
Table 1. Structure and refinement statistics for PaDHQase (PDB entry 4l8l).
Values in parentheses are for the highest resolution shell.
| Data | |
| Space group | F23 |
| Unit-cell parameters (, ) | a = b = c = 125.39, = = = 90 |
| Resolution () | 31.41.74 (1.81.74) |
| R merge † (%) | 10.0 (21.5) |
| Completeness (%) | 99 (100) |
| Multiplicity | 41.8 (6.96) |
| I/(I) | 31.2 (3.5) |
| Refinement | |
| R factor‡ (%) | 16.2 (35.4) |
| R free § (%) | 19.8 (43.9) |
| Correlation coefficients | |
| F o F c | 0.969 |
| F o F c, free | 0.959 |
| Components of model | |
| Amino-acid residues | 138 |
| Water | 96 |
| Mean B factor (2) | 23.59 |
| R.m.s. deviation from ideal | |
| Bond length () | 0.029 |
| Bond angles () | 2.114 |
| Chiral (3) | 0.159 |
| Ramanchandran plot, residues in (%) | |
| Favored regions | 97.0 |
| Allowed regions | 3.0 |
| Outlier regions | 0 |
| PROCHECK ¶ | |
| Side-chain outliers (%) | 1.8 |
| RSZR outliers (%) | 0.7 |
†
R
merge = 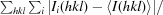
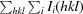 , where I
i(hkl) and I(hkl) are the intensity of the ith measurement of I(hkl) and the mean intensity of the reflection with indices hkl, respectively.
, where I
i(hkl) and I(hkl) are the intensity of the ith measurement of I(hkl) and the mean intensity of the reflection with indices hkl, respectively.
‡
R
cryst = 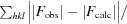
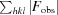 , where F
obs are observed and F
calc are calculated structure-factor amplitudes.
, where F
obs are observed and F
calc are calculated structure-factor amplitudes.
§
The R free set consisted of a randomly chosen 5% of reflections.
¶
PROCHECK validation was used (Laskowski et al., 1993 ▶). RSZR is the root-mean-square of all Z scores.