Vibrio parahaemolyticus. J. Infect. Dis. 2003;187:1085–1096. doi: 10.1086/368257. [DOI] [PubMed] [Google Scholar]
- 2.Ritchie J.M., Rui H., Zhou X., Iida T., Kodoma T., Ito S., Davis B.M., Bronson R.T., Waldor M.K. Inflammation and disintegration of intestinal villi in an experimental model for Vibrio parahaemolyticus-induced diarrhea. PLoS Pathog. 2012;8:e1002593. doi: 10.1371/journal.ppat.1002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park K.-S., Ono T., Rokuda M., Jang M.-H., Okada K., Iida T., Honda T. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect. Immun. 2004;72:6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou X., Gewurz B.E., Ritchie J.M., Takasaki K., Greenfeld H., Kieff E., Davis B.M., Waldor M.K. A Vibrio parahaemolyticus T3SS effector mediates pathogenesis by independently enabling intestinal colonization and inhibiting TAK1 activation. Cell Rep. 2013;3:1690–1702. doi: 10.1016/j.celrep.2013.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hiyoshi H., Kodama T., Saito K., Gotoh K., Matsuda S., Akeda Y., Honda T., Iida T. VopV, an F-Actin-binding type III secretion effector, is required for Vibrio parahaemolyticus-induced enterotoxicity. Cell Host Microbe. 2011;10:401–409. doi: 10.1016/j.chom.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Makino K., Oshima K., Kurokawa K., Yokoyama K., Uda T., Tagomori K., Iijima Y., Najima M., Nakano M., Yamashita A., et al. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet. 2003;361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- 7.Broberg C.A., Calder T.J., Orth K. Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect. 2011;13:992–1001. doi: 10.1016/j.micinf.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiyoshi H., Kodama T., Iida T., Honda T. Contribution of Vibrio parahaemolyticus virulence factors to cytotoxicity, enterotoxicity, and lethality in mice. Infect. Immun. 2010;78:1772–1780. doi: 10.1128/IAI.01051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izutsu K., Kurokawa K., Tashiro K., Kuhara S., Hayashi T., Honda T., Iida T. Comparative genomic analysis using microarray demonstrates a strong correlation between the presence of the 80-kilobase pathogenicity island and pathogenicity in Kanagawa phenomenon-positive Vibrio parahaemolyticus strains. Infect. Immun. 2008;76:1016–1023. doi: 10.1128/IAI.01535-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gotoh K., Kodama T., Hiyoshi H., Izutsu K., Park K.-S., Dryselius R., Akeda Y., Honda T., Iida T. Bile acid-induced virulence gene expression of Vibrio parahaemolyticus reveals a novel therapeutic potential for bile acid sequestrants. PLoS ONE. 2010;5:e13365. doi: 10.1371/journal.pone.0013365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kodama T., Gotoh K., Hiyoshi H., Morita M., Izutsu K., Akeda Y., Park K.-S., Cantarelli V.V., Dryselius R., Iida T., et al. Two regulators of Vibrio parahaemolyticus play important roles in enterotoxicity by controlling the expression of genes in the Vp-PAI region. PLoS ONE. 2010;5:e8678. doi: 10.1371/journal.pone.0008678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou X., Shah D.H., Konkel M.E., Call D.R. Type III secretion system 1 genes in Vibrio parahaemolyticusare positively regulated by ExsA and negatively regulated by ExsD. Mol. Microbiol. 2008;69:747–764. doi: 10.1111/j.1365-2958.2008.06326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gode-Potratz C.J., Chodur D.M., McCarter L.L. Calcium and iron regulate swarming and type III secretion in Vibrio parahaemolyticus. J. Bacteriol. 2010;192:6025–6038. doi: 10.1128/JB.00654-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCarter L. The multiple identities of Vibrio parahaemolyticus. J. Mol. Microbiol. Biotechnol. 1999;1:51–57. [PubMed] [Google Scholar]
- 15.Stewart B.J., McCarter L.L. Lateral flagellar gene system of Vibrio parahaemolyticus. J. Bacteriol. 2003;185:4508–4518. doi: 10.1128/JB.185.15.4508-4518.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gode-Potratz C.J., Kustusch R.J., Breheny P.J., Weiss D.S., McCarter L.L. Surface sensing in Vibrio parahaemolyticus triggers a programme of gene expression that promotes colonization and virulence. Mol. Microbiol. 2010;79:240–263. doi: 10.1111/j.1365-2958.2010.07445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russell A.B., Peterson S.B., Mougous J.D. Type VI secretion system effectors: poisons with a purpose. Nat. Rev. Micro. 2014;12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho B.T., Dong T.G., Mekalanos J.J. A view to a kill: the bacterial type VI secretion system. Cell Host Microbe. 2014;15:9–21. doi: 10.1016/j.chom.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y., Yang H., Li J., Zhang P., Wu B., Zhu B., Zhang Y., Fang W. Putative type VI secretion systems of Vibrio parahaemolyticus contribute to adhesion to cultured cell monolayers. Arch Microbiol. 2012;194:827–835. doi: 10.1007/s00203-012-0816-z. [DOI] [PubMed] [Google Scholar]
- 20.Salomon D., Gonzalez H., Updegraff B.L., Orth K. Vibrio parahaemolyticus type VI secretion system 1 is activated in marine conditions to target bacteria, and is differentially regulated from system 2. PLoS ONE. 2013;8:e61086. doi: 10.1371/journal.pone.0061086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gode-Potratz C.J., McCarter L.L. Quorum sensing and silencing in Vibrio parahaemolyticus. J. Bacteriol. 2011;193:4224–4237. doi: 10.1128/JB.00432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ritchie J.M., Rui H., Bronson R.T., Waldor M.K. Back to the future: studying cholera pathogenesis using infant rabbits. MBio. 2010;1:e00047. doi: 10.1128/mBio.00047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritchie J.M., Waldor M.K. Vibrio cholerae interactions with the gastrointestinal tract: lessons from animal studies. 2009;337:37–59. doi: 10.1007/978-3-642-01846-6_2. [DOI] [PubMed] [Google Scholar]
- 24.Mandlik A., Livny J., Robins W.P., Ritchie J.M., Mekalanos J.J., Waldor M.K. RNA-Seq-based monitoring of infection-linked changes in Vibrio cholerae gene expression. Cell Host Microbe. 2011;10:165–174. doi: 10.1016/j.chom.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreira R.B.R., Chodur D.M., Antunes L.C.M., Trimble M.J., McCarter L.L. Output targets and transcriptional regulation by a cyclic dimeric GMP-responsive circuit in the Vibrio parahaemolyticus Scr network. J. Bacteriol. 2012;194:914–924. doi: 10.1128/JB.05807-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morales V.M., Bäckman A., Bagdasarian M. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene. 1991;97:39–47. doi: 10.1016/0378-1119(91)90007-x. [DOI] [PubMed] [Google Scholar]
- 27.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardner P.P., Daub J., Tate J.G., Nawrocki E.P., Kolbe D.L., Lindgreen S., Wilkinson A.C., Finn R.D., Griffiths-Jones S., Eddy S.R., et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haas B.J., Chin M., Nusbaum C., Birren B.W., Livny J. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes. BMC Genom. 2012;13:734. doi: 10.1186/1471-2164-13-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conesa A., Gotz S., Garcia-Gomez J.M., Terol J., Talon M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 32.Carver T., Harris S.R., Berriman M., Parkhill J., McQuillan J.A. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou X., Ritchie J.M., Hiyoshi H., Iida T., Davis B.M., Waldor M.K., Kodama T. The hydrophilic translocator for Vibrio parahaemolyticus, T3SS2, is also translocated. Infect. Immun. 2012;80:2940–2947. doi: 10.1128/IAI.00402-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ono T., Park K.-S., Ueta M., Iida T., Honda T. Identification of proteins secreted via Vibrio parahaemolyticus type III secretion system 1. Infect. Immun. 2006;74:1032–1042. doi: 10.1128/IAI.74.2.1032-1042.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun F., Zhang Y., Wang L., Yan X., Tan Y., Guo Z., Qiu J., Yang R., Xia P., Zhou D. Molecular characterization of direct target genes and cis-acting consensus recognized by Quorum-sensing regulator AphA in Vibrio parahaemolyticus. PLoS ONE. 2012;7:e44210. doi: 10.1371/journal.pone.0044210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomelsky M. Cyclic dimeric GMP-mediated decisions in surface-grown Vibrio parahaemolyticus: a different kind of motile-to-sessile transition. J. Bacteriol. 2012;194:911–913. doi: 10.1128/JB.06695-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen G.A., Ahmad R., Hjerde E., Fenton C.G., Willassen N.-P., Haugen P. Expression profiling reveals Spot 42 small RNA as a key regulator in the central metabolism of Aliivibrio salmonicida. BMC Genom. 2012;13:37. doi: 10.1186/1471-2164-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beisel C.L., Storz G. The base-pairing RNA Spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli. Mol. Cell. 2011;41:286–297. doi: 10.1016/j.molcel.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanabe T., Funahashi T., Nakao H., Maki J., Yamamoto S. The Vibrio parahaemolyticus small RNA RyhB promotes production of the siderophore vibrioferrin by stabilizing the polycistronic mRNA. J. Bacteriol. 2013;195:3692–3703. doi: 10.1128/JB.00162-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sudarsan N., Lee E.R., Weinberg Z., Moy R.H., Kim J.N., Link K.H., Breaker R.R. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science. 2008;321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X., Massol R.H., Nakamura F., Chen X., Gewurz B.E., Davis B.M., Lencer W.I., Waldor M.K. Remodeling of the intestinal brush border underlies adhesion and virulence of an enteric pathogen. MBio. 5:e01639-14. doi: 10.1128/mBio.01639-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.