

Abstract
Subtype-selective agonists of the neurotensin receptor NTS2 represent a promising option for the treatment of neuropathic pain, as NTS2 is involved in the mediation of μ-opioid-independent anti-nociceptive effects. Based on the crystal structure of the subtype NTS1 and previous structure–activity relationships (SARs) indicating a potential role for the sub-pocket around Tyr11 of NT(8–13) in subtype-specific ligand recognition, we have developed new NTS2-selective ligands. Starting from NT(8–13), we replaced the tyrosine unit by β2-amino acids (type 1), by heterocyclic tyrosine bioisosteres (type 2) and peptoid analogues (type 3). We were able to evolve an asymmetric synthesis of a 5-substituted azaindolylalanine and its application as a bioisostere of tyrosine capable of enhancing NTS2 selectivity. The S-configured test compound 2 a, [(S)-3-(pyrazolo[1,5-a]pyridine-5-yl)-propionyl11]NT(8–13), exhibits substantial NTS2 affinity (4.8 nm) and has a nearly 30-fold NTS2 selectivity over NTS1. The (R)-epimer 2 b showed lower NTS2 affinity but more than 600-fold selectivity over NTS1.
Keywords: neurotensin, NTS2, subtype selectivity, tyrosine analogues, β2-amino acids
Introduction
The neurotensin system is involved in a variety of physiological and pathophysiological processes that are mediated mainly via the two G protein-coupled receptors (GPCRs) NTS1 and NTS2.1 Because the NTS1 subtype is primarily responsible for the regulation of the dopaminergic system,2 selective ligands could serve as interesting new targets for the treatment of schizophrenia or Parkinsons disease.2, 3 As an extension, the NTS2 receptor is responsible for μ-opioid-independent anti-nociceptive effects of neurotensin.4 Thus, NTS2-selective agonists could serve as well-tolerated therapeutic agents for the treatment of neuropathic pain.
Recent progress in GPCR crystallography led to the elucidation of the crystal structure of the neurotensin receptor NTS1 bound to NT(8–13) (H-Arg-Arg-Pro-Tyr-Ile-Leu-OH), the fully active C-terminal hexapeptide of the tridecapeptide neurotensin.,5, 6 Careful analysis of the receptor–ligand complex offers invaluable insights into the binding mode of the peptide agonist on a molecular level. The crystal structures facilitate a structural understanding on the importance of single residues of NT(8–13) originally derived from experimental studies. Thus, Tyr11 has been shown to be crucial for receptor binding, as the replacement by alanine resulted in a 5400-fold reduction of ligand affinity.7 This effect can be explained by the crystal structure indicating stabilization of Tyr11 by extensive van der Waals interactions and hydrogen bonds (Figure 1 A,C). Besides an attractive interaction of the backbone carbonyl unit with Thr226 of extracellular loop 2 (EL2) and a water-mediated contact to His348 in transmembrane helix 7, the side chain hydroxyl group of Tyr11 forms a hydrogen bond to the backbone carbonyl of the N-terminal residue Leu55 of NTS1, which participates in stabilizing a conformation of the N-terminus lining the ligand binding pocket (Figure 1 A). An ionic interaction between His133 of extracellular loop 1 (EL1) and the N-terminal residue Asp56 seems to conformationally restrict the N-terminal region of NTS1, thereby forming a pre-organized subpocket tightly packing against Tyr11. Interestingly, the corresponding residues comprising this salt bridge are absent in NTS2 (NTS1: Asp56/His133, NTS2: Gly24/Tyr99, Figure 1 B), which makes it tempting to assume a less well-organized architecture of this microdomain in NTS2 tolerating selectivity-inducing variations of the peptidic ligand in position 11. In agreement with this observation, the affinity of the [11Ala]NT(8–13) mutant of NT(8–13) towards NTS2 was only moderately affected (70-fold) compared to its NTS1 binding (5400-fold decrease).7 This indicates a minor contribution of Tyr11 to the binding energy of NT(8–13) to NTS2 suggesting a potential role of the microdomain around Tyr11 in subtype selectivity. Consequently, structural manipulation of Tyr11 including the use of a β3-homotyrosine, tyramine-derived peptoid moieties or d-amino acids induced substantial decrease of NTS1 binding.7 Because such modifications were largely tolerated by NTS2, this approach led to NTS2-selective ligands.4b, 8
Figure 1.
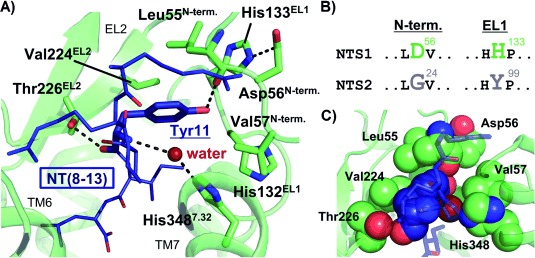
Interactions within the crystal structure of rat NTS1 around Tyr11 of NT(8–13) (PDB ID: 4GRV). A) The NTS1 receptor is shown as green ribbons, with important residues stabilizing the conformation around Tyr11 of NT(8–13) (dark-blue) depicted as sticks. In addition, a structural water molecule is indicated as a dark-red dot. Three hydrogen bonds of Tyr11 to residues of NTS1 are visualized: one via its side chain to Leu55 and two via its backbone and oxygen atoms to His348 (mediated by a water molecule) and Thr226, respectively. B) Parts of aligned sequences of NTS1 (rat) and NTS2 (human) are visualized, which point out sequence differences between distinct parts of the N-terminal regions and EL1. C) A space-filling representation of the interactions between Tyr11 and residues of NTS1 is shown, with the respective residues highlighted as dark-blue (Tyr11) and green (NTS1) spheres. A slightly modified perspective compared to panel A) was used for clarity. Again, the structural water molecule mediating the hydrogen bond between Tyr11 and His348 is shown as a dark-red sphere.
Encouraged by these observations, we envisioned to investigate NT(8-13)-derived target compounds incorporating a set of Tyr11 modifications including β2-homotyrosines of type 1 (Figure 2), heterocyclic tyrosine bioisosteres of type 2 and the corresponding peptoid analogue (type 3). In this paper, we present the asymmetric syntheses of new unnatural amino acids, their incorporation into NT(8–13), and receptor binding studies of the novel target compounds of types 1, 2 and 3.
Figure 2.
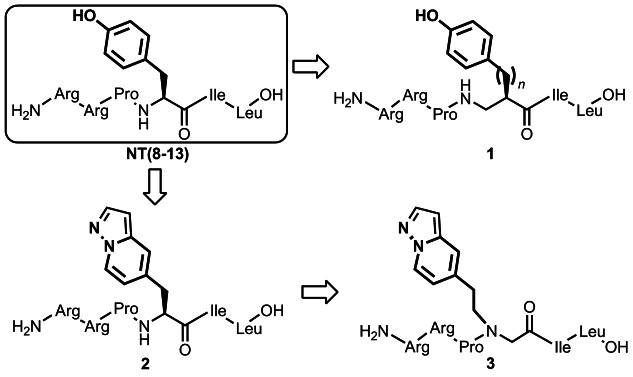
NT(8–13) derivatives chosen for enhancing NTS2 selectivity.
Results and Discussion
Syntheses of the NT(8-13)-analogues
Our plan for the syntheses of NT(8-13) derivatives 1–3 (Figure 2) required the preparation of the corresponding three modified amino acids 4, 5 and 6 (Figure 3) and subsequent solid-phase-supported incorporation of these building blocks to give the respective peptides.
Figure 3.

Synthesized amino acid modifications.
Insertion of β-amino acids is known as a promising strategy leading to peptide analogues with interesting structural and functional behaviour.9 As an example, β-peptides or mixed α/β-peptides are more resistant against proteases and are able to form stable secondary structures with chain lengths as short as four residues.10 Alternating sequences of β3- and β2-amino acids results in unique frameworks for example, left handed 314-helices, helices consisting of alternating 10- and 12-membered hydrogen-bonded rings, or turn structures.11 This concept was successfully used in the development of more potent open-chain mimics of the tetrapeptide somatostatin.12 In addition, terminal homologation of NT(8–13) using β-amino acids resulted in derivatives with an enhanced stability.13 In this context, we were interested in the effect of replacing Tyr11 in NT(8–13) by corresponding β2-amino acids. Therefore, the two enantiomers of β2-homotyrosine described by Sebesta et al.14 have been used, and the two new β2-amino acids 4 a and 4 b, with an additional homologation in the side chain, have been synthesized (Scheme 1). Preparation of the two β2-amino acids 4 a and 4 b was conducted according to the procedure described by Seebach and co-workers,14 which is based on the use of 4-isopropyl-5,5-diphenyloxazolidin-2-one (DIOZ),15 a modified Evans oxazolidinone-auxiliary.16
Scheme 1.
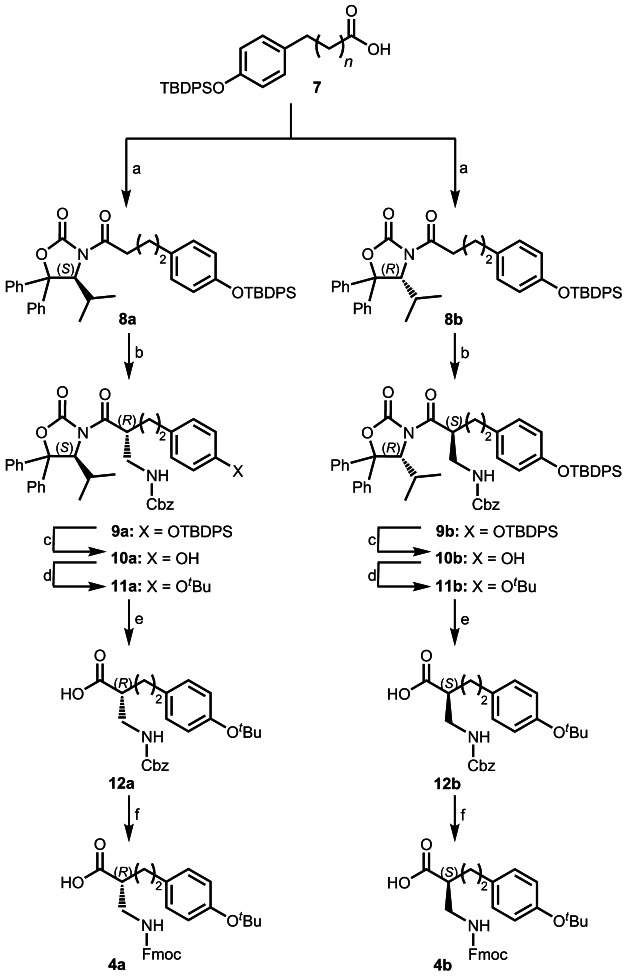
Reagents and conditions: a) (1) NEt3, PivCl, THF, −30 °C, 1.5 h; (2) LiCl, (4S)-isopropyl-5,5-diphenyloxazolidin-2-one (affording 8 a) or (4R)-isopropyl-5,5-diphenyloxazolidin-2-one (affording 8 a), −30 °C→RT in 14 h, 8 a: 99 %, 8 b: 80 %; b) (1) TiCl4, NEt3, CH2Cl2, −15 °C, 30 min; (2) CbzNHCH2OBn, TiCl4, 0 °C, 4 h, 9 a: 61 %, 9 b: 53 %; c) TBAF, THF, RT, 4 h, 10 a: 86 %, 10 b: 88 %; d) isobutene, CF3SO3H, CH2Cl2, −30 °C, 5.5 h, 11 a: 51 %,11 b: 58 %; e) 1 n NaOH, MeOH/THF (1:1), RT, 3 h, quant.; f) (1) H2, Pd/C, MeOH, RT, 2 h; (2) Fmoc-OSu, aq NaHCO3/dioxane, RT, 16 h, 4 a: 72 %, 4 b: 62 %.
N-Acylation of the oxazolidinone moiety was achieved with silyl-protected 3-(4-hydroxyphenyl)propionic acid 7 by the mixed anhydride method17 affording 8 a/b in 80–99 % yield (see Scheme 1). Next, amidomethylation of the Ti-enolate of acyl-DIOZ-derivatives 8 was carried out using benzyl N-[(benzyloxy)methyl]carbamate18 as an electrophile and proceeded with 53–67 % yield and 95 % diastereoselectivity, as determined by 1H NMR spectroscopy. After removal of the O-silyl protection by treatment with TBAF, the resulting free phenolic hydroxyl group was protected with a tert-butyl substituent, which is necessary for solid-phase peptide synthesis (SPPS), to afford the corresponding tert-butoxy-derivatives 11 a/b in 47–51 % yield over two steps. The diastereomer of interest was isolated in >99 % purity (preparative HPLC). Cleavage of the auxiliary proceeded smoothly to yield the carboxylic acid derivatives 12 a/b. Deprotection of the amino group and final 9-fluorenylmethoxycarbonyl (Fmoc) protection provided the desired enantiomerically pure β2-homo-amino acids 4 a/b (see Scheme 1).
Introduction of an additional nitrogen atom into the indole moiety of tryptophane leads to azatryptophane derivatives allowing to probe tryptophane-induced receptor–ligand interactions.19 We envisioned the synthesis of the 5-substituted azaindolyl alanine 5 as a promising tyrosine bioisostere mimicking the H-bond accepting properties of the hydroxyphenyl moiety of Tyr11 but not rendering H-bond donor function. Both enantiomers of this amino acid (5 a and 5 b) were synthesized as Fmoc-protected building blocks starting from 5-hydroxymethyl-pyrrazolo[1,5-a]pyridine (Scheme 2).20 Activation by 2,4,6-trichloro-1,3,5-triazine gave the chloromethyl-substituted electrophile 13, which was employed for a diastereoselective, nBuLi-promoted alkylation with both enantiomers of Schöllkopfs bislactimether21 affording the (2S)- and (2R)-dihydropyrazine derivatives 14 a and 14 b, respectively. Hydrolysis under acidic conditions followed by Fmoc-protection of the resulting amino acid provided the enantiomerically pure azaindolyl alanine derivatives 5 a and 5 b in 45 % overall yield.
Scheme 2.

Reagents and conditions: a) (1) 2,4,6-trichloro-1,3,5-triazine, DMF, RT, 75 min, 71 %; b) (1) (2R)-2,5-dihydro-3,6-dimethoxy-2-isopropylpyrazine (affording 14 a) or (2S)-2,5-dihydro-3,6-dimethoxy-2-isopropylpyrazine (affording 14 b), nBuLi (2.5 m in hexane), −78 °C, 15 min; (2) 13, THF, −78 °C, 1.5 h, 14 a: 91 %, 14 b: 72 %; c) 0.2 n HCl, RT, 4 h, 15 a: 81 %, 15 b: 80 %; d) Fmoc-OSu, aq NaHCO3/dioxane, RT, 16 h, 5a: 85 %, 5b: 73 %.
Replacement of the Tyr11 residue of NT(8–13) by a tyramine-derived peptoid moiety showed a strong impact on subtype selectivity.7 Because we aimed to investigate a bioisosteric exchange by a 5-substituted azaindolyl unit, synthesis of the building block 6 was intended (Scheme 3). Amination of vinyl-pyridine with ammonium chloride and a subsequent tert-butyloxycarbonyl (Boc) protection gave the pyridine derivative 17. Subsequent N-amination with O-(2,4-dinitrophenyl)-hydroxylamine22 afforded the corresponding N-aminopyridinium salt 18, which was subjected to a 1,3-dipolar cycloaddition with methyl propiolate and potassium carbonate under oxidative conditions. The thus formed pyrazolo[1,5-a]pyridinyl-3-carboxylate 19 was converted to the amino derivative 20 by treatment with aqueous hydrobromic acid. N-Alkylation with ethyl bromoacetate furnished the N-alkyl glycine derivative 21. Ester hydrolysis and subsequent protection using 9-fluorenylmethyl N-succinimidyl carbonate (Fmoc-OSu) gave access to the peptoid building block 6.
Scheme 3.

Reagents and conditions: a) NH4Cl, MeOH/H2O, NaOH, reflux, 17 h, 55 %; b) Boc2O, tBuOH, RT, 17 h, 85 %; c) O-(2,4-dinitrophenyl)-hydroxylamine, CH2Cl2/CH3CN, RT, 24 h, 77 %; d) (1) methyl propiolate, K2CO3, DMF, RT, 0.5 h; (2) air, RT, 16 h, 63 %; e) 48 % aq HBr, reflux, 2 h, 80 %; f) ethyl-2-bromoacetate, NEt3, THF, RT, 14 h, 60 %; g) (1) 1 n NaOH, MeOH, RT, 2 h; (2) Fmoc-OSu, aq NaHCO3/dioxane, RT, 20 h, 97 %.
SPPS was performed starting from Fmoc-leucinyl-loaded Wang resin (Scheme 4), and microwave irradiation was applied to accelerate Fmoc deprotection and amino acid coupling. Standard acylations were performed with (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) as coupling agent, whereas the β2-amino acids 4 a/b and (R)/(S)-β2-homotyrosine were incorporated using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU). Azaindolylalanine derivative coupling (5 a/b) was carried out with the help of N,N′-diisopropylcarbodiimide (DIC)/1-hydroxybenzotriazole (HOAt), affording the corresponding peptides 1 a–d, 2 a/b and 3.
Scheme 4.

Reagents and conditions: a) piperidine/DMF, μ-wave; b) standard coupling of the amino acids: Fmoc-AA-OH, PyBOP, DIPEA, HOBt, DMF, μ-wave; except for 4 a/b, Fmoc-(R)-β2 hTyr(tBu)-OH, Fmoc-(S)-β2 hTyr(tBu)-OH and Fmoc-Pro-OH: Fmoc-AA-OH, HATU, DIPEA, μ-wave; except for 5 a/b: Fmoc-AA-OH, DIC, HOAt, μ-wave; c) TFA/phenol/H2O/TIS, RT, 3 h.
Biological investigations
Radioligand binding studies were conducted to evaluate peptides 1 a–d, 2 a,b and peptide-peptoid hybrid 3 for their NTS1 and NTS2 affinity (Table 1). Binding data were determined utilizing the radioligand [3H]neurotensin and Chinese hamster ovary (CHO) cells stably expressing human NTS1. [3H]NT(8–13) was used for binding assays investigating human NTS2, which was transiently transfected in human embryonic kidney (HEK 293) cells.
Table 1.
Receptor binding data of the peptide-derivatives 1 a–d, 2 a, 2 b and 3 for the human NTS1 and NTS2 receptor in comparison with the reference agent NT(8–13) and 22.
| Compd | Sequence | Ki [nm][a] | Selectivity | |
|---|---|---|---|---|
| NTS1[b] [3H]neurotensin | NTS2[c] [3H]NT[8-13] | Ratio Ki [NTS1]/Ki [NTS2] | ||
| NT(8–13)[d] | 0.24±0.024 | 1.2±0.17[e] | 0.20 | |
| 22[d] |  |
31 000±7000 | 8.0±0.94 | 3900 |
| 1 a |  |
1700±780[f] | 2500±1800[f] | 0.68 |
| 1 b |  |
2500±490[f] | 3600±1500[f] | 0.69 |
| 1 c |  |
79 000±21 000 | 5600±810 | 14 |
| 1 d |  |
19 000±2400 | 5400±2200 | 3.5 |
| 2 a | 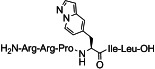 |
130±36 | 4.8±1.1 | 27 |
| 2 b | 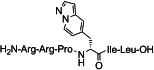 |
52 000±12 000 | 83±19 | 630 |
| 3 |  |
31 000±18 000 | 520±170 | 60 |
[a] Ki values in [nm±SEM] are the means of 3–9 individual experiments each done in triplicate. [b] Membranes from CHO cells stably expressing human NTS1. [c] Homogenates from HEK 293 cells transiently expressing human NTS2. [d] Data from ref. 7. [e] KD value. [f] Ki values in [nm±SD] are the means of two individual experiments each done in triplicate.
Binding data of the α/β-peptides 1 a–d revealed that the modification of the peptide backbone, which was obtained by the insertion of a CH2-group between the Cα-atom and the amine function of Tyr11, led to a decrease in affinity at both neurotensin receptor subtypes compared to the reference compound NT(8–13). Only a slightly increased NTS2 selectivity could be achieved with the (R)-β2-homotyrosine derivative 1 c. In contrast, peptide 2 a bearing an (S)-azaindolylalanine residue exhibits NTS2 affinity in the single-digit nanomolar range (4.8 nm) and almost 30-fold selectivity over NTS1. Interestingly, the corresponding (R)-epimer 2 b showed a more than 600-fold NTS2 selectivity and a Ki value of 83 nm. Noteworthy, our selectivity determination is based on binding and not on functional assays. Compared to hitherto reported NTS2-selective peptides using (R)-fluorophenyltyrosine8 or (R)-naphthylalanine4b as surrogates for Tyr11, 2 b revealed superior subtype selectivity. The peptide-peptoid hybrid 3 exhibited only weak binding affinity for NTS1 and NTS2 indicating that the hydroxyphenyl substituent of the tyramine substructure of the lead compound 22 cannot be successively replaced by a 5-substituted azaindole unit.
Computational chemistry
Radioligand binding studies showed an enhanced subtype selectivity of heterocyclic tyrosine bioisosteres of type 2 a compared to the parent compound NT(8–13). To help understanding the influence of this side chain modification on the subtype-selective binding of 2 a, we performed molecular dynamics (MD) simulations in a lipidic bilayer environment employing homology models of human NTS1 and NTS2 (based on the crystal structure of the agonist-bound rat NTS1) in complex with the peptide agonist NT(8–13) and compound 2 a.
As discussed above in more detail, the crystal structure of rat NTS1 shows that the side chain of Tyr11 is hydrogen bonded to the backbone oxygen of residue Leu55 located on the N terminus (Figure 1 A). The N-terminal domain thereby adopts a distinct conformation that features van der Waals interactions to the arene moiety of Tyr11. The conformation of the N-terminal sequence of the receptor is further stabilized by intramolecular interactions with the rat NTS1 residues Asp56 and His133EL1. In agreement with these observations, MD simulations on the human NTS1 receptor coupled to NT(8–13) revealed an analogous hydrogen bond between the side chain of Tyr11 and the backbone carbonyl of Leu54 (N terminus), which again leads to formation of a tightly packed sub-pocket around Tyr11 (Figure 4 A,C, figure S1 A in the Supporting Information). In addition to this hydrogen bond, the hydroxyl moiety of Tyr11 is alternately hydrogen bonded to the backbone carbonyl of His131EL1, thus participating in the stabilization of the conformation of Tyr11 (Figure 4 A, figure S1 B). In contrast, our MD simulations employing the human NTS1 receptor coupled to 2 a revealed a conformation of the azaindole moiety missing any hydrogen bonding to residues located at the N terminus of NTS1 (Figure 4 A). As a result, 2 a loses stabilizing hydrogen bonds and van der Waals interactions to the N-terminal region of NTS1 around its azaindole moiety (Figure 4 E), which we suggest is likely to be the reason for its reduced affinity.
Figure 4.

Ligand–receptor interactions within the MD simulations. The NTS1 and the NTS2 receptor are indicated as green and grey ribbons, respectively. Important amino acids of the receptors interacting with the ligands are indicated as sticks (A,B) and spheres (C–E). The ligand NT(8–13) is shown in dark-blue (A,C) and light-blue (B,D), whereas test compound 2 a is shown in orange (A,E) and dark-yellow (B,F).
For the human NTS2 subtype, we observed a different conformation around the tyrosine side chain of NT(8–13), which forms a hydrogen bond to the backbone oxygen of residue His98EL1, thereby occupying a more deeply buried sub-pocket compared to NTS1 (Figure 4 B,D, figure S1 C). Thus, only a minor contribution to the stabilization of the conformation of Tyr11 seems to arise from the N-terminal portion of the receptor at NTS2. In analogy to the model of the NT(8-13)–NTS2 complex, we found a similar orientation for the bioisosteric azaindole moiety of compound 2 a compared to the hydroxyphenyl unit of NT(8–13). Although there is no hydrogen bond between the tyrosine side chain of NT(8–13) and residues of EL1, the heteroaromatic system of compound 2 a seems to be sufficiently stabilized by substantial van der Waals interactions, which is in agreement with our biological data showing only a moderate reduction of affinity for 2 a compared to NT(8–13) at NTS2 (Figure 4 F).
Conclusions
Starting from NT(8–13) as lead compound, we herein report the synthesis and biological evaluation of peptides containing structural variations at position 11, which include a replacement of the tyrosine unit by β2-amino acid residues (type 1) as well as incorporation of heterocyclic tyrosine bioisosteres (type 2) and the corresponding peptoid analogue of type 3. Taking advantage of Seebachs β2-amino acid synthesis and Schöllkopfs bis-lactim ether methodology, we were able to assess the new unnatural amino acids 4 and 5, respectively, which could be incorporated into the parent peptide NT(8-13) exchanging the residue Tyr11. Receptor binding data of corresponding α/β-peptides 1 a–d suggest that backbone homologation in this position by introducing β2-amino acids is hardly tolerated by the neurotensin receptors, probably due to a loss of backbone interactions, an unfavourable backbone conformation or repulsive steric interactions with the protein residues surrounding the proline residue of the ligand. However, bioisosteric replacement of the hydrogen-bond donating hydroxyphenyl side chain of Tyr11 by a hydrogen-bond accepting azaindole moiety with an extended aromatic moiety was well tolerated by NTS2 but not by the NTS1 subtype. Comparative computational studies performed on the S-configured test compound 2 a and NT(8–13) suggested possible structural consequences of such side chain modifications on ligand–receptor interactions, which finally culminate in an altered selectivity profile. Interestingly, peptide 2 b with an R-configured azaindolylalanine at position 11 showed reduced binding affinity but more than 600-fold subtype selectivity for NTS2 over NTS1. Taking advantage of the newly presented NTS2-selective chemical tools and lead compounds, further structural evolution may lead to nonpeptidic drug candidates for the treatment of neuropathic pain.
Experimental Section
Syntheses of peptides 1–3
The peptide synthesis was done according to standard protocols as described below. The synthesis was performed starting from commercially available Fmoc-Leu-Wang resin. α-Amino acids were incorporated as their commercially available derivatives: Fmoc-Ile-OH, Fmoc-Pro-OH and Fmoc-Arg(Pbf)-OH. Elongation of the peptide chain was done by repetitive cycles of Fmoc deprotection and subsequent coupling of the amino acid (AA) derivative with the help of microwave irradiation (Discover microwave oven, CEM Corp.), performed in silanized glass tubes. Remark: the development of overpressure was avoided by using N,N-dimethylformamide (DMF) as the solvent and intermittent cooling. Fmoc deprotection was performed by treatment of the resin with 20 % piperidine in DMF (1×, microwave irradiation: 8×5 s, 100 W), followed by washings with DMF (5×). Peptide coupling was done following methods 1–3. After the last acylation step the N-terminal Fmoc-residue was deprotected, the resin was rinsed with CH2Cl2 (5×) and dried in vacuo. The cleavage from the resin was performed using a mixture of trifluoroacetic acid (TFA)/phenol/H2O/triisopropylsilane (TIS) (88:5:5:2) for 3 h, followed by filtration of the resin. After evaporation of the solvent in vacuo and precipitation in tert-butylmethylether, the crude peptides were purified using preparative RP-HPLC: Agilent 1100 preparative series, column: Zorbax Eclipse XDB-C8, 21.2×150 mm, 5 μm particles [C8], employing solvent systems, linear gradient and flow rate (FR) as specified below or VP 250/32 NUCLEODUR C18 HTec 5 μm particles [C18] employing solvent systems, linear gradient and FR as specified below.
After separation, peptides were lyophilized and peptide purity and identity were assessed by analytical HPLC (Agilent 1100 analytical series, equipped with QuatPump and VWD detector, column: Zorbax Eclipse XDB-C8 analytical column, 4.6×150 mm, 5 μm, FR: 0.5 mL min−1) coupled to a Bruker Esquire 2000 mass detector equipped with an ESI trap. System 1 (S1): x–y % CH3OH in H2O+0.1 % HCO2H (S1C: 10–55 % in 18 min, 55–95 % in 2 min, 95–95 % in 2 min; S1D: 10–65 % in 18 min, 65–95 % in 2 min, 95–95 % in 2 min), System 2 (S2): x–y % CH3CN in H2O + 0.1 % HCO2H (S2B: 3–40 %, in 26 min, 40–95 % in 2 min, 95–95 % in 2 min; S2C: 3–55 %, in 26 min, 65–95 % in 2 min, 95–95 % in 2 min).
Method 1: Peptide coupling was done employing Fmoc-AA/PyBOP/DIPEA (5 equiv each) and 1-hydroxybenzotriazole (HOBt, 7.5 equiv) dissolved in a minimum amount of DMF (irradiation: 20×10 s, 50 W). For introduction of the β2h-AA (3 equiv), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU, 5 equiv) and N,N-diisopropylethylamine (DIPEA, 10 equiv) were employed (irradiation: 20×10 s, 50 W). In between each irradiation step, cooling of the reaction mixture to a temperature of −10 °C was achieved by sufficient agitation in an EtOH/ice bath.
Method 2: Peptide coupling was done employing Fmoc-AA/PyBOP/DIPEA (5 equiv each) and HOBt (7.5 equiv) dissolved in a minimum amount of DMF (irradiation: 20×10 s, 50 W). For introduction of azaindolylalanines 5 a/b, Fmoc-AA/DIC/HOAt (4.3 equiv each) in DMF were employed using 10 min preactivation time. In between each irradiation step, cooling of the reaction mixture to a temperature of −10 °C was achieved by sufficient agitation in an EtOH/ice bath.
Method 3: Peptide coupling was done employing Fmoc-AA/PyBOP/DIPEA (5 equiv each) and HOBt (7.5 equiv) dissolved in a minimum amount of DMF (irradiation: 20×10 s, 50 W). Peptoid building block 6 was coupled in the following manner: Fmoc-Peptoid/PyBOP/DIPEA (3 equiv each) and HOBt (4.5 equiv) dissolved in a minimum amount of DMF (irradiation: 20×10 s, 50 W). The following Fmoc-Pro-OH (5 equiv) was coupled (2×) with HATU (5 equiv) and DIPEA (10 equiv) in DMF. In between each irradiation step, cooling of the reaction mixture to a temperature of −10 °C was achieved by sufficient agitation in an EtOH/ice bath.
[11(R)-2-[(Amino)methyl]-4-[4-(hydroxy)phenyl]-1-oxobutyl]-NT(8–13) (1 a): The peptide was synthesized according to method 1. Purification [C8]: eluent: CH3CN (A) and 0.1 % HCOOH in H2O (B) applying a linear gradient starting from 5 % A in 95 % B to 35 % A in 65 % B in 12.0 min; FR: 10.0 mL min−1; tR=9.7 min; purity: S1C: >99 % (tR=17.0 min); S2B: >99 % (tR=16.4 min); MS (ESI): m/z 845.6 [M+H]+; HRMS-ESI: m/z [M+H]+ calcd for C40H68N12O8: 845.5353, found: 845.5356.
[11(S)-2-[(Amino)methyl]-4-[4-(hydroxy)phenyl]-1-oxobutyl]-NT(8–13) (1 b): The peptide was synthesized according to method 1. Purification [C8]: eluent: CH3CN (A) and 0.1 % HCOOH in H2O (B) applying a linear gradient starting from 5 % A in 95 % B to 35 % A in 65 % B in 12.0 min; FR: 10.0 mL min−1; tR=10.3 min; purity: S1C: >99 % (tR=17.2 min); S2B: >99 % (tR=16.4 min); MS (ESI): m/z 845.6 [M+H]+; HRMS-ESI: m/z [M+H]+ calcd for C40H68N12O8: 845.5356, found: 845.5377.
[11(R)-β2 hTyr]-NT(8–13) (1 c): The peptide was synthesized according to method 1 using 10 a as β2-amino acid. Purification [C8]: eluent: CH3CN (A) and 0.1 % HCOOH in H2O (B) applying a linear gradient starting from 5 % A in 95 % B to 40 % A in 60 % B in 14.0 min; FR: 10.0 mL min−1; tR=12.3 min; purity: S1C: >99 % (tR=16.9 min); S2B: >99 % (tR=14.8 min); MS (ESI): m/z 831.6 [M+H)]+; HRMS-ESI: m/z [M+H]+ calcd for C39H66N12O8: 831.5199, found: 831.5201.
[11(S)-β2 hTyr]-NT(8–13) (1 d): The peptide was synthesized according to method 1. Purification [C8]: eluent: CH3CN (A) and 0.1 % HCOOH in H2O (B) applying a linear gradient starting from 5 % A in 95 % B to 35 % A in 65 % B in 12.0 min; FR: 10.0 mL min−1; tR=9.1 min; purity: S1C: >99 % (tR=13.9 min); S2B: >99 % (tR=14.0 min); MS (ESI): m/z 831.6 [M+H]+; HRMS-ESI: m/z [M+H]+ calcd for C39H66N12O8 :831.5199, found: 831.5215.
[(S)-3-(Pyrazolo[1,5-a]pyridine-5-yl)-propionyl11]NT(8–13) (2 a): The peptide was synthesized according to method 2. Purification [C8]: eluent: MeOH (A) and 0.1 % HCOOH in H2O (B) applying a linear gradient starting from 10 % A in 90 % B to 40 % A in 60 % B in 14.0 min, then 40 % A in 60 % to 40 % A in 60 % for 2 min; FR: 10.0 mL min−1; tR=12.4 min (95.5 %) [(R)-3-(pyrazolo[1,5-a]pyridine-5-yl)-propionyl11-epimer: 4.5 %]; purity: S1D: >99 % (tR=12.9 min); S2C: 98.0 % (tR=12.7 min); HRMS-ESI: m/z [M+H]+ calcd for C39H65N14O7: 841.5161, found: 841.5163.
[(R)-3-(Pyrazolo[1,5-a]pyridine-5-yl)-propionyl11]NT(8–13) (2 b): The peptide was synthesized according to method 2. Purification [C8]: eluent: MeOH (A) and 0.1 % HCO2H in H2O (B) applying a linear gradient starting from 10 % A in 90 % B to 40 % A in 60 % B in 14.0 min, then 40 % A in 60 % to 40 % A in 60 % for 2 min; FR: 10.0 mL min−1; tR=15.0 min (87.4 %) [(S)-3-(pyrazolo[1,5-a]pyridine-5-yl)-propionyl11-epimer: 12.6 %]; purity: S1D: >99.2 % (tR=14.8 min); S2C: 99.3 % (tR=14.2 min); HRMS-ESI: m/z [M+H]+ calcd for C39H65N14O7: 841.5161, found: 841.5154.
[11N-{5-(2-Aminoethyl)pyrazolo[1,5-a]pyridine}-Gly]NT(8–13) (3): The peptide was synthesized according to method 3. Purification [C18]: eluent: CH3CN (A) and 0.1 % HCOOH in H2O (B) applying a linear gradient starting from 5 % A in 95 % B to 30 % A in 70 % B in 12.0 min; FR: 32.0 mL min−1; tR=10.6 min; purity: S1C: >99 % (tR=15.1 min); S2B: >99 % (tR=16.0 min); MS (ESI): m/z 855.6 [M+H]+; HRMS-ESI: m/z [(M+2 H)/2]2+ calcd for C40H66N14O7H2: 428.2692, found: 428.2690.
Syntheses of compounds 4–6
Reagents and dry solvents were obtained from commercial sources and were used as received. Reactions were conducted under dry N2. Evaporations of product solutions were done in vacuo with a rotatory evaporator. Column chromatography was performed using 60 μm silica gel. For thin-layer chromatography (TLC), silica gel 60 μm plates were used (UV, I2 or ninhydrin detection). Preparative RP-HPLC was performed employing Agilent 1100 preparative series, column Zorbax Eclipse XDB-C8, 21.2 mm×150 mm, 5 μm particles [C8], employing solvent systems, linear gradient and FR as specified below or VP 250/32 NUCLEODUR C18 HTec 5 μm particles [C18] employing solvent systems, linear gradient and FR as specified below. Melting temperatures (mp) are uncorrected. IR spectra were registered from a thin film on a NaCl crystal. NMR data were acquired with the help of a 360 MHz or a 600 MHz spectrometer, if not stated otherwise in CDCl3; 13C NMR spectra were recorded at 90 MHz if not stated otherwise in CDCl3. Chemical shifts (δ) are given relative to tetramethylsilane (TMS) in parts per million (ppm) relative to TMS (=0). Mass spectra were acquired using electronic ionization (EI), atmospheric pressure chemical ionization (APCI) or electron spray ionization (ESI) techniques. EI high-resolution mass spectra were measured with a JOEL GCmate II spectrometer. ESI-TOF high mass accuracy and resolution experiments were performed on a Bruker maXis MS (Bruker Daltonics, Bremen, Germany) in the laboratory of the Chair of Bioinorganic Chemistry (Prof. Dr. Ivana Ivanović-Burmazović), FAU. CHN analyses were conducted in the analytical laboratory of the Division of Organic Chemistry, FAU. HPLC analysis revealed a purity >95 % for all SAR compounds. Purity was assessed by analytical HPLC (Agilent 1100 analytical series, equipped with QuatPump and VWD detector, column: Zorbax Eclipse XDB-C8 analytical column, 4.6×150 mm, 5 μm, FR: 0.5 mL min−1); System 1 (S1): x–y % CH3OH in H2O+0.1 % HCO2H (S1 A: 4–15 % in 18 min, 90–95 % in 2 min, 95–95 % in 2 min, S1B: 10–90 % in 18 min, 90–95 % in 2 min, 95–95 % in 2 min), System 2 (S2): x–y % CH3CN in H2O+0.1 % HCO2H (S2 A: 3–85 % in 26 min, 85–95 % in 2 min, 95–95 % in 2 min).
(R)-2-[({[(9 H-Fluoren-9-yl)methoxy]carbonyl}amino)methyl]-4-[4-(tert-butoxy)-phenyl]-butanoic acid (4 a): To a solution of 12 a (0.5 g, 1.3 mmol) in MeOH (20 mL), Pd/C (0.06 g, 10 wt %) was added. The flask was evacuated, flushed with H2 (3×), and the mixture war stirred at RT for 2 h. The catalyst was filtered off with the help of celite and washed thoroughly with MeOH. The solvent was evaporated, and the resulting colorless oil was diluted with saturated aq NaHCO3 (23 mL). A solution of 9-fluorenylmethyl N-succinimidyl carbonate (Fmoc-OSu, 0.38 g, 1.3 mmol) in dioxane (8 mL) was added, and the mixture was stirred at RT for 12 h. The solvent was evaporated, H2O was added, and the aqueaous phase was washed with Et2O (1×). The aqueous layer was acidified with 2 n HCl and extracted with CH2Cl2 (3×). The combined organic extracts were dried (MgSO4), flitered and evaporated. Flash column chromatography (EtOAc/hexane, 1:2+0.5 % HCO2H) afforded 4 a as colorless solid (0.4 g, 72 %): mp: 71–73 °C; [α]D31=−4.0 (c=0.27 in CHCl3); 1H NMR (360 MHz, CDCl3): δ=7.74 (d, J=7.5 Hz, 2 H), 7.58 (d, J=7.2 Hz, 2 H), 7.45–7.35 (m, 2 H), 7.34–7.28 (m, 2 H), 7.10–7.00 (m, 2 H), 6.92–6.83 (m, 2 H), 6.55, 5.28 (2×bs, 1 H), 4.58–4.47, 4.46–4.37 (2 m, 2 H), 4.28–4.16 (m, 1 H), 3.51–3.37, 3.30–3.07 (2 m, 2 H), 2.73–2.58 (m, 2 H), 2.57–2.36 (m, 1 H), 2.04–1.94, 1.88–1.76, 1.68–1.54 (3 m, 2 H), 1.32 ppm (s, 9 H); 13C NMR (90 MHz, CDCl3): δ=179.4, 156.6, 153.5, 143.8, 141.3, 135.8, 128.8, 128.6, 127.8, 127.7, 127.1, 127.0, 125.0, 124.2, 120.0, 78.3, 67.2, 66.8, 47.2, 45.0, 42.5, 41.9, 32.5, 32.4, 31.5, 31.2, 28.8 ppm; IR (NaCl): ν=3337, 2977, 1709, 1507, 1450, 1365, 1235, 1160, 1105, 911, 851 cm−1; MS (ESI): m/z 510.3 [M+Na]+, HRMS-ESI: m/z [M+Na]+ calcd for C30H33NO5: 510.2251, found: 510.2245.
(S)-2-[({[(9 H-Fluoren-9-yl)methoxy]carbonyl}amino)methyl]-4-[4-(tert-butoxy)phenyl]butanoic acid (4 b): Compound 4 b was synthesized according to the procedure described for compound 4 a starting with 12 b (0.40 g, 1.0 mmol). Compound 4 b was obtained as a colorless solid (0.3 g, 62 %). Spectroscopic characterization data were identical to those obtained for the enantiomer 4 a. [α]D31=+4.1 (c=0.27 in CHCl3).
(S)-2-(9 H-Fluoren-9-ylmethoxycarbonylamino)-3-(pyrazolo[1,5-a]pyridine-5-yl)-propionic acid (5 a): A solution of Fmoc-OSu (99 mg, 0.29 mmol) in dioxane (1.5 mL) was added to a solution of 15 a (72 mg, 0.22 mmol) in a saturated aq NaHCO3 (2 mL) at RT. The solution was adjusted to pH 9.0–9.5, and stirring at RT was performed for 16 h. After the addition of H2O, the resulting suspension was extracted with CH2Cl2 and the organic layer was discarded. Thereafter, the aqueous layer was adjusted to pH 2 with a saturated aq citric acid and extracted with CH2Cl2 (5×). The combined organic extracts were dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (CH2Cl2/MeOH, 99:1+0.5 % HCO2H) afforded 5 a as a colorless solid (82 mg, 85 %): mp: 187–193 °C (decomposition); [α]D21=−18.1 (c=0.52 in DMF); 1H NMR (600 MHz, [D6]DMSO): δ=12.84 (s, 1 H), 7.51 (s, 1 H), 8.57 (d, J=7.2 Hz, 1 H), 7.93 (d, J=2.3 Hz, 1 H), 7.84–7.88 (m, 2 H), 7.78 (d, 1 H, J=8.4 Hz), 7.56–7.63 (m, 2 H), 7.51 (s, 1 H), 7.36–7.44 (m, 2 H), 7.24–7.28 (m, 1 H), 7.18–7.22 (m, 1 H), 6.81 (dd, J=7.2, 1.8 Hz, 1 H), 6.49 (d, J=2.3 Hz, 1 H), 4.27 (ddd, J=10.5, 8.4, 4.3 Hz, 1 H), 4.13–4.22 (m, 4 H), 3.12 (dd, J=13.8, 4.3 Hz, 1 H), 2.91 ppm (dd, J=13.8, 10.5 Hz, 1 H); 13C NMR (150 MHz, [D6]DMSO): δ=173.0, 115.9, 143.7, 143.6, 141.8, 140.6, 139.4, 133.9, 128.1, 127.6, 127.0, 126.9, 125.2, 125.1, 120.1, 117.2, 113.7, 96.1, 65.6, 54.7, 46.5, 35.8 ppm; IR (KBr): ν=3413, 3320, 1713, 1692 cm−1; MS (APCI): m/z 428 [M+H]+; HRMS-ESI: m/z [M]+ calcd for C25H21N3O4: 427.1532, found: 427.1532; HPLC (220 nm) system S1B: tR=21.0 min (96.7 %), system S2 A: tR=22.0 min (96.7 %).
(R)-2-(9 H-Fluoren-9-ylmethoxycarbonylamino)-3-(pyrazolo[1,5-a]pyridine-5-yl)-propionic acid (5 b): Compund 5 b was prepared according to the protocol for the synthesis of 5 a starting with 15 b (0.07 g, 0.22 mmol). Compound 5 b was obtained as a colorless solid (0.067 g, 73 %). Spectroscopic characterization data were identical to those obtained for the enantiomer 5 a, starting from 15 b. [α]D23=+14.0 (c=0.95 in DMF); HPLC (220 nm) system S1B: tR=20.7 min (98.1 %), system S2 A: tR=21.9 min (97.9 %).
N-(9-Fluorenylmethyloxycarbonyl)-N-{5-(2-aminoethyl)pyrazolo[1,5-a]pyridine}-glycine (6): To a solution of 21 (0.36 g, 1.44 mmol) in MeOH (7 mL), 1 n NaOH (3.5 mL) was added, and stirring was performed at RT for 3 h. The mixture was concentrated in vacuo, and the residue was diluted with a saturated aq NaHCO3. After cooling to 0 °C, a solution of Fmoc-OSu (0.52 g, 1.53 mmol) in dioxane (2 mL) was added, and the mixture was stirred at RT for 19 h. The solvent was evaporated, H2O was added and the aqueous phase was washed with Et2O (1×). Thereafter, the aqueous layer was acidified with 2 n HCl and extracted with CH2Cl2 (3×). The combined organic extracts were dried (MgSO4), filtered and concentrated in vacuo yielding 6 as colorless solid (0.62 g, 97 %): mp: 71–73 °C; 1H NMR (600 MHz, CDCl3): δ=8.49a, 8.42b (2×d, Ja=7.0 Hz, Jb=7.0 Hz, 1 H), 7.94–7.88 (m, 1 H), 7.72 (d, J=7.5 Hz, 2 H), 7.58a, 7.53b (2×d, Ja=7.4 Hz, Jb=7.5 Hz, 2 H), 7.40–7.29, 7.25–7.22, 7.02–6.97 (3 m, 5 H), 6.62a, 6.24b (2×d, Ja=6.3 Hz, Jb=5.9 Hz, 1 H), 6.41 (d, J=1.3 Hz, 1 H), 4.70a, 4.46b (2×d, Ja=5.0 Hz, Jb=6.4 Hz, 2 H), 4.24–4.18 (m, 1 H), 3.89, 3.85 (2 s, 2 H), 3.63–3.56, 3.28–3.17 (2 m, 2 H), 2.92–2.87, 2.49–2.42 ppm (2 m, 2 H); 13C NMR (150 MHz, CDCl3): δ=172.9, 156.4, 155.8, 143.8, 141.4, 141.3, 140.1, 128.2, 127.8, 127.7, 127.2, 127.0, 124.9, 124.6, 120.0, 116.9, 116.7, 113.7, 113.5, 96.4, 67.7, 66.8, 49.9, 49.7, 49.3, 47.4, 47.2, 34.0, 33.9 ppm; IR (NaCl): ν=3438, 2952, 1699, 1647, 1477, 1447, 1218, 1180, 1123, 759, 739 cm−1; MS (ESI): m/z 442.2 [M+H]+; HRMS-ESI: m/z [M+Na]+ calcd for C26H23N3O4: 464.1581, found: 464.1575.
4-{4-[(tert-Butyl)diphenylsilyl]phenyl}butanoic acid (7): To a solution of 4-(4-hydroxyphenyl)butanoic acid23 (3.13 g, 17 mmol) and imidazole (5.2 g, 76 mmol) in DMF (45 mL), tert-butyl-diphenylsilylchloride (9.9 mL, 38 mmol) was added dropwise and stirred at RT for 4.5 h. After addition of brine (450 mL), the reaction mixture was extracted with Et2O (3×). The combined organic extracts were washed with ice-cold 1 n HCl and brine, dried (MgSO4), filtered and concentrated in vacuo. The residue was redissolved in MeOH/THF (1:1, 144 mL) and 10 % aq K2CO3 (22.5 mL) and stirred at RT for 1 h. After evaporation, the residue was diluted with brine (180 mL), acidified with 1 n HCl and extracted with Et2O (3×). Organic extracts were washed with brine, dried (MgSO4), filtered and concentrated in vacuo. Crude product was purified by flash column chromatography (hexane/Et2O, 4:1+0.5 % HCO2H) to yield acid 7 as a colorless oil (6.9 g, 98 %): 1H NMR (390 MHz, CDCl3): δ=7.73–7.68 (m, 4 H), 7.43–7.39 (m, 2 H), 7.37–7.33 (m, 4 H), 6.90–6.86 (m, 2 H), 6.69–6.66 (m, 2 H), 2.52 (t, J=7.6 Hz, 2 H), 2.29 (t, J=7.6 Hz, 2 H), 1.92–1.86 (m, J=7.6 Hz, 2 H), 1.09 ppm (s, 9 H); 13C NMR (150 MHz, CDCl3): δ=179.3, 153.8, 133.5, 133.1, 129.8, 129.1, 127.7, 119.5, 34.1, 33.1, 26.5, 26.3, 19.4 ppm; IR(NaCl): ν=3431, 3072, 2931, 2856, 1705, 1608, 1500, 1428, 1254, 1117, 922, 822 cm−1; MS(ESI): m/z 441.7 [M+Na]+.
(S)-3-{4-[4-(tert-Butyl)diphenylsilyl)phenyl]-1-oxobutyl}-4-isopropyl-5,5-diphenyl-oxazolidin-2-one (8 a): A solution of 7 (1.34 g, 2 mmol) and NEt3 (1.11 mL, 7.9 mmol) in THF (15 mL) was cooled to −30 °C, and pivaloyl chloride (PivCl, 0.4 mL, 3.2 mmol) was added dropwise. The white suspension was stirred for 1.5 h, and LiCl (0.15 g, 3.5 mmol) and (4S)-4-(1-methylethyl)-5,5-diphenyl-2-oxazolidinone (0.86 g, 3.2 mmol) were added and stirred overnight, allowing to warm to RT. The reaction mixture was diluted with Et2O and saturated aq NH4Cl. The organic phase was washed with 1 n HCl (2×), 1 n NaOH (1×) and brine (1×), dried (MgSO4), filtered and evaporated. Flash column chromatography (EtOAc/hexane, 1:4) afforded 8 a as a colorless solid (2.06 g, 99 %): mp: 57–60 °C; [α]D25=−95.4 (c=1.05 in CHCl3); 1H NMR (600 MHz, CDCl3): δ=7.74–7.66 (m, 4 H), 7.49–7.17 (m, 18 H), 6.80–6.74 (m, 2 H), 6.65–6.59 (m, 2 H), 5.35 (d, J=3.4 Hz, 1 H), 2.91–2.77, 2.77–2.64 (2 m, 2 H), 2.45–2.37 (m, 2 H), 2.01–1.91 (m, 1 H), 1.82–1.72 (m, 2 H), 1.09 (s, 9 H), 0.86 (d, J=6.8 Hz, 3 H), 0.74 ppm (d, J=6.8 Hz, 3 H); 13C NMR (150 MHz, CDCl3): δ=172.9, 153.7, 153.0, 142.4, 138.2, 135.5, 133.8, 133.2, 129.8, 129.0, 128.9, 128.6, 128.4, 127.9, 127.7, 125.9, 125.6, 119.4, 89.3, 64.5, 34.4, 34.0, 29.8, 26.6, 26.3, 21.7, 19.4 ppm; IR (NaCl): ν=3072, 2930, 2932, 2857, 1791, 1703, 1607, 1500, 1452, 1429, 1362, 1318, 1254, 1175, 1118, 998, 919, 843, 822 cm−1; MS (ESI): m/z 705.2 [M+Na]+; HRMS-ESI: m/z [M+Na]+ calcd for C44H47NO4Si: 704.3167, found: 704.3171.
(R)-3-{4-[4-(tert-Butyl)diphenylsilyl)phenyl]-1-oxobutyl}-4-isopropyl-5,5-diphenyloxazolidin-2-one (8 b): Compound 8 b was synthesized according to the procedure described for 8 a starting from 7 (3.0 g, 7.2 mmol) and (4R)-4-(1-methylethyl)-5,5-diphenyl-2-oxazolidinone (1.9 g, 6.8 mmol). Compound 8 b was obtained as a colorless solid (3.7 g, 80 %). Spectroscopic characterization data were identical to those obtained for the enantiomer 8 a: [α]D25= +94.6 (c=1.07 in CHCl3).
(S)-3-((R)-2-{[(Benzyloxy)carbonyl]amino})methyl-4-[4-(tert-butyl)diphenylsilyl)-phenyl]-1-oxobutyl)-4-isopropyl-5,5-diphenyloxazolidin-2-one (9 a): To a solution of 8 a (3.6 g, 5.0 mmol) in CH2Cl2 (27 mL), TiCl4 (0.86 mL, 5.8 mmol) and then NEt3 (0.86 mL, 5.8 mmol) was added at −15 °C. The resulting dark-red solution was stirred at −15 °C for 0.5 h. Benzyl-N-[(benzyloxy)-methyl]carbamate (CbzNHCH2OBn; 1.64 g, 5.8 mmol) and TiCl4 (0.67 mL, 5.8 mmol) were dissolved in CH2Cl2 (13 mL) in an ice bath, and the resulting solution was added to the mixture via cannula. The reaction mixture was stirred at 0 °C for 4.5 h, treated with saturated aq NaHCO3 and CH2Cl2. The organic extract was washed with 1 n HCl (2×), 1 n NaOH (1×) and brine (1×), dried (MgSO4), filtered and evaporated. Flash column chromatography (EtOAc/hexane, 1:4) afforded 9 a as a colorless solid (2.72 g, 61 %, d.r.: 19): mp: 59–61 °C; 1H NMR (600 MHz, CDCl3): δ=7.74–7.69 (m, 4 H), 7.47–7.26 (m, 18 H), 7.16 (dd, J=7.9, 7.9 Hz, 2 H), 7.02 (dd, J=7.4, 7.4 Hz, 1 H), 6.55–6.50 (m, 2 H), 6.42–6.35 (m, 2 H), 5.34 (d, J=3.4 Hz, 1 H), 5.18 (bs, 1 H), 5.06 (s, 2 H), 3.79–3.71 (m, 1 H), 3.59–3.47, 3.39–3.32 (2 m, 2 H), 2.03–1.86 (m, 3 H), 1.67–1.59 (m, 2 H), 1.09 (s, 9 H), 0.83 (d, J=7.1 Hz, 3 H), 0.70 ppm (d, J=6.9 Hz, 3 H); 13C NMR (150 MHz, CDCl3): δ=174.5, 156.2, 153.6, 153.0, 142.2, 137.7, 136.5, 135.5, 133.4, 133.2, 133.1, 129.8, 128.9, 128.7, 128.70, 128.6, 128.5, 128.4, 128.1, 128.0, 128.00, 127.7, 125.7, 125.3, 119.3, 89.6, 66.7, 65.3, 42.9, 42.5, 31.7, 31.3, 29.6, 26.5, 25.9, 21.6, 19.4 ppm; IR (NaCl): ν=2931, 2858, 1783, 1722, 1607, 1509, 1450, 1428, 1362, 1316, 1252, 1175, 1113, 1051, 1000, 913, 822 cm−1; MS (ESI): m/z 868.1 [M+Na]+; HRMS-ESI: m/z [M+Na]+ calcd for C53H56N2O6Si: 867.3800, found: 867.3792.
(R)-3-((S)-2-{[(Benzyloxy)carbonyl]amino})methyl-4-[4-(tert-butyl)diphenylsilyl)-phenyl]-1-oxobutyl)-4-isopropyl-5,5-diphenyloxazolidin-2-one (9 b): Compound 9 b was synthesized according to the procedure described for 9 a starting with 8 b (3.5 g, 5.0 mmol). Compound 9 b was obtained as a colorless solid (2.3 g, 53 %, d.r.: 19). Spectroscopic characterization data were identical to those obtained for the enantiomer 9 a.
(S)-3-[(R)-2-({[(Benzyloxy)carbonyl]amino})methyl-4-(4-hydroxyphenyl)-1-oxobutyl]-4-isopropyl-5,5-diphenyloxazolidin-2-one (10 a): A mixture of 9 a (1.3 g, 1.5 mmol) in THF (12 mL) and tetra-n-butylammonium fluoride (TBAF, 1.8 mL, 1 m in THF) was stirred at RT for 4 h. The mixture was diluted with EtOAc, washed with H2O (1×) and brine (1×). The combined organic extracts were dried (MgSO4), filtered and evaporated. Flash column chromatography (EtOAc/hexane, 1:3→1:2) afforded 10 a as colorless solid (0.8 g, 86 %, d.r.: 19): mp: 75–76 °C; 1H NMR (360 MHz, CDCl3): δ=7.56–7.50 (m, 2 H), 7.45–7.27 (m, 12 H), 7.22–7.16 (m, 1 H), 6.68–6.61 (m, 2 H), 6.59–6.54 (m, 2 H), 5.39 (d, J=3.5 Hz, 1 H), 5.24 (bs, 1 H), 5.15 (s, 1 H), 5.10–5.00 (m, 2 H), 3.86–3.79 (m, 1 H), 3.59–3.50, 3.41–3.34 (2 m, 2 H), 2.04–1.93 (m, 3 H), 1.76–1.63, 1.48–1.35 (2 m, 2 H), 0.85 (d, J=7.0 Hz, 3 H), 0.72 ppm (d, J=6.7 Hz, 3 H); 13C NMR (150 MHz, CDCl3): δ=174.6, 156.3, 153.7, 153.1, 142.2, 137.7, 136.4, 133.0, 129.2, 129.0, 128.8, 128.5, 128.4, 128.1, 128.0, 125.8, 125.7, 125.5, 115.1, 89.8, 66.7, 65.3, 43.0, 42.5, 31.6, 30.9, 29.6, 21.7 ppm; IR (NaCl): ν=3381, 2965, 1782, 1701, 1614, 1515, 1450, 1362, 1317, 1212, 1176, 1052, 989, 912 cm−1; MS (ESI): m/z 615.4 [M+Na]+; HRMS-ESI: m/z [M+Na]+ calcd for C37H38N2O6: 629.2622, found: 629.2619.
(R)-3-((S)-2-{[(Benzyloxy)carbonyl]amino})methyl-4-[4-hydroxyphenyl]-1-oxobutyl)-4-isopropyl-5,5-diphenyloxazolidin-2-one (10 b): Compound 10 b was synthesized according to the procedure described for 10 a starting with 9 b (2.2 g, 2.6 mmol). Compound 10 b was obtained as a colorless solid (1.4 g, 88%, d.r.: 19). Spectroscopic characterization data were identical to those obtained for the enantiomer 10 a.
(S)-3-[(R)-2-({[(Benzyloxy)carbonyl]amino})methyl-4-(4-(tert-butoxy)phenyl)-1-oxobutyl]-4-isopropyl-5,5-diphenyloxazolidin-2-one (11 a): Isobutene (1.0 mL) was dissolved in CH2Cl2 (1 mL) at −78 °C. At −30 °C first CF3SO3H (6 μL, 0.04 mmol) and afterwards a solution of 10 a (0.21 g, 0.35 mmol) in CH2Cl2 (4 mL) was added, and the mixture was stirred at −30 °C for 5 h. NEt3 (6 μL, 0.04 mmol) was added, and the solution was allowed to warm slowly to RT, before the solvent was evaporated. Purification via preparatice HPLC [C18]: eluent: MeOH (A) and 0.1 % HCO2H in H2O (B) applying a linear gradient starting from 70 % A in 30 % B to 100 % A in 20.0 min, FR: 32.0 mL min−1 afforded 11 a as a colorless solid (0.113 g, 51 %): mp: 54–57 °C; [α]D27=−79.8 (c=0.29 in CHCl3); 1H NMR (360 MHz, CDCl3): δ=7.56–7.47 (m, 2 H), 7.44–7.26 (m, 12 H), 7.23–7.14 (m, 1 H), 6.80–6.72 (m, 2 H), 6.64–6.57 (m, 2 H), 5.38 (d, J=3.5 Hz, 1 H), 5.16 (bs, 1 H), 5.07 (s, 2 H), 3.89–3.76 (m, 1 H), 3.61–3.48, 3.45–3.34 (2 m, 2 H), 2.13–1.89 (m, 3 H), 1.77–1.60, 1.51–1.39 (2 m, 2 H), 1.31 (s, 9 H), 0.85 (d, J=7.0 Hz, 3 H), 0.72 ppm (d, J=6.7 Hz, 3 H); 13C NMR (90 MHz CDCl3): δ=174.5, 156.2, 153.3, 153.1, 142.3, 137.7, 136.0, 129.0, 128.8, 128.49, 128.47, 128.4, 128.1, 128.05, 125.7, 125.4, 124.0, 89.7, 78.12, 66.7, 65.3, 43.0, 42.6, 32.0, 31.4, 29.6, 28.8, 21.7 ppm; IR (NaCl): ν=3378, 3062, 2976, 2932, 1783, 1720, 1507, 1451, 1364, 1316, 1236, 1174, 1052, 990, 911 cm−1; MS (ESI): m/z 685.5 [M+Na]+; HRMS-ESI: m/z [M+Na]+ calcd for C41H46N2O6: 685.3248, found: 685.3249.
(R)-3-[(S)-2-({[(Benzyloxy)carbonyl]amino})methyl-4-(4-(tert-butoxy)phenyl)-1-oxobutyl]-4-isopropyl-5,5-diphenyloxazolidin-2-one (11 b): Compound 11 b was synthesized according to the procedure described for 11 a starting with 10 b (1.0 g, 1.6 mmol). Purification via preparative HPLC [C18]: eluent: MeOH (A) and 0.1 % HCOOH in H2O (B) applying a linear gradient starting from 70 % A in 30 % B to 100 % A in 20.0 min, FR: 32.0 mL min−1 afforded 11 b as a colorless solid (0.62 g, 58 %). Spectroscopic characterization data were identical to those obtained for the enantiomer 11 a. [α]D27=+80.4 (c=0.30 in CHCl3).
(R)-2-({[(Benzyloxy)carbonyl]amino})methyl-4-(4-(tert-butoxy)phenyl)-butanoic acid (12 a): To a solution of 11 a (1.0 g, 1.6 mmol) in MeOH/THF (1:1, 8.4 mL, 0.2 m) 1 n NaOH (3.0 mL) was added, and the mixture was stirred at RT for 2.5 h. The solvent was evaporated, Et2O was added, and the resulting suspension was stirred at RT for 15 min and filtered. The residue was washed with 1 n NaOH, H2O, Et2O and pentane and dried in high vacuum affording the auxiliary as colorless solid. The filtrate was diluted with Et2O. The aqueous phase was separated, washed with Et2O (1×), acidified with 6 n HCl (pH 1–2) and extracted with EtOAc (3×). The combined organic extracts were dried (MgSO4), filtered and concentrated in vacuo. The resulting colorless oil 12 a (0.64 g, quant.) was pure enough for next reaction. For analytical purposes, a sample was purified using preparative HPLC [C18]: eluent: CH3CN (A) and 0.1 % HCO2H in H2O (B) applying a linear gradient starting from 50 % A in 50 % B to 95 % A in 5 % B in 18.0 min, FR: 32.0: [α]D26=−3.0 (c=0.22 in CHCl3); 1H NMR (600 MHz, CDCl3): δ=7.39–7.29 (m, 5 H), 7.09–6.98 (m, 2 H), 6.93–6.82 (m, 2 H), 5.23 (bs, 1 H), 5.10 (s, 2 H), 3.56–3.20 (m, 2 H), 2.73–2.55 (m, 3 H), 2.02–1.71 (m, 2 H), 1.32 ppm (s, 9 H); 13C NMR (150 MHz, CDCl3): δ=171.2, 156.5, 153.5, 136.4, 135.8, 128.8, 128.5, 128.2, 128.1, 124.2, 78.3, 67.4, 66.9, 45.2, 44.9, 42.4, 41.6, 32.5, 31.1, 28.8 ppm; IR (NaCl): ν=3330, 3033, 2977, 1710, 1507, 1455, 1365, 1236, 1161, 1016, 897, 851 cm−1; MS (ESI): m/z 422.7 [M+H]+; HRMS-ESI: m/z [M+Na]+ calcd for C23H29NO5: 422.1938, found: 422.1943.
(S)-2-({[(Benzyloxy)carbonyl]amino})methyl-4-(4-(tert-butoxy)phenyl)-butanoic acid (12 b): Compound 12 b was synthesized according to the procedure described for compound 12 a starting with 11 b (1.0 g, 1.5 mmol). Compound 12 b was obtained as a colorless oil (0.62 g, quant.). Spectroscopic characterization data were identical to those obtained for the enantiomer 9 a. [α]D26=+2.9 (c=0.22 in CHCl3).
5-Chloromethyl-pyrazolo[1,5-a]pyridine (13): A solution of 2,4,6-trichloro-1,3,5-triazine (660 mg, 3.58 mmol) in DMF (1.5 mL) was stirred at RT for 1 h. Subsequently, a solution of 5-hydroxymethyl-pyrazolo[1,5-a]pyridine20 (542 mg, 3.66 mmol) in CH2Cl2 (10 mL) was added. After stirring for 15 min, H2O (0.5 mL) was added, and the mixture was transferred into a separation funnel. After washing with saturated aq Na2CO3, diluted HCl and brine, the organic layer was dried (Na2SO4), filtered and concentrated in vacuo. Flash column chromatography (CH2Cl2/MeOH 99:1) afforded 11 as a colorless solid (430 mg, 71 %): mp: 67 °C; 1H NMR (360 MHz, CDCl3): δ=8.46 (ddd, J=7.3, 0.9, 0.9 Hz, 1 H), 7.97 (d, J=2.3 Hz, 1 H), 7.53 (dd, J=2.0, 0.9 Hz, 1 H), 6.78 (dd, J=7.3, 2.0 Hz, 1 H), 6.53 (dd, J=2.3, 0.9 Hz, 1 H), 4.59 ppm (s, 2 H); 13C NMR (150 MHz, CDCl3): δ=142.5, 139.5, 132.9, 128.9, 117.2, 112.0, 97.6, 45.3 ppm; IR (film, NaCl): ν=1644, 1527, 1515 cm−1; MS (APCI): m/z 167, 169 [M+H]+.
(2S,5 R)-2,5-Dihydro-3,6-dimethoxy-5-isopropyl-2-[(pyrazolo[1,5-a]pyridin-5-yl)methyl]pyrazine (14 a): A solution of (2R)-2,5-dihydro-3,6-dimethoxy-2-isopropylpyrazine (453 μL, 466 mg, 2.53 mmol) in THF (7 mL) was cooled to −78 °C, then a 2.5 m solution of n-butyllithium in hexane (1.01 mL, 2.53 mmol) was added and stirring was continued for 15 min (solution 1). 5-Chloromethyl-pyrazolo[1,5-a]pyridine (13, 420 mg, 2.53 mmol) was dissolved in THF (7 mL), cooled to −88 °C and then quickly added to solution 1 via syringe. After 3 h of stirring at −78 °C, H2O was added, the solution was concentrated in vacuo, and the residue was redissolved in Et2O and washed with brine. Combined organic extracts were dried (MgSO4), filtered and concentrated in vacuo, and the residue was purified by flash column chromatography (petrol ether/EtOAc, 95:5) affording 14 a as a colorless solid (725 mg, 91 %): [α]D23= +44.4 °C (c=0.79 in CH2Cl2); 1H NMR (CDCl3, 600 MHz, spectrum also shows single resonances of approx. 2.3 % of the (2R,5R)-epimer, which are not listed, e.g. an isopropyl methyl at 0.28 ppm): δ=8.31 (dd, J=7.0, 7.0, 1.0 Hz, 1 H), 7.89 (d, J=2.3 Hz, 1 H), 7.27 (dd, J=2.0, 1.0 Hz, 1 H), 6.57 (dd, J=7.2, 2.0 Hz, 1 H), 6.39 (dd, J=2.3, 1.0 Hz, 1 H), 4.33 (ddd, J=5.8, 4.2, 3.8 Hz, 1 H), 3.68 (s, 3 H), 3.74 (s, 3 H), 3.59 (dd, J=3.8, 3.4 Hz, 1 H), 3.14 (dd, J=13.4, 4.2 Hz, 1 H), 3.09 (dd, J=13.4, 5.8 Hz, 1 H), 2.17 (dqq, J=7.0, 7.0, 3.4 Hz, 1 H), 0.96 (d, J=7.0 Hz, 3 H), 0.64 ppm (d, J=7.0 Hz, 3 H); 13C NMR ([D6]DMSO, 90 MHz): δ=164.1, 162.1, 141.9, 140.1, 133.8, 127.4, 117.0, 114.5, 96.0, 60.6, 56.1, 52.4, 52.3, 39.4, 31.6, 18.9, 16.5 ppm; IR (film, NaCl): ν=2944, 1696, 1643 cm−1; MS (APCI): m/z 315 [M+H]+; HRMS-ESI: m/z [M]+ calcd for C17H22N4O2: 314.1743, found: 314.1744.
(2 R,5S)-2,5-Dihydro-5-isopropyl-3,6-dimethoxy-2-[(pyrazolo[1,5-a]pyridin-5-yl)methyl]pyrazine (14 b): Compund 14 b was prepared according to the protocol for the synthesis of 14 a, starting from (2S)-2,5-dihydro-3,6-dimethoxy-2-isopropylpyrazine (0.35 g, 1.88 mmol). Compound 14 b was obtained as a colorless solid (0.42 g, 72 %). Spectroscopic characterization data were identical to those obtained for the enantiomer 14 a. [α]D23=−43.9 (c=0.82 in CH2Cl2).
(S)-2-Amino-3-(pyrazolo[1,5-a]pyridine-5-yl)-propionic acid hydrochloride (15 a): A solution of 14 a (0.4 g, 1.28 mmol) in 0.2 n HCl (14 mL) was stirred at RT for 4 h. Afterwards the aqueous solution was extracted with Et2O (3×) and the organic layer was discarded. The solution was concentrated in vacuo at RT, then pH 11 was adjusted using aq ammonia (25 %) and extraction with Et2O was performed. The combined organic layers were dried (MgSO4), filtered, concentrated in vacuo, dried very thoroughly for 2 days using high vacuum to remove the valin methyl ester for the most part. The resulting crude, oily methyl ester (still slightly contaminated with valine) was then stirred in 6 n HCl (25 mL) at 70 °C for 3.5 h. After cooling, the solvent was removed in vacuo at RT to afford crude 15 a as a colorless solid (249 mg, 81 %, crude, slightly contaminated with valine), which was used for the next reaction without further purification. For characterization, a small amount of substance was purified by preparative HPLC [C8]: eluent: MeOH (A) and 0.1 % TFA in H2O (B) applying a linear gradient starting from 4 % A in 96 % B to 15 % A in 85 % B in 18.0 min, FR: 10.0 mL min−1. mp: 197–198 °C (decomposition), [α]D20=−9.4 (c=1.01 in H2O); 1H NMR (360 MHz, D2O): δ=8.60 (d, J=7.5, 0.5 Hz, 1 H), 8.13 (s, 1 H), 7.71 (dddd, J=2.0, 0.5 Hz, 1 H), 7.02 (dd, J=7.3, 2.0 Hz, 1 H), 6.76 (dd, J=2.8, 0.8 Hz, 0.13 H), 4.42 (dd, J=7.5, 6.0 Hz, 1 H), 3.44 (dd, J=14.7, 6.0 Hz, 1 H), 3.32 ppm (dd, J=14.7, 7.5 Hz, 1 H); 13C NMR (150 MHz, D2O): δ=171.8, 140.9, 140.1, 134.2, 128.4, 119.8, 116.0, 98.2, 54.0, 35.8 ppm; IR (KBr): ν=3429, 1732, 1679 cm−1; MS (APCI): m/z 206 [M+H]+; HRMS-ESI: m/z [M]+ calcd for C10H11N3O2: 205.0851, found: 205.0850; HPLC (220 nm) system S1 A: tR=9.6 min (>99 %, sample purified by preparative HPLC).
(R)-2-Amino-3-(pyrazolo[1,5-a]pyridine-5-yl)-propionic acid hydrochloride (15 b): Compound 15 b was prepared according to the protocol for the synthesis of 15 a, starting from 14 b (0.40 g, 1.28 mmol). Compound 15 b was obtained as a colorless solid (0.25 g, 80 %). Spectroscopic characterization data were identical to those obtained for the enantiomer 15 a. [α]D25=+6.9 (c=1.01 in H2O); HPLC (220 nm) system S1 A: tR=9.8 min (>99 %, sample purified by preparative HPLC, see 15 a).
4-(2-Aminoethyl)pyridine24 (16): A solution of NH4Cl (30.5 g, 0.57 mol) and 4-vinyl pyridine (30 g, 0.29 mol) in H2O (77 mL) and MeOH (40 mL) was heated to reflux overnight. After cooling to 0 °C, NaOH (23.6 g, 0.50 mol) was added slowly and the aqueous phase was extracted with CH2Cl2 (3×). The combined organic extracts were dried (MgSO4), filtered and evaporated. Flash column chromatography (CH2Cl2/MeOH, 93:7 + NH4OH) afforded 15 as a yellow oil (15.7 g, 0.13 mol): 1H NMR (360 MHz, CD3OD): δ=8.53–8.50 (m, 2 H), 7.15–7.12 (m, 2 H), 3.03–2.98 (m, 2 H), 2.79–2.69 (m, 2 H), 1.36 ppm (bs, 2 H); 13C NMR (90 MHz, CD3OD): δ=149.7, 148.8, 124.2, 42.5, 39.3 ppm; IR (NaCl): ν=3359, 2938, 2867, 1941, 1605, 1558, 1473, 1418, 1386, 1325, 1221, 1070, 1032, 1001, 810 cm−1; MS (ESI): m/z 123.1 [M+H]+.
4-[2-N-(tert-Butyloxycarbonyl)aminoethyl]pyridine25 (17): To a solution of 4-(2-aminoethyl)pyridine (16, 2 g, 16 mmol) in tert-butanol (33 mL), a solution of di-tert-butyldicarbonate (3.9 mL, 18 mmol) in tert-butanol (7 mL) was added dropwise and stirred at RT for 16 h. Et2O was added, and the organic phase was washed with saturated aq NaHCO3 (2×) and brine (1×), dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (EtOAc/hexane, 3:1+2 % NEt3) afforded 17 as a yellow oil (2.94 g, 83 %): 1H NMR (600 MHz, CDCl3): δ=8.54–8.48 (m, 2 H), 7.17–7.11 (m, 2 H), 4.65 (bs, 1 H), 3.45–3.34 (m, 2 H), 2.90–2.75 (m, 2 H), 1.43 ppm (s, 9 H); 13C NMR (150 MHz, CDCl3): δ=155.7, 149.9, 148.0, 124.1, 79.5, 40.8, 35.6, 28.4 ppm; IR (NaCl): ν=3341, 3029, 2977, 2932, 1709, 1604, 1523, 1455, 1416, 1391, 1365, 1274, 1252, 1172, 1051, 965, 810 cm−1; MS (ESI): m/z 223.0 [M+H]+.
N-Amino-(4-[2-N-(tert-butyloxy-carbonyl)aminoethyl]pyridinium-2,4-dinitrophenolate (18): To a solution of O-(2,4-dinitrophenyl)hydroxylamine22 (4.9 g, 24.6 mmol) in CH2Cl2 (40 mL) and CH3CN (6 mL), 17 (5 g, 22.5 mmol) was added, and the solution was stirred at RT for 26 h. After addition of Et2O, the resulting solid was filtered, washed with Et2O and dried in high vacuum overnight yielding 18 as an orange solid (7.26 g, 77 %): mp: 141–145 °C; 1H NMR (360 MHz, [D6]DMSO): δ=8.73–8.64 (m, 2 H), 8.59 (d, J=3.2 Hz, 1 H), 8.28 (bs, 2 H), 7.88–7.82 (m, 2 H), 7.78 (dd, J=9.8, 3.2 Hz, 1 H), 6.94 (bs, 1 H), 6.32 (d, J=9.8 Hz, 1 H), 3.32–3.23 (m, 2 H), 2.98–2.85 (m, 2 H), 1.33 ppm (s, 9 H); 13C NMR (90 MHz, [D6]DMSO): δ=170.2, 155.5, 154.2, 138.2, 136.1, 128.2, 127.4, 127.3, 126.5, 125.0, 77.8, 39.8, 34.7, 28.1 ppm; IR (NaCl): ν=3421, 2255, 2129, 1655, 1318, 1050, 1026, 1004, 825 cm−1; MS (ESI): m/z 238.0 [M+H]+.
5-{2-[N-(tert-Butyloxycarbonyl)amino]ethyl}pyrazolo[1,5-a]pyridine-3-carboxylic acid methylester (19): To a solution of 18 (3.0 g, 7.1 mmol) in DMF (25 mL), K2CO3 (2.16 g, 15.7 mmol) and methylpropiolate (0.76 mL, 8.5 mmol) were added, and the mixture was stirred at RT for 0.5 h. Stirring was continued for another 14 h at RT while a stream of air was bubbled through the mixture. After filtration and evaporation of the solvent, the residue was treated with a saturated aq NaHCO3 and extracted with Et2O (3×). The combined organic extracts were washed with 1 n HCl (1×) and H2O (2×), dried (MgSO4), filtered and concentrated in vacuo. Crude product was purified by flash column chromatography (EtOAc/hexane, 1:3+2 % NEt3) to yield 19 as a yellowish solid (1.42 g, 63 %): mp: 95–101 °C; 1H NMR (360 MHz, CDCl3): δ=8.44 (d, J=7.1 Hz, 1 H), 8.36 (s, 1 H), 7.95 (d, J=0.9 Hz, 1 H), 6.83 (d, J=6.6 Hz, 1 H), 4.65 (bs, 1 H), 3.91 (s, 3 H), 3.55–3.40 (m, 2 H), 2.99–2.85 (m, 2 H), 1.42 ppm (s, 9 H); 13C NMR (150 MHz, CDCl3): δ=163.9, 155.8, 145.1, 140.9, 139.8, 129.0, 117.9, 115.3, 103.0, 79.6, 51.1, 40.9, 36.0, 28.3 ppm; IR (NaCl): ν=3363, 2976, 1703, 1646, 1532, 1438, 1367, 1271, 1237, 1216, 1173, 1117, 1050 cm−1; MS (APCI): m/z 320.2 [M+H]+; HRMS-ESI: m/z [M+Na]+ calcd for C16H21N3O4: 342.1424, found: 342.1433.
5-(2-Aminoethyl)pyrazolo[1,5-a]pyridine (20): A solution of 19 (0.5 g, 1.57 mmol) in 48 % aq HBr (20 mL) was heated to reflux for 2 h. The cooled mixture was alkalized with 5 n NaOH and extracted with CH2Cl2 (3×). The combined organic extracts were dried (MgSO4), filtered and concentrated in vacuo. The resulting orange oil 20 was pure enough for the next reaction (0.2 g, 80 %): 1H NMR (360 MHz, CDCl3): δ=8.40 (d, J=7.1 Hz, 1 H), 7.91 (d, J=2.2 Hz, 1 H), 7.36–7.31 (m, 1 H), 6.60 (dd, J=7.1, 1.8 Hz, 1 H), 6.41 (dd, J=2.2, 0.8 Hz, 1 H), 3.05–3.00 (m, 2 H), 2.79–2.74 ppm (m, 2 H); 13C NMR (90 MHz, CDCl3): δ=142.1, 140.2, 135.6, 128.3, 116.7, 113.3, 95.9, 42.5, 39.4 ppm; IR (NaCl): ν=3361, 2928, 2866, 1644, 1579, 1513, 1439, 1339, 1258, 1227, 1184, 920 cm−1; MS (APCI): m/z 162.0 [M+H]+.
N-{5-(2-Aminoethyl)pyrazolo[1,5-a]pyridine}glycine-ethylester (21): Compund 20 (0.5 g, 3.1 mmol) was dissolved in THF (10 mL) and cooled to 0 °C. At this temperature, NEt3 (0.78 mL, 5.6 mmol) and ethyl bromoacetate (0.31 mL, 2.8 mmol) were added, and the mixture was stirred at RT for 4 h. After filtration of the resulting solid, the solvent was evaporated. The crude product was purified by flash column chromatography (EtOAc) to yield 21 as a yellow oil (0.39 g, 60 %): 1H NMR (360 MHz, CDCl3): δ=8.39 (d, J=7.2 Hz, 1 H), 7.91 (d, J=2.3 Hz, 1 H), 7.37–7.35 (m, 1 H), 6.62 (dd, J=7.2, 1.9 Hz, 1 H), 6.41 (dd, J=2.3, 0.8 Hz, 1 H), 4.18 (q, J=7.2 Hz, 2 H), 3.42 (s, 2 H), 2.96–2.91 (m, 2 H), 2.85–2.80 (m, 2 H), 1.71 (bs, 1 H), 1.26 ppm (t, J=7.2 Hz, 3 H); 13C NMR (150 MHz, CDCl3): δ=172.2, 142.1, 140.1, 135.5, 128.2, 116.5, 113.2, 95.9, 60.7, 50.8, 49.5, 35.8, 14.2 ppm; IR (NaCl): ν=3448, 3328, 3101, 2980, 2935, 2856, 1736, 1644, 1514, 1474, 1438, 1371, 1340, 1257, 1184, 1146, 1028, 918, 864 cm−1; MS (APCI): m/z 248.1 [M+H]+.
Receptor binding experiments
Receptor binding data were determined according to protocols as described previously.7, 26 In detail, NTS1 binding was measured using homogenates of membranes from CHO cells stably expressing human NTS1 at a final concentration of 1–2 μg/well and the radioligand [3H]neurotensin (specific activity 101 Ci mmol−1; PerkinElmer, Rodgau, Germany) at a concentration of 0.50 nm. Specific binding of the radioligand was determined at KD values of 0.37–0.69 nm and a Bmax of 6200–9300 fmol mg−1 protein. Nonspecific binding was determined in the presence of 10 μm neurotensin. NTS2 binding was done using homogenates of membranes from HEK 293 cells, which were transiently transfected with the pcDNA3.1 vector containing the human NTS2 gene (Missouri S&T cDNA Resource Center (UMR), Rolla, MO, USA) by the calcium phosphate method.27 Membranes were incubated at a final concentration of 6–20 μg/well together with 0.50 nm of [3H]NT(8–13) (specific activity 136 Ci mmol−1; custom synthesis of [leucine-3H]NT(8–13) by GE Healthcare, Freiburg, Germany) at KD values in the range from 0.83–1.35 nm and a Bmax of 250–670 fmol mg−1 protein. Nonspecific binding was determined in the presence of 10 μm NT(8–13), and the protein concentration was generally determined by the method of Lowry using bovine serum albumin as standard.28
Data analysis: Data analysis of the competition curves from the radioligand binding experiments was accomplished by non-linear regression analysis using the algorithms in PRISM5.0 (GraphPad Software, San Diego, CA, USA). EC50 values derived from the resulting dose–response curves were transformed into the corresponding Ki values utilizing the equation of Cheng and Prusoff.29
Computational chemistry
We used the crystal structure of the rat NTS1 receptor in complex with NT(8–13) (PDB ID: 4GRV)5 as a template to create homology models of human NTS1 and human NTS2. The amino acid sequences of rat and human NTS1 and human NTS2 were retrieved from the SWISS-PROT database30 and aligned using ClustalX31 (Gonnet series matrix with a gap open penalty of 10 and a gap extension penalty of 0.2) (Figure S2). The third intracellular loop was omitted in the alignment, as it was absent in the crystal structure. Based on the final alignment and the crystal structure of rat NTS1 as a template, we created 10 models of human NTS1 and human NTS2 using MODELLER 9v432 and manually selected one model each.
The ligand NT(8–13) was positioned in the binding sites of human NTS1 and human NTS2 as indicated in the crystal structure of rat NTS1. For the test compound 2 a, we replaced the hydroxyphenyl side chain of NT(8–13) at position 11 by an azaindole moiety, while retaining the conformation of the main chain atoms. We refer to the custom-made residue at position 11 as PPS. The ligand–receptor complexes were subsequently submitted to energy minimization using the SANDER module of AMBER10 as described.33 The all-atom force field ff99SB34 was used for standard amino acid residues within NT(8–13), 2 a, human NTS1 and human NTS2. For the custom-made residue PPS, we used ff99SB for the main chain atoms and the general AMBER force field (GAFF)35 for the side chain. Charges for PPS were calculated using Gaussian 0936 at the HF/6–31(d,p) level and a manually conducted RESP37 procedure, in which we adapted the charges of the azaindole moiety while keeping the charges of the main chain atoms at their values taken from the ff99SB force field (figure S3). A formal charge of +2 was defined for NT(8–13) and 2 a. We applied 2500 steps of steepest descent minimization, followed by 7500 steps of conjugate gradient minimization. The minimization steps were carried out in a water box with periodic boundary conditions and a nonbonded cut-off of 10.0 Å.
The AMBER parameter topology and coordinate files for the minimized complexes were converted into GROMACS38 input files and inserted into a lipidic bilayer of DOPC residues as described.39 The charges of the simulation systems were neutralized by adding 12 and 13 chlorine atoms to the NTS1- and the NTS2-complexes, respectively. The simulation systems were submitted to a short simulation run of 10 ns each, with restraints of 1.0 kcal mol−1 Å-2 applied on the main chain atoms of the proteins, and subsequent production molecular-dynamics simulation runs of 400 ns and 350 ns for the NT(8–13)- and the 2 a-complexes, respectively, using the GROMACS 4.5.2 simulation package as described.40 The analysis of the trajectories was performed using PTRAJ of the AMBER package and figures were prepared using PyMOL.41
Acknowledgments
We thank Andreas Gadelmeier and Dr. Wolfgang Utz (Chair of Food Chemistry, Friedrich Alexander University) for their help with the preparation of the custom-made residue for the MD simulations. We thank Dr. Peter Kolb (Philipps University Marburg) for helpful discussions, Florian Haga for conducting chemical syntheses as well as Prof. Ivana Ivanovic-Burmazovic and Maximilan Dörr (Friedrich Alexander University) for high-resolution ESI mass spectra.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1a.Tanaka K, Masu M, Nakanishi S. Neuron. 1990;4:847–854. doi: 10.1016/0896-6273(90)90137-5. [DOI] [PubMed] [Google Scholar]
- 1b.Mazella J, Botto JM, Guillemare E, Coppola T, Sarret P, Vincent JP. J. Neurosci. 1996;16:5613–5620. doi: 10.1523/JNEUROSCI.16-18-05613.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c.Tyler-McMahon BM, Boules M, Richelson E. Regul. Pept. 2000;93:125–136. doi: 10.1016/s0167-0115(00)00183-x. [DOI] [PubMed] [Google Scholar]
- 2.Binder EB, Kinkead B, Owens MJ, Nemeroff CB. Pharmacol. Rev. 2001;53:453–486. [PubMed] [Google Scholar]
- 3a.Boules M, Shaw A, Fredrickson P, Richelson E. CNS Drugs. 2007;21:13–23. doi: 10.2165/00023210-200721010-00002. [DOI] [PubMed] [Google Scholar]
- 3b.Fuxe K, Von Euler G, Agnati LF, Merlo Pich E, OConnor WT, Tanganelli S, Li XM, Tinner B, Cintra A, Carani C. Ann. N. Y. Acad. Sci. 1992;668:186–204. doi: 10.1111/j.1749-6632.1992.tb27350.x. [DOI] [PubMed] [Google Scholar]
- 3c.Kasckow J, Nemeroff CB. Regul. Pept. 1991;36:153–164. doi: 10.1016/0167-0115(91)90053-j. [DOI] [PubMed] [Google Scholar]
- 4a.Clineschmidt BV, McGuffin JC, Bunting PB. Eur. J. Pharmacol. 1979;54:129–139. doi: 10.1016/0014-2999(79)90415-1. [DOI] [PubMed] [Google Scholar]
- 4b.Boules M, Liang Y, Briody S, Miura T, Fauq I, Oliveros A, Wilson M, Khaniyev S, Williams K, Li Z, Qi Y, Katovich M, Richelson E. Brain Res. 2010;1308:35–46. doi: 10.1016/j.brainres.2009.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic-Jeremic J, Shah P, Shiloach J, Tate CG, Grisshammer R. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a.Egloff P, Hillenbrand M, Klenk C, Batyuk A, Heine P, Balada S, Schlinkmann KM, Scott DJ, Schutz M, Pluckthun A. Proc. Natl. Acad. Sci. USA. 2014;111:E655–662. doi: 10.1073/pnas.1317903111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b.Hughes FM, Jr, Shaner BE, May LA, Zotian L, Brower JO, Woods RJ, Cash M, Morrow D, Massa F, Mazella J, Dix TA. J. Med. Chem. 2010;53:4623–4632. doi: 10.1021/jm100092s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Einsiedel J, Held C, Hervet M, Plomer M, Tschammer N, Hubner H, Gmeiner P. J. Med. Chem. 2011;54:2915–2923. doi: 10.1021/jm200006c. [DOI] [PubMed] [Google Scholar]
- 8.Pratsch G, Unfried JF, Einsiedel J, Plomer M, Hubner H, Gmeiner P, Heinrich MR. Org. Biomol. Chem. 2011;9:3746–3752. doi: 10.1039/c1ob05292f. [DOI] [PubMed] [Google Scholar]
- 9.Seebach D, Beck AK, Bierbaum DJ. Chem. Biodiversity. 2004;1:1111–1239. doi: 10.1002/cbdv.200490087. [DOI] [PubMed] [Google Scholar]
- 10.Seebach D, Overhand M, Kuhnle FNM, Martinoni B, Oberer L, Hommel U, Widmer H. Helv. Chim. Acta. 1996;79:913–941. [Google Scholar]
- 11a.Seebach D, Beck AK, Capone S, Deniau G, Groselj U, Zass E. Synthesis. 2009:1–32. [Google Scholar]
- 11b.Lelais G, Seebach D. Biopolymers. 2004;76:206–243. doi: 10.1002/bip.20088. [DOI] [PubMed] [Google Scholar]
- 12a.Seebach D, Rueping M, Arvidsson PI, Kimmerlin T, Micuch P, Noti C, Langenegger D, Hoyer D. Helv. Chim. Acta. 2001;84:3503–3510. [Google Scholar]
- 12b.Gademann K, Kimmerlin T, Hoyer D, Seebach D. J. Med. Chem. 2001;44:2460–2468. doi: 10.1021/jm010816q. [DOI] [PubMed] [Google Scholar]
- 13a.Seebach D, Lukaszuk A, Patora-Komisarska K, Podwysocka D, Gardiner J, Ebert MO, Reubi JC, Cescato R, Waser B, Gmeiner P, Hubner H, Rougeot C. Chem. Biodiversity. 2011;8:711–739. doi: 10.1002/cbdv.201100093. [DOI] [PubMed] [Google Scholar]
- 13b.Sparr C, Purkayastha N, Yoshinari T, Seebach D, Maschauer S, Prante O, Hubner H, Gmeiner P, Kolesinska B, Cescato R, Waser B, Reubi JC. Chem. Biodiversity. 2013;10:2101–2121. doi: 10.1002/cbdv.201300331. [DOI] [PubMed] [Google Scholar]
- 14.Sebesta R, Seebach D. Helv. Chim. Acta. 2003;86:4061–4072. [Google Scholar]
- 15a.Sprecher H, Pletscher S, Mori M, Marti R, Gaul C, Patora-Komisarska K, Otchertianova E, Beck AK, Seebach D. Helv. Chim. Acta. 2010;93:90–110. [Google Scholar]
- 15b.Hintermann T, Seebach D. Helv. Chim. Acta. 1998;81:2093–2126. [Google Scholar]
- 16a.Evans DA, Bartroli J, Shih TL. J. Am. Chem. Soc. 1981;103:2127–2129. [Google Scholar]
- 16b.Evans DA, Weber AE. J. Am. Chem. Soc. 1986;108:6757–6761. [Google Scholar]
- 17.Ho GJ, Mathre DJ. J. Org. Chem. 1995;60:2271–2273. [Google Scholar]
- 18.Brocklehurst CE, Furegati M, Muller-Hartwieg JCD, Ossola F, La Vecchia L. Helv. Chim. Acta. 2010;93:314–323. [Google Scholar]
- 19a.Gmeiner P, Sommer J. Arch. Pharm. 1988;321:505–507. doi: 10.1002/ardp.19883210902. [DOI] [PubMed] [Google Scholar]
- 19b.Fauq AH, Hong F, Cusack B, Tyler BM, Ping-Pang Y, Richelson E. Tetrahedron: Asymmetry. 1998;9:4127–4134. [Google Scholar]
- 19c.Cusack B, Boules M, Tyler BM, Fauq A, McCormick DJ, Richelson E. Brain Res. 2000;856:48–54. doi: 10.1016/s0006-8993(99)02363-x. [DOI] [PubMed] [Google Scholar]
- 20.Bettinetti L, Schlotter K, Hubner H, Gmeiner P. J. Med. Chem. 2002;45:4594–4597. doi: 10.1021/jm025558r. [DOI] [PubMed] [Google Scholar]
- 21.Schöllkopf U, Groth U, Deng C. Angew. Chem. Int. Ed. Engl. 1981;20:798–799. [Google Scholar]
- Angew. Chem. 1981;93:793–795. [Google Scholar]
- 22.Legault C, Charette AB. J. Org. Chem. 2003;68:7119–7122. doi: 10.1021/jo034456l. [DOI] [PubMed] [Google Scholar]
- 23.Boden P, Eden JM, Hodgson J, Horwell DC, Hughes J, McKnight AT, Lewthwaite RA, Pritchard MC, Raphy J, Meecham K, Ratcliffe GS, Suman-Chauhan N, Woodruff GN. J. Med. Chem. 1996;39:1664–1675. doi: 10.1021/jm950892r. [DOI] [PubMed] [Google Scholar]
- 24.Steinmetzer T, Schweinitz A, Sturzebecher A, Donnecke D, Uhland K, Schuster O, Steinmetzer P, Muller F, Friedrich R, Than ME, Bode W, Sturzebecher J. J. Med. Chem. 2006;49:4116–4126. doi: 10.1021/jm051272l. [DOI] [PubMed] [Google Scholar]
- 25.Yoshizawa H, Kubota T, Itani H, Ishitobi H, Miwa H, Nishitani Y. Bioorg. Med. Chem. 2004;12:4211–4219. doi: 10.1016/j.bmc.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Hübner H, Haubmann C, Utz W, Gmeiner P. J. Med. Chem. 2000;43:756–762. doi: 10.1021/jm991098z. [DOI] [PubMed] [Google Scholar]
- 27.Jordan M, Schallhorn A, Wurm FM. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 29.Cheng Y, Prusoff WH. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 30.Bairoch A, Apweiler R. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 32.Šali A, Blundell TL. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 33.Hiller C, Kling RC, Heinemann FW, Meyer K, Hubner H, Gmeiner P. J. Med. Chem. 2013;56:5130–5141. doi: 10.1021/jm400520c. [DOI] [PubMed] [Google Scholar]
- 34.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 36. M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009.
- 37.Bayly CI, Cieplak P, Cornell W, Kollman PA. J. Phys. Chem. B. 1993;97:10269–10280. [Google Scholar]
- 38a.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 38b.Hess B, Kutzner C, van der Spoel D, Lindahl E. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 39.Möller D, Kling RC, Skultety M, Leuner K, Hubner H, Gmeiner P. J. Med. Chem. 2014;57:4861. doi: 10.1021/jm5004039. [DOI] [PubMed] [Google Scholar]
- 40.Goetz A, Lanig H, Gmeiner P, Clark T. J. Mol. Biol. 2011;414:611. doi: 10.1016/j.jmb.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 41. Schrodinger, LLC, 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary