
FIGURE 2.
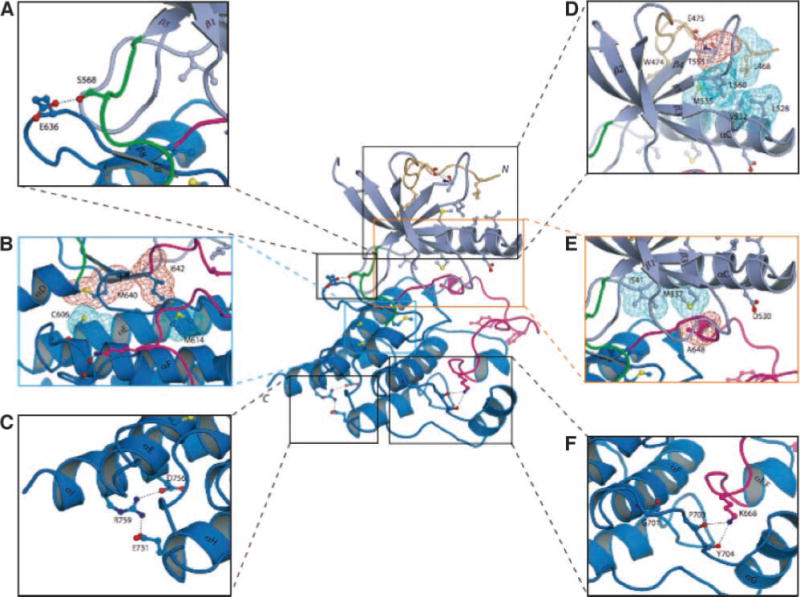
FGFR2c kinase domain mutations identified in melanoma tumors/cell lines mapped onto the crystal structures of the FGFR2 kinase domain suggest that these mutations should impair the kinase activity of FGFR2. The positions of the mutated residues are mapped onto the crystal structure of unphosphorylated wild-type FGFR2 kinase domain (PDB entry: 2PSQ; ref. 15). The coloring of the intracellular tyrosine kinase domain is as follows: the NH2-terminal lobe of kinase is in light blue; the COOH-terminal lobe is in bright blue; the activation loop is in magenta; the kinase hinge region is in green; and the NH2-terminal tail of the kinase is colored wheat. Note that ATP (not shown) binds in the cleft between the N-lobe and C-lobe of the kinase domain. To assist the viewer, certain β strands and α helices, which are relevant for explanation of the effects of the melanoma mutations, are labeled. A. E636, which maps to the sharp turn between β7 and β8 strands, makes hydrogen bond with S563 in the kinase hinge region. Mutation of this residue to lysine could influence the relative disposition of the N-lobe to the C-lobe, and hence alter the tyrosine kinase activity of FGFR2. B. The M640 and I642 side chains (red mesh) point into the inner hydrophobic core of the kinase domain and engage C606 and M614 (cyan mesh). Mutation of either M640 or I642 to smaller hydrophobic residues should weaken the extent of these core hydrophobic contacts and lead to a reduction in kinase activity. C. The hydrogen bonds between R759 and side chains of negatively charged E731 (in helix αH) and D756 (in helix αI) are shown. In addition to the loss of these hydrogen bonds, introduction of a stop codon at this location will also truncate the helix αI by one helical turn. These structural changes should reduce the stability of the kinase domain, specifically making the kinase domain temperature sensitive and ultimately leading to receptor loss of function. D. Close-up view of the region where E475 is located. The side chain of E475 engages in two strong hydrogen bonds with the side chain and backbone atom of T555 (located in the loop between β4 and β5 strands), which facilitate the conformation of the β4–β5 loop as well of the region preceding E475 itself. This in turn promotes the hydrophobic contacts between L468, T555 (the methyl group), and L560 from these regions and L528, V532, and M535 in the catalytically important αC helix. To emphasize this point, the side chains of these hydrophobic residues are shown as colored mesh. E. Close-up view of the region where D530 and A648 are located. In unphosphorylated wild-type FGFR2K structure, D530 does not play any role. However, in the A-loop phosphorylated activated FGFR2K structure (PDB entry: 2PVF; ref. 15), the side chain of D530 engages in salt bridge with R664 in the A-loop and contributes to A-loop conformation in the active state (not shown). The side chain of A648, located at the beginning of the A-loop, packs against the hydrophobic side chains of M537 in the αC helix. Replacement of A648 with threonine is expected to lead to loss of function because the larger side chain of threonine will impede αC helix from nearing sufficiently close enough to the A-loop in the active FGFR2K. F. Close-up view of the kinase region where G701 is located. G701 plays a role in stabilizing the conformation of the loop between the helices αF and αG, which is in hydrogen-bonding contacts with K668 in the A-loop. The G701S mutation should destabilize the αF-αG loop and indirectly affect the A-loop conformation.