

Abstract
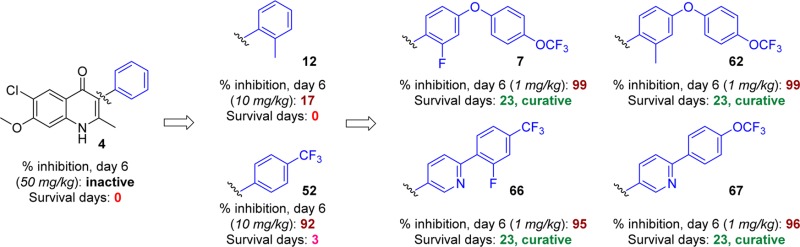
The continued proliferation of malaria throughout temperate and tropical regions of the world has promoted a push for more efficacious treatments to combat the disease. Unfortunately, more recent remedies such as artemisinin combination therapies have been rendered less effective due to developing parasite resistance, and new drugs are required that target the parasite in the liver to support the disease elimination efforts. Research was initiated to revisit antimalarials developed in the 1940s and 1960s that were deemed unsuitable for use as therapeutic agents as a result of poor understanding of both physicochemical properties and parasitology. Structure–activity and structure–property relationship studies were conducted to generate a set of compounds with the general 6-chloro-7-methoxy-2-methyl-4(1H)-quinolone scaffold which were substituted at the 3-position with a variety of phenyl moieties possessing various properties. Extensive physicochemical evaluation of the quinolone series was carried out to downselect the most promising 4(1H)-quinolones, 7, 62, 66, and 67, which possessed low-nanomolar EC50 values against W2 and TM90-C2B as well as improved microsomal stability. Additionally, in vivo Thompson test results using Plasmodium berghei in mice showed that these 4(1H)-quinolones were efficacious for the reduction of parasitemia at >99% after 6 days.
Introduction
Malaria continues its devastating impact on the health of human populations in tropical regions, with over 200 million cases of malaria and over 600 000 deaths from malaria each year.1−3 The most impacted region is sub-Saharan Africa, which accounts for an estimated 90% of all deaths, occurring primarily in children less than 5 years old. Of the five Plasmodium species which cause human disease, the two most prevalent are P. falciparum and P. vivax. These strains have become increasingly more difficult to control and treat due to the emergence of multi-drug resistance as well as a lack of preventative drugs for the populations at highest risk, in particular children and pregnant women. During the past three decades, P. falciparum has developed resistance to every commonly available antimalarial, including the most recent findings of reduced parasite clearance with the artemisinin combination therapies (ACTs).4−8 The rapid spread of these resistant parasites has significantly hindered the successful treatment of patients to the point that some drugs have been rendered virtually useless in many parts of the world.
In an effort to curb artemisinin resistance, the World Health Organization promoted the use of ACTs within endemic countries, by which two or more antimalarials with different modes of action are taken simultaneously.5 Unfortunately, disturbing reports of parasite resistance to the ACTs, which were originally considered to be reliable treatments for malaria, have emanated in recent studies. History has shown that once resistance has manifested itself to one compound, the resistance usually is conferred to the entire chemotype class. Therefore, much effort has been taken to modify existing drugs to counteract the induced resistance through structural modifications.9,10
Recent understanding of the mechanism of action and resistance to current drugs suggests that previously discovered leads remain viable candidates, provided renewed efforts overcome chemotype-specific hurdles.11,12 Efforts to surmount these hurdles have been supplemented by advances in library screening, hit-to-lead optimization, physicochemical understanding of biologically active compounds, and refined understanding of mechanisms of action. One specific example of this challenge is endochin (1), which in the 1940s was identified to be a causal prophylactic (kills growing liver-stage parasites) and potent erythrocytic-stage agent in avian malaria models. Despite its promise as an antimalarial agent, the further development of endochin-like agents languished because of inadequate preclinical models and a poor understanding of parasite biochemistry (Figure 1).13 Approximately 25 years later, Casey tested a focused series of 3-alkenyl- and 3-alkyl-2-methyl-4(1H)-quinolones, but no antimalarial activity was observed in the utilized preclinical screen (Rane single-dose rodent malaria model).14,15 Evaluation of the coccidiostat quinolone ester identified as ICI 56,780 (2), a 4(1H)-quinolone structurally related to 1, displayed causal prophylactic (single dose of 30 mg/kg subcutaneous) and blood schizonticidal activity (ED50 = 0.05 mg/kg) in rodent malaria models (Figure 1). Compound 2 was later found to have anti-relapse activity in P. cynomolgi-infected rhesus monkeys (10–30 mg/kg subcutaneous).16 Unfortunately, a high degree of resistance to this compound was obtained after one passage in P. berghei-infected mice. This, in conjunction with a lack of oral bioavailability, led to the abandonment of the quinolone ester series.17
Figure 1.

Common antimalarials and 4(1H)-quinolone scaffolds.
Since the aforementioned studies were conducted over 20 years ago without adequate evaluation in current preclinical efficacy models and without assessment of drug-like properties, several laboratories recently revisited the optimization and development of 4(1H)-quinolone-based antimalarials.18−29 Starting from compound 1, detailed SAR and structure–property relationship (SPR) studies conducted in our laboratory identified the substituent at the 3-position to be of highest priority for in vitro activity. Furthermore, a remarkable synergistic effect, improving the antimalarial activity by a factor of 30 and eliminating the cross-resistance to atovaquone, was identified for compounds substituted with a chloro substituent at the 6-position and a methoxy group at the 7-position. 3-Ethyl- and 3-phenyl-substituted 6-chloro-7-methoxy-2-methyl-4(1H)-quinolones (3 and 4) were among the most promising compounds, as both not only displayed low-nanomolar EC50 values against the multi-drug-resistant isolates W2 and TM90-C2B of P. falciparum but also possessed acceptable physicochemical properties.20
Independent optimization of 1 by Riscoe and co-workers led to the development of the polyethylene glycol carbonate prodrug 5, which was orally bioavailable with an ED50 value of 11 mg/kg against P. yoelii infections.21 Our combined efforts to further optimize the 4(1H)-quinolone compound series yielded ELQ-300 (6) and P4Q-391 (7), of which the translational team from Medicines for Malaria Venture selected compound 6 to undergo preclinical development.22,30 Both compounds 6 and 7 were highly efficacious against both the blood and the liver stages of the malaria parasite, as well as active against the forms critical for disease transmission. Both compounds were shown to selectively inhibit Plasmodium cytochrome bc1 complex over mammalian bc1 due to the 6-chloro-7-methoxy substitution pattern of the 4(1H)-quinolone’s benzenoid ring.22 Herein, we report the design, synthesis, physicochemical and pharmacokinetic evaluation, and testing for in vitro and in vivo antimalarial activity of a 3-aryl-6-chloro-7-methoxy-4(1H)-quinolone library (12–67) that lead the way to the identification of frontrunner compounds 6 and 7.
Results and Discussion
Synthetic Chemistry
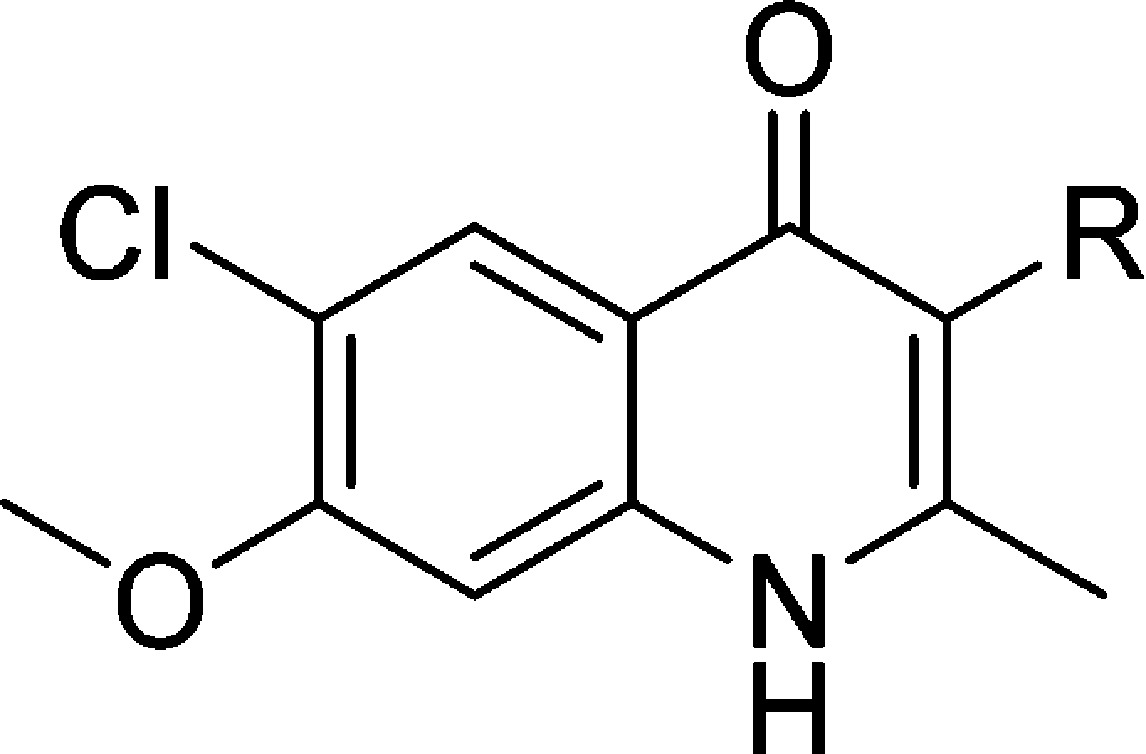
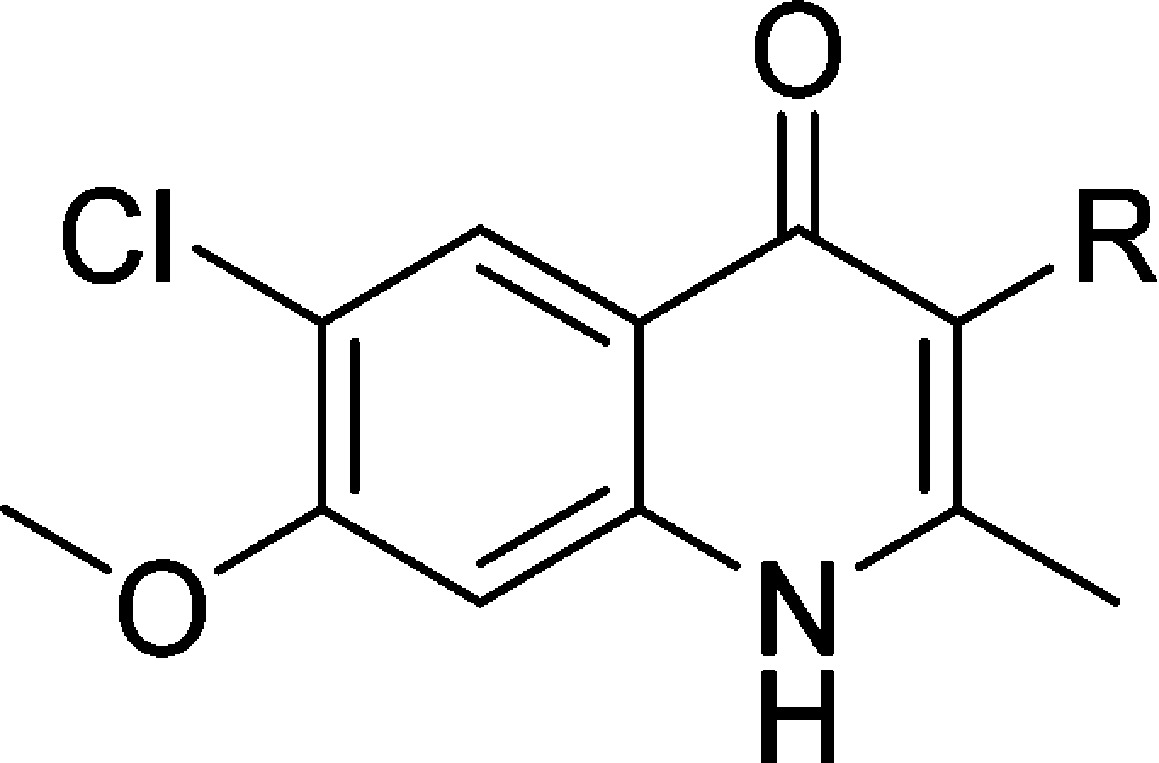
In our efforts to optimize the antimalarial 4(1H)-quinolones, 3-ethyl- and 3-phenyl-substituted analogues 3 and 4 were initially prepared with the intention of evaluating their in vitro and in vivo efficacy. According to previous reports, the β-keto esters 2-ethyl- and 2-phenyl-substituted ethyl acetoacetate were reacted with 4-chloro-3-methoxyaniline (8) via Conrad–Limpach cyclization, providing compounds 3 and 4, respectively, in good yields (Scheme 1). Further optimization focused on 3-aryl-substituted compounds due to the favorable antimalarial activity and physiochemical properties as well as the synthetic tractability of 3-phenyl-4(1H)-quinolone 4 as opposed to 3-ethyl-4(1H)-quinolone 3. These investigations were conducted with four initial sub-series of compounds probing the 3-position with alkylphenyl, heteroaryl, para-substituted aryl, and fluoroaryl moieties followed by a fifth sub-series focusing on the optimization of these most promising 3-aryl moieties. Syntheses of these analogues were carried out in a linear fashion beginning with 4(1H)-quinolone 9 (Scheme 2). Regioselective iodination produced intermediate 3-iodo-4(1H)-quinolone 10. Upon Suzuki–Miyaura cross-coupling of the iodo intermediate 10 with the corresponding boronic acid, 4(1H)-quinolones 7 and 12–67 could be prepared.31 Since a broad range of Suzuki adducts was desired, myriad changes in cross-coupling conditions were employed (see the Supporting Information for details). As previously reported, intermediate 11 was also used in the synthesis of analogues 7 and 12–67, when progress was halted due to sluggish reaction times, poor yields, and/or difficult purification schemes.22
Scheme 1. Synthesis of 6-Chloro-7-methoxy-2-methyl-3-substituted 4(1H)-Quinolones.

Reagents and conditions: (a) corresponding β-keto-ester, HOAc, PhH, reflux, 18 h, then Ph2O, reflux, 12 min.
Scheme 2. Synthesis of 6-Chloro-7-methoxy-2-methyl-3-aryl-4(1H)-quinolones.

Reagents and conditions: (a) I2, Na2CO3, MeOH, rt. (b) ArB(OH)2, SPHOS, K3PO4, toluene or DMF, 80–100 °C, 0.5–48 h. (c) EtI, Cs2CO3, DMF, 5 h, rt. (d) HBr, reflux, 15 min.
Antimalarial Activity
All synthesized compounds were tested, as previously reported, against the clinically relevant multi-drug-resistant P. falciparum strains W2 (chloroquine and pyrimethamine resistant) and TM90-C2B (chloroquine, mefloquine, pyrimethamine, and atovaquone resistant).20,31,32 The human malaria parasites are grown in vitro in dilute human erythrocytes in RPMI 1640 media containing 10% heat-inactivated plasma, and the potency for each 4(1H)-quinolone has been calculated against the individual strains as the 50% effective concentration (EC50).32 The emergence of resistance and cross-resistance with atovaquone is a concern for new antimalarials that target the parasite mitochondria (e.g., atovaquone), thus the resistance index (RI) of each compound was calculated as the ratio of the effective concentrations for W2 and TM90-C2B (RI = EC50(TM90-C2B)/EC50(W2)) (see the Supporting Information for details). Compounds with RI = 0.3–3.0 were considered acceptable with regard to risk of cross-resistance with atovaquone, whereas compounds with RI > 10 or RI < 0.1 are likely to have clinically relevant levels of cross-resistance, thus making them unsuitable for development as new antimalarials.33,34
Structure–Activity Relationship (SAR) Studies
Initial SAR studies were carried out to determine the best-suited benzenoid ring substituents resulting in compounds containing the 6-chloro-7-methoxy substituent pattern that not only provided a synergistic effect on antimalarial activity but also narrowed the RI and improved the solubility in select analogues.20 Furthermore, despite 3-phenyl-4(1H)-quinolone (4) being approximately 20-fold less potent as compared to 3-alkyl and 3-alkenyl analogues, it was considered to be a more robust platform for development due to improved hepatic microsomal stability and slightly increased aqueous solubility. Several sub-series of 4(1H)-quinolones were designed to cover a wide chemical space, improving antimalarial potency and addressing physicochemical liabilities such as aqueous solubility and microsomal stability in parallel.
The first sub-series of analogues, consisting of 3-alkylphenyl-4(1H)-quinolones (12–18, Table 1), was designed to test for steric effects. While 3-phenyl-4(1H)-quinolone 4 displayed an approximately 2-fold improvement in antimalarial activity against W2 and TM90-C2B when compared to the 3-ethyl-substituted congener 3, additional gains in potency were observed with compounds modified at the 4(1H)-quinolone’s 3-position with simple ortho-alkyl- or para-alkyl-substituted phenyl moieties. In comparison to reference compound 4, the addition of a methyl substituent to the ortho- or para-position of 4(1H)-quinolones, as in 12 and 13, improved the potency against TM90-C2B by at least 2-fold. In contrast, for the strain W2, only compound 12 displayed an improvement in potency. Combining multiple methyl groups such as 3-(2,4-dimethylphenyl)- and 3-mesityl-substituted 4(1H)-quinolones (14 and 15, respectively) did not show any further improvements in potency. Increasing the steric effects with groups such as ethyl-, isopropyl-, and tert-butyl provided compounds 16–18, of which only 3-(4-isopropyl)phenyl-4(1H)-quinolone (17) turned out to be equipotent to the most potent 4(1H)-quinolone 12. In summary, compounds 12 and 17 were the most potent analogues in this sub-series, with EC50 values of 5.83 and 6.65 nM against W2 and 4.03 and 2.67 nM against TM90-C2B, respectively, thus yielding acceptable RI values.
Table 1. 3-Alkylphenyl-4(1H)-quinolonesa.
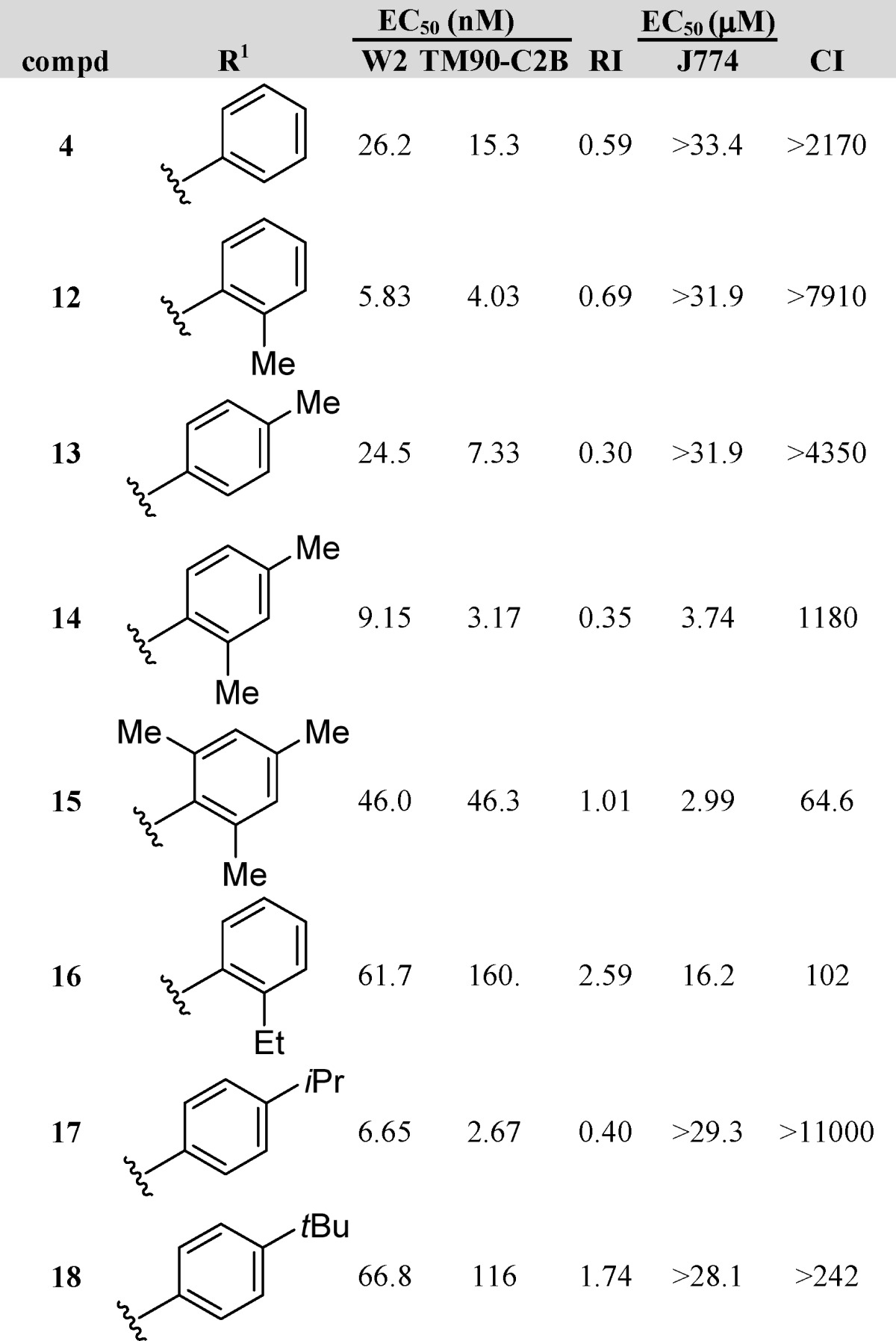
Dihydroartemisinin (DHA), chloroquine (CQ), and atovaquone (ATO) are internal controls for each in vitro assay: DHA, 5.5 nM W2 and 5.9 nM TM90-C2B; CQ, 229 nM for TM90-C2B and 421 nM for W2; and ATO, 1.4 nM W2 and 18.4 μM TM90-C2B.
We next synthesized and tested a sub-series of 3-heteroaryl-4(1H)-quinolones (19–32, Table 2), probing both steric and electronic factors. We anticipated that the incorporation of a heteroaromatic residue would elicit an enhancement of solubility. 3-Pyridyl analogues 19–21 possessed EC50 values of 128–928 nM against W2 and 120–219 nM against TM90-C2B. Of the three compounds, 4(1H)-quinolone 20, possessing a nitrogen at the meta-position, proved to be most active against TM90-C2B, whereas the best activity against W2 was shown by analogue 21, with the nitrogen at the para-position. Pyrimidine analogue 22, containing two nitrogens both at the meta-positions, lacked antimalarial activity altogether. Alteration of pyridine 20 with a para-N,N-dimethylamino group yielded 4(1H)-quinolone 23, which showed slightly improved activity against TM90-C2B; however, the potency against W2 remained unaffected. Similar results were obtained with the N-methylpiperazino-substituted pyridine 24, a contiguous extension of analogue 23, displaying poor activities with EC50 values no lower than 536 nM for TM90-C2B. 3-Furanyl analogues 25 and 26 displayed excellent EC50 values of 8.43 and 1.85 nM against TM90-C2B and 34.2 and 13.3 nM for W2, respectively, with 26 being the most potent analogue in the sub-series. These two analogues highlight the breadth of design opportunities, given the considerable improvement in antimalarial activity despite a relatively minor structural modification over reference compound 4. Isoxazole 27 was prepared next and shown to be inactive, with poor EC50 values of 1060 nM against W2 and 629 nM against TM90-C2B. Finally, a selection of 4(1H)-quinolones substituted with bicyclic systems at the 3-position were tested. Benzo-oxadiazole 28 had poor activity against W2 and an unacceptable RI value of 0.04, yet it had decent activity against TM90-C2B, with an EC50 of 80.6 nM. Benzofuran 29 and benzothiophene 30 possessed EC50 values of 24.8–138 nM against TM90-C2B, with analogue 29 displaying nearly equipotent activity against both strains W2 and TM90-C2B and, thus, an RI of nearly 1.0. Similarly, favorable RI values were determined for 4(1H)-quinolones 31 and 32, although both compounds were less potent than reference compound 4.
Table 2. 3-Heteroaryl-4(1H)-quinolonesa.
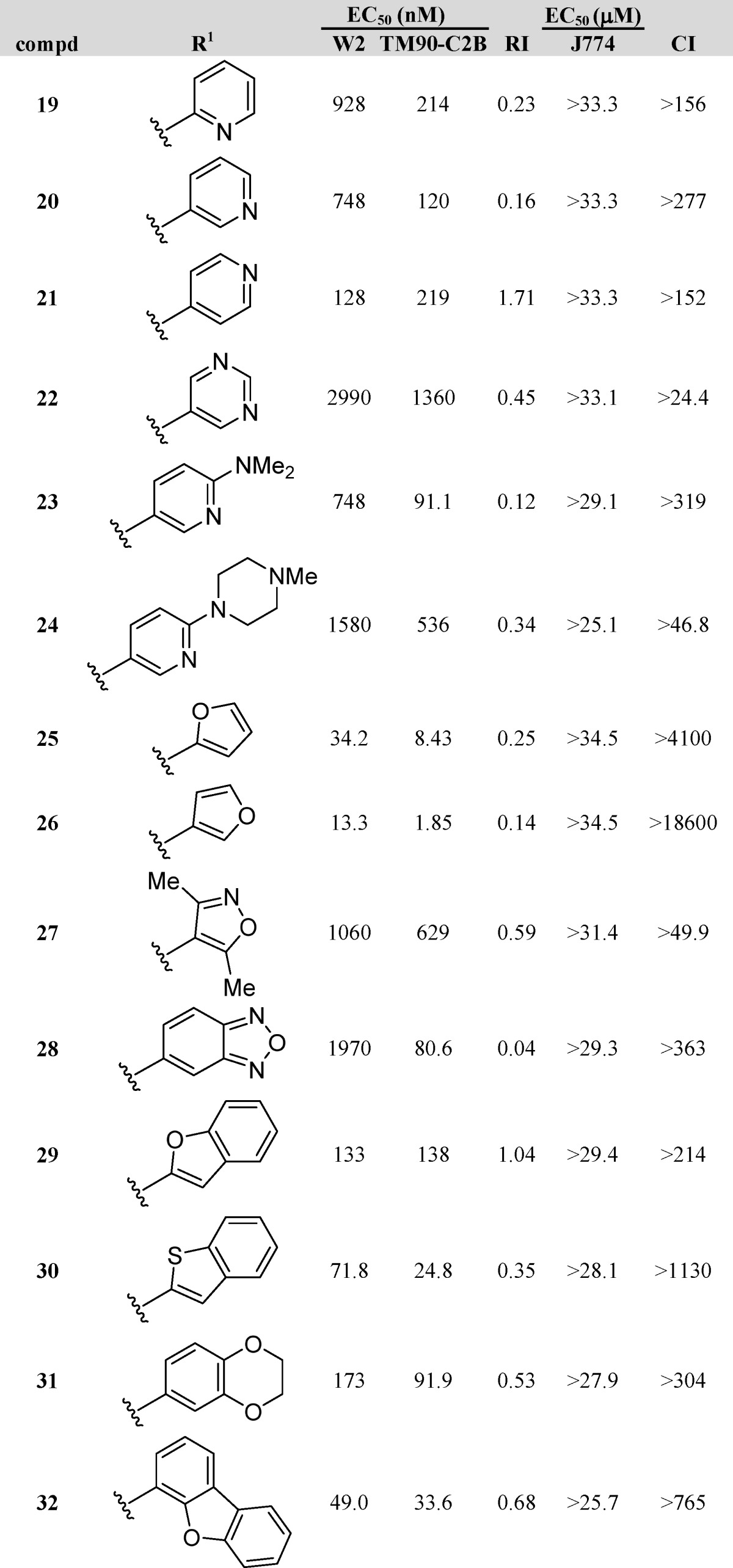
Dihydroartemisinin (DHA), chloroquine (CQ), and atovaquone (ATO) are internal controls for each in vitro assay: DHA, 5.5 nM W2 and 5.9 nM TM90-C2B; CQ, 229 nM for TM90-C2B and 421 nM for W2; and ATO, 1.4 nM W2 and 18.4 μM TM90-C2B.
As compounds 12 and 17 have shown to be slightly more potent than reference compound 4, a sub-series of 3-phenyl-substituted 4(1H)-quinolones was prepared, in which the phenyl ring has been mono-substituted with the intent of identifying structural modifications that improve the aqueous solubility or the metabolic stability while increasing or maintaining the antimalarial activity (Table 3). Analogous to GSK’s successful development of their pyridone prodrug, this sub-series was also designed with the secondary objective of identifying a chemical handle to potentially develop a 4(1H)-quinolone phosphate prodrug.35
Table 3. 3-(4-Substituted)aryl-4(1H)-quinolonesa.
Dihydroartemisinin (DHA), chloroquine (CQ), and atovaquone (ATO) are internal controls for each in vitro assay: DHA, 5.5 nM W2 and 5.9 nM TM90-C2B; CQ, 229 nM for TM90-C2B and 421 nM for W2; and ATO, 1.4 nM W2 and 18.4 μM TM90-C2B.
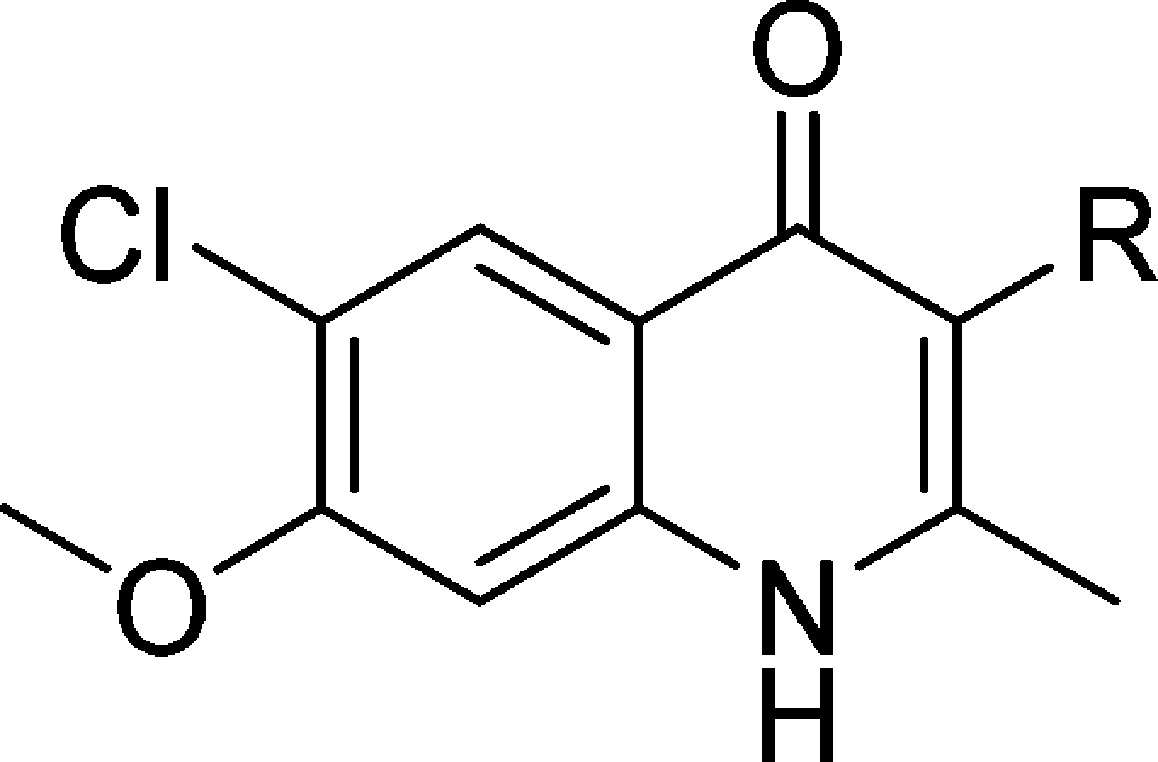
A set of compounds (33–48, Table 3) with hydrogen bond-accepting potential, containing various functional groups, including ethers, carbonyls, amines, and sulfonyls, was prepared and tested. Methyl ether 33 and isopropyl ether 34, with EC50 values of 11.3 and 12.6 nM, respectively, against TM90-C2B, were slightly more potent than reference 4, whereas their potency against W2 dropped by a factor of 2. While carbonyl analogues 35–38 showed no improvement over 4 in activity, methyl ester 38 appeared to be equipotent with 4 and possessed an excellent RI value approaching 1. Similar to the ethers 33 and 34, the antimalarial activity of N,N-dimethylaniline 39 was unaffected for TM90-C2B and W2. N,N-Dimethylsulfonamide 40 was nearly inactive, with EC50 values in the lower micromolar range. Noting that analogues 33, 34, and 39 all possessed similar activities against both W2 and TM90-C2B and all possessed non-hydrogen-bearing heteroatoms at the para-position of the 3-phenyl substituent, curiosity arose as to the efficacy of analogue 41 with hydrogen-bonding potential. Thus, synthesis and testing of phenol 41 resulted in a compound that was 50 times less potent against W2 and on average 35 times less potent against TM90-C2B. An interesting trend was realized with carbinol-bearing 3-phenyl-4(1H)-quinolones. Analogues 42–44, with carbinol appendages at the ortho-, meta-, and para-positions of the 3-phenyl ring, possessed EC50 values ranging from 93.9 nM to 5.40 μM. The general trend that resulted was that the para-substituted 3-phenyl-4(1H)-quinolone (44) was much more active than the ortho-substituted isomer (42), while the meta-isomer (43) exhibited low activities.
Finally, a set of 4(1H)-quinolones was prepared whose 3-substituent consisted of two aromatic rings. Several of these analogues were inspired by results obtained with GSK’s pyridone series (Table 3).19 Strikingly, the EC50 values of these analogues were in the low single digit nanomolar ranges, with most of the analogues also possessing acceptable RI values. Biaryl 45 and biaryl ether 46 were very potent, displaying EC50 values of 1.78 and 2.40 nM for W2 and 4.33 and 1.31 nM for TM90-C2B, respectively. Replacement of the terminal phenyl ring of compound 46 by a benzyl substituent provided phenoxybenzyl ether 47, which was approximately equipotent against TM90-C2B and approximately 5-fold less potent against W2. Benzyloxyphenyl ether 48, an isomer of compound 47 differing only in the position of the ether oxygen, was even more active, with EC50 values of 0.638 nM for TM90-C2B and 1.30 nM for W2.
To address possible microsomal lability within the 3-phenyl-4(1H)-quinolones, a sub-series of fluorinated analogues (49–58, Table 4) was prepared and tested. The majority of these analogues displayed good potency against both strains, with EC50 values in the low nanomolar ranges. Monofluorophenyl-4(1H)-quinolone 49 and bis-fluoro analogues 50 and 51 were slightly less potent than reference compound 4. Trifluoromethylphenyl analogue 52 emerged as one of the most potent compounds among the entire library, with EC50 values of 6.25 nM against W2 and 5.98 nM against TM90-C2B, while the bis-trifluoromethylphenyl derivative 53 possessed poor activity. Combination of a fluoro and a trifluoromethyl moiety on the 3-phenyl substituent of the 4(1H)-quinolone (54) did not significantly improve the activity over compound 4. In contrast, compounds 55 and 56, with a 3-phenyl moiety substituted with one or two fluoro substituents and one alkoxy group, demonstrated poor antimalarial activities altogether. Finally, ortho-trifluoromethoxyphenyl analogue 57 was devoid of antimalarial activity, while the para-trifluoromethoxyphenyl analogue 58 possessed excellent EC50 values of 7.72 nM against W2 and 7.76 nM against TM90-C2B. This compound was extremely promising as it exhibited not only single-digit nanomolar activities but a nearly perfect RI of 1.0.
Table 4. 3-Fluoroaryl-4(1H)-quinolonesa.
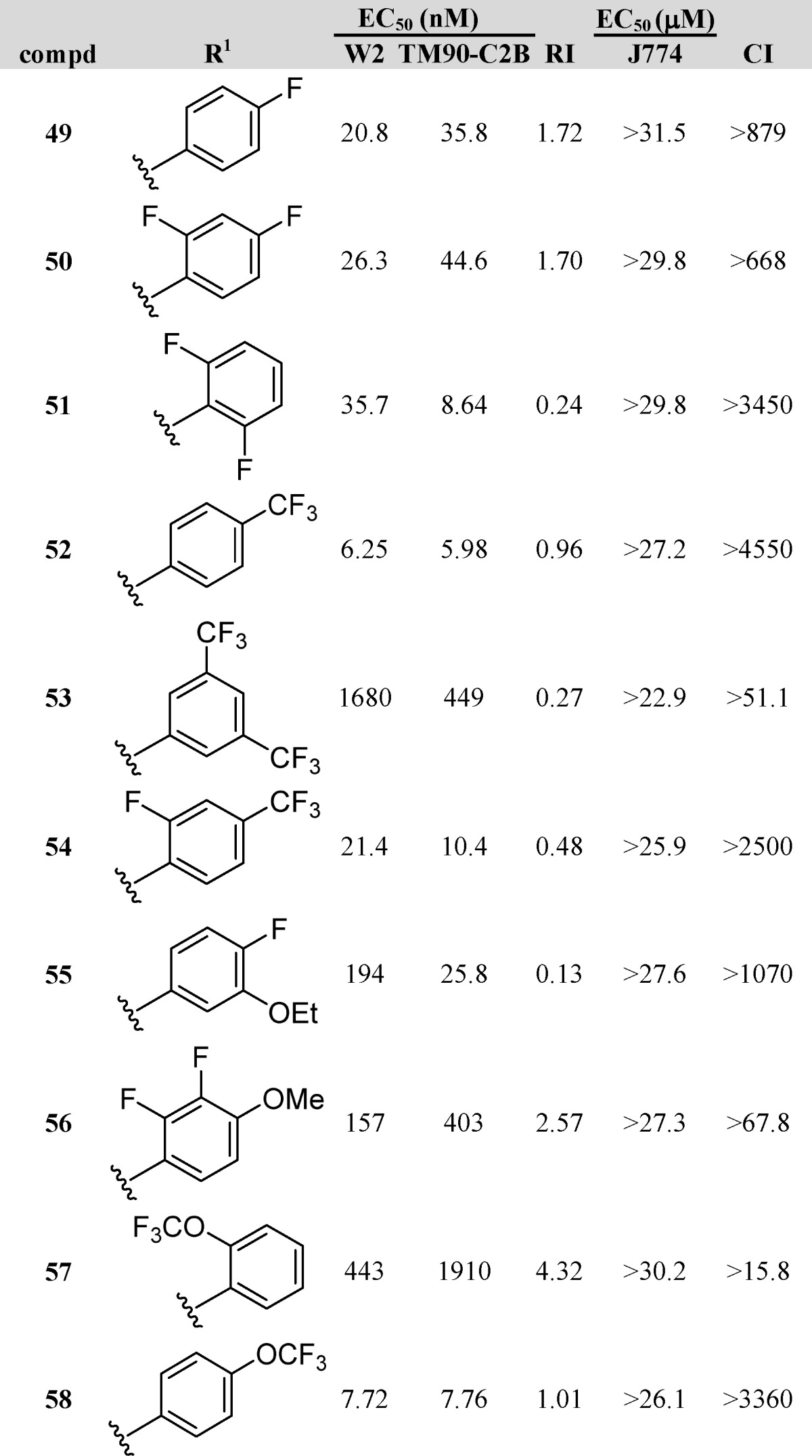
Dihydroartemisinin (DHA), chloroquine (CQ), and atovaquone (ATO) are internal controls for each in vitro assay: DHA, 5.5 nM W2 and 5.9 nM TM90-C2B; CQ, 229 nM for TM90-C2B and 421 nM for W2; and ATO, 1.4 nM W2 and 18.4 μM TM90-C2B.
Physicochemical and ADME Properties
In parallel to the in vitro antimalarial activity testing, standard physicochemical and ADME properties were determined to identify potential compound liabilities. All compounds were routinely assessed for aqueous solubility and partition coefficient (log D) using HPLC-based protocols, which have been previously described and utilized for quinolone-based antimalarials.20 Passive transcellular permeability was determined using the standard parallel artificial membrane permeability assay (PAMPA).20 Plasma protein binding was determined by ultracentrifugation (see Supporting Information for method details) for selected compounds that were progressed to in vivo pharmacokinetic studies. Finally, a large set of the most promising 4(1H)-quinolones was tested for human microsomal stability following a previously reported protocol.22 Briefly, the compounds were incubated with human liver microsomes, and the metabolic reaction was quenched at various time points over the incubation period by the addition of acetonitrile. The relative loss of compound was fitted to an exponential decay function to determine the first-order rate constant and in vitro intrinsic clearance.22 The distribution coefficients of all analogues were in the acceptable range (1 < log D < 4). The majority of the more potent compounds also possessed the greatest lipophilicity, with an average log D7.4 = 3.6 (see Supporting Information for more details). This represents an improvement over the 4(1H)-quinolones of the initial study,20 as the most promising compounds possessed log D7.4 values which are reduced by approximately 1 unit, while their potency increased by an estimated factor of 10. The permeability of the majority of the library was high (Pe > 50 × 10–6 cm/s), with the exception of a few analogues, such as the pyridyl analogues 20 and 22, the isoxazole 27, the phenol 41, and the benzyl alcohol analogues 42–44.
Despite improvement in many physiochemical properties, the majority of the compounds possessed poor aqueous solubility. As previously proposed,20 the poor aqueous solubility of the 4(1H)-quinolones possibly derives from a strong lattice energy and high melting point due to strong intermolecular hydrogen bonds within the crystal lattice. Compounds 12 and 14, possessing an ortho-methyl-substituted phenyl moiety at the 3-position, displayed slightly improved aqueous solubility over reference compound 4 while maintaining their in vivo activity. In contrast, heteroaromatic compounds such as 21, 22, 24, and 27 with improved aqueous solubility showed a decrease in potency, suggesting ionizable groups to be incompatible for this chemotype.
Human liver microsomal stability was considered to be important for the selection of the candidates to be tested for in vivo efficacy studies. Within the first sub-series of 4(1H)-quinolones, the 3-ethyl analogue 3 displayed a high intrinsic clearance (CLint) of 56 μL/(min·mg), whereas the 3-aryl-substituted 4(1H)-quinolone 4 had a moderate CLint of 17 μL/(min·mg). Analogues bearing a heterocyclic moiety at the 3-position exhibited improved stability over 4. While CLint values were less than 15 μL/(min·mg) for pyridines 20 and 21, furan analogues 25 and 26 were significantly less stable, making them unattractive for in vivo efficacy testing. Isoxazole 27 highlighted the potential for a heteroaryl-substituted 4(1H)-quinolone with minimal degradation after 250 min. Similarly, phenol 41, phenyl ethers 33 and 34, and benzyl alcohols 43 and 44 were minimally metabolized, indicating substitution at the para-position of the 3-aryl group to be critical for stability. As expected, of the fluorinated analogues, trifluoromethylphenyl and trifluoromethoxyphenyl analogues 52 and 58 were shown to be highly stable.
Screening for in Vivo Efficacy
Nine 4(1H)-quinolones with potent in vitro activity against P. falciparum were selected to undergo screening for in vivo efficacy using a rodent malaria model (Table 5). Physicochemical and ADME criteria such as aqueous solubility, microsomal stability, and compound availability were also taken into account for the selection process. Compounds 12 and 24 were chosen for the in vivo efficacy screening due to the increased aqueous solubility derived from their out-of-plane or ionizable 3-substituent. Among the selected candidates were 4(1H)-quinolones 33, 34, 45, 46, 50, and 58, which possessed moderate to high microsomal stability. The screen involved treating mice with a single dose of compound at a concentration of 50 mg/kg on day 1 post-exposure (PE) and then assessing parasitemia on days 3 and 6 PE. Compounds with >50% inhibition of parasitemia on both days were considered to be active. In preliminary studies, a PEG400 formulation was shown to enhance exposure; thus, the same vehicle was indiscriminately utilized for all the compounds tested in this assay.22
Table 5. Results of the in Vivo Efficacy Screening.
| inhibition
(%) |
||
|---|---|---|
| compd | day 3 PE | day 6 PE |
| 4 | 0.0 | 0.0 |
| 12 | 9.0 | 0.0 |
| 24 | 0.0 | 0.0 |
| 33 | 53.6 | 26.9 |
| 34 | 41.4 | 26.8 |
| 45 | 85.7 | 61.0 |
| 46 | 83.9 | 69.0 |
| 50 | 44.3 | 29.1 |
| 58 | 60.0 | 28.8 |
| amodiaquine | 95.5 | 99.9 |
| artesunate | 97.0 | 81.0 |
| atovaquone | 96.3 | 99.8 |
As expected, the reference antimalarial compounds amodiaquine (AMO), artesunate (AS), and atovaquone (ATOV) had >95% inhibition of parasitemia on days 3 and 6 PE. Biphenyl and biaryl ether 4(1H)-quinolones 45 and 46 were the most active compounds, displaying over 80% inhibition on day 3 PE and over 60% inhibition on day 6 PE. Reduced in vivo efficacy, averaging 52% inhibition on day 3 PE and 28% inhibition on day 6 PE, was observed for compounds 33, 34, 50, and 58. Compounds 12 and 24, which were among the most soluble compounds in this test series, were poorly active or not active at all.
From this initial in vivo efficacy screening, it was concluded that the four analogues 33, 34, 50, and 58 were physicochemically similar to compounds 45 and 46, and the decreased in vivo activity was related primarily to the 10-fold reduction of in vitro potency. On the other hand, compound 12, being nearly equipotent to 3-(4-trifluoromethoxy)phenyl-4(1H)-quinolone 58 in the in vitro efficacy testing, was not efficacious in vivo. This result is likely due to its drastically decreased microsomal stability. These results prompted a significant paradigm shift, as microsomal stability and aqueous solubility were now considered to be equally important in the further optimization of in vivo efficacious 4(1H)-quinolones.
Disruption of the 3-Aryl-4(1H)-quinolones’ Molecular Planarity and Symmetry
Compound 12 was slightly more active in vitro than 4; nevertheless, it displayed minor suppressive antimalarial activity in vivo. This observation was attributed primarily to the slightly better solubility of 12 by the out-of-plane aryl moiety due to the additional ortho-methyl group. In order to support our hit-to-lead optimization efforts, we decided to investigate why these frontrunner compounds displayed such differences in the in vivo efficacy assays. Quantum mechanics (QM) torsional profile calculations and single-crystal X-ray diffraction (XRD) studies were conducted with the objective of determining structural modifications responsible for the improvement in solubility of 4(1H)-quinolone 12.
As the main structural difference between compounds 4 and 12 is the absence or presence of the ortho-methyl group on the 3-phenyl ring, the rotational barriers along the C3–C12 bond of both 3-aryl-4(1H)-quinolones 4 and 12 were first established using QM calculations following methodology previously reported (Figure 2).36 Relaxed dihedral angle scans (starting with a 0° angle when the methyl groups of the quinolone and ortho-methyl-3-phenyl moieties are directed toward each other) at torsion angle increments of 15° were carried out employing the ab initio HF/6-31G** method using the Jaguar program (version 7.9, Schrödinger, Inc.). Maestro software (version 9.2, Schrödinger, Inc.) was used for model building and visualization and to launch the Jaguar program. The structures were subjected to geometry optimization prior to the torsion scan using the same level of theory. The torsional profile was then obtained by plotting 24 energy points versus the dihedral angles. Previously, the calculated rotational barrier ΔErot = 20 kcal/mol was reported as a suitable threshold between atropisomers and non-atropisomers.37 As previously reported, the accuracy of QM calculations to predict atropisomerism is 86% relative to experimental values; therefore, QM calculations are a practical tool for this purpose. Based on the QM energy profile obtained for compound 4, the lowest energy barrier that allowed the C3–C12 bond rotation was ΔErot = 11.9 kcal/mol. This result suggests that the torsional rotation half-time at room temperature was in the order of milliseconds with a free rotation of the C3–C12 bond. In contrast, the minimal energy barrier of compound 12 has been calculated to be ΔErot = 23.2 kcal/mol, implying that the half-time for the same bond rotation was in the range of hours. According to the classification proposed by LaPlante and co-workers,36 compound 12 could be categorized as showing Class 2 atropisomerism, as the rotation of the 3-phenyl ring around the C3–C12 bond was significantly slower. Importantly, Class 2 atropisomers can be developed as a mixture of stereoisomers as long as the racemization is faster relative to the in vivo elimination rates.
Figure 2.
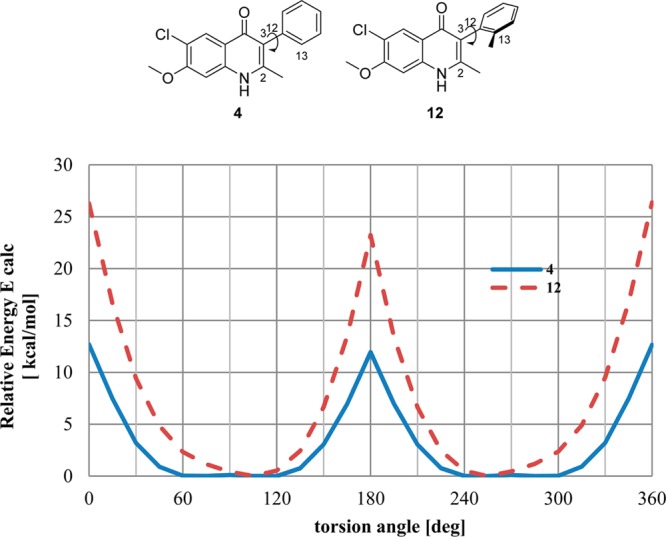
Quantum mechanics energy profile for 4 and 12 around the C3–C12 bond. Relaxed dihedral angle scans with a torsional angle increment of 15° were carried out employing a HF/6-31G** method using the Jaguar application in the Maestro suite (version 9.2, Schrödinger, Inc.).
Previously, it has been reported that disruption of the molecular planarity is an effective method to increase the aqueous solubility up to 350-fold.38 The expected orthogonal orientation of the 3-aryl substituent relative to the 4(1H)-quinolone plane for 12 and 4 (confirmed by QM calculations) could therefore be a likely cause of the increase in aqueous solubility. In order to explain the solubility differences between 4 and 12, XRD experiments have been conducted to study the crystal packing for both compounds. For compound 4, an asymmetric unit contains one molecule, whereas two different conformers are found in the asymmetric unit of analogue 12 (Figure 3A). In both structures, molecular chains are observed due to the presence of hydrogen bond interactions between the quinolone nitrogen and the quinolone oxygen (Figure 3B). The hydrogen bond distances between the quinolone oxygen and nitrogen atoms are 2.800 Å for compound 4, and 2.687 and 2.808 Å for compound 12 (with two molecules in the asymmetric unit). For 4 and 12, the 3-aryl substituents were nearly orthogonal to the 4(1H)-quinolone plane, with torsion angles of −81.1° and +73.3° for compound 12 and −69.96° for compound 4 (Figure 3A). In both cases, π–π interactions between quinolone planes were significantly disrupted, most likely because of the presence of weak CH···O and CH···π interactions with the methyl group as the H-bond donor (Figure 3C). For compound 12, the additional methyl group of the 3-aryl substituent forms a weak CH···O bond with an oxygen of the 7-methoxy group, leading to crystalline pockets with 3.4% free space in the crystal structure (yellow circles in Figure 3C,D) and a Kitaigorodski packing index (KPI) of 68%. This is in stark contrast to the crystal packing of compound 4, with a higher KPI of 71.4%, lacking any free space. Therefore, the potentially weaker interactions between molecules arising from the ortho-methyl-3-phenyl substituent, which probably correlates with a looser packing of the molecules within the crystal, are possibly the reason for the slightly higher aqueous solubility of compound 12 over its analogue 4. The XRD studies performed in conjunction with QM calculations strongly support the notion that the disruption of dense molecular crystal packing of 4(1H)-quinolones improves the aqueous solubility. At the molecular level, weak and strong intermolecular interactions have to be taken into account in order to explain and expand this observation in detail for each case at study.
Figure 3.

Crystal structures of compounds 4 and 12. (A) Asymmetric units, conformation, and numbering schemes. (B) Hydrogen-bonding schemes. (C) Packing schemes showing the disruption of π···π interactions through weak CH···O and CH···π interactions (dotted lines). The crystalline pocket in 12 is marked as a yellow sphere. Some of the molecules are omitted for clarity. (D) Packing schemes. Crystalline voids can be seen in the crystal structure of 12.
Lead Optimization
With activity data and physicochemical results in hand for an array of compounds bearing a wide variety of aryl substituents at the 3-position, a number of potential agents were envisioned that would retain the solubility of compounds similar to 12 while maintaining antimalarial potency and microsomal stability of the para-substituted phenyls at the 3-position (7, 59–67, Table 6). Initially, compounds 59–61 were synthesized in efforts to incorporate both the 3-substituted phenyl with an ortho-methyl and a para-substituent. Each of the three compounds maintained its activities against both W2 and TM90-C2B and showed improved log D values of 2.69–2.88. Solubility, however, plummeted in reference to compound 12. Microsomal stability of the three analogues showed a marked improvement over the para-fluorophenyl analogue by at least 2-fold, with analogue 60 showing virtually no degradation. An attempt to expound on the promising results for biaryl ether 46 led to the synthesis of analogous biaryl ethers possessing both an ortho-substituent on the proximal ring and a para-substituent on the distal ring. Analogues 62 and 7 were synthesized and tested. While activities for analogue 7 remained essentially unchanged, analogue 62 showed sub-nanomolar activities against both W2 and TM90-C2B. Additionally, both analogues showed improvements in solubility and log D. Microsomal stability improved from a CLint of 19 μL/(min·mg) for compound 46 to the point that no degradation was measured.
Table 6. Optimized 3-Aryl-4(1H)-quinolonesa.
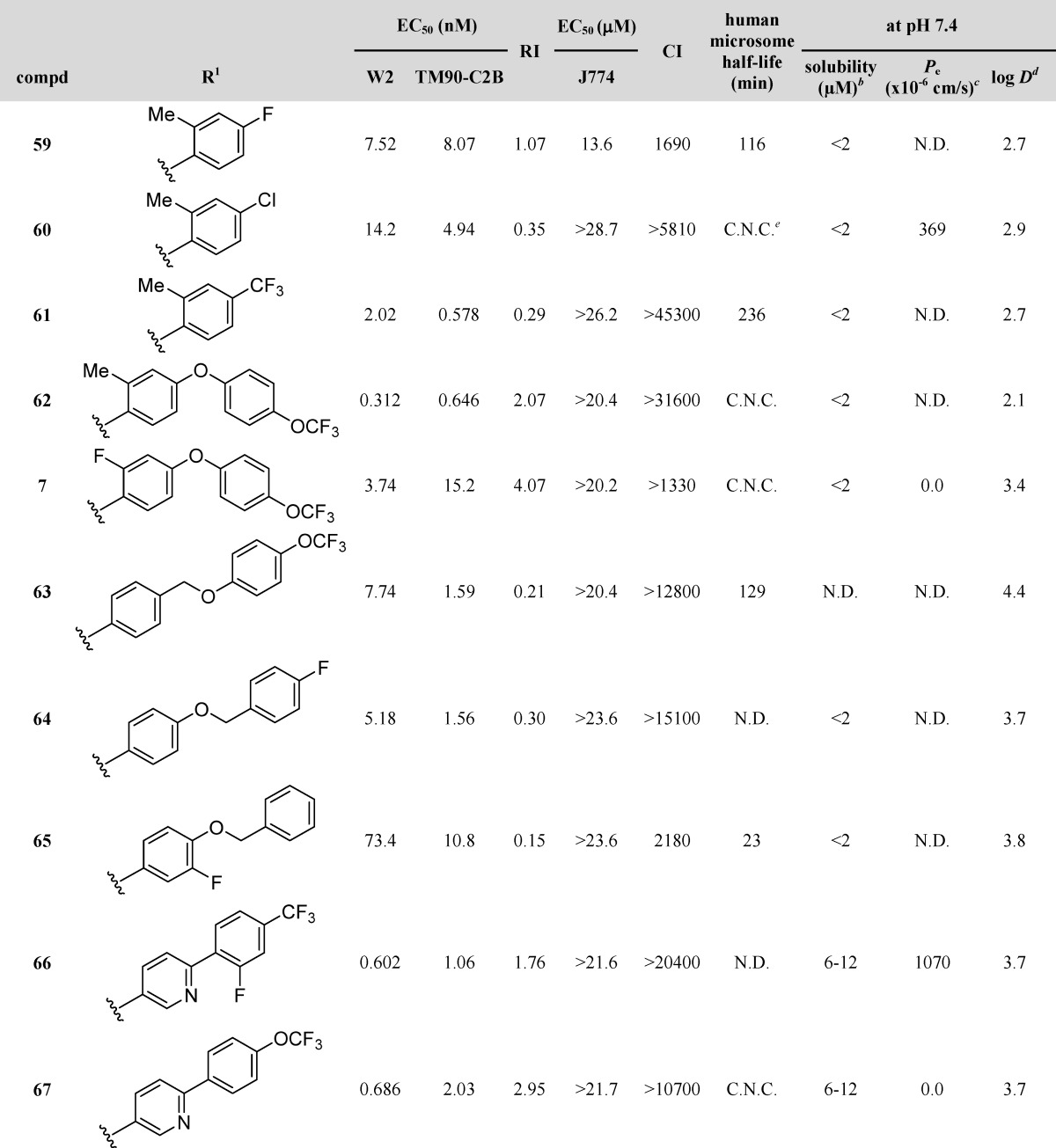
Dihydroartemisinin (DHA), chloroquine (CQ), and atovaquone (ATO) are internal controls for each in vitro assay: DHA, 5.5 nM W2 and 5.9 nM TM90-C2B; CQ, 229 nM for TM90-C2B and 421 nM for W2; and ATO, 1.4 nM W2 and 18.4 μM TM90-C2B.
Standards for the solubility assay include carbamazepine and albendazole. Solubility for carbamazepine at pH 7.4, 4.0, and 2.0 is 95 μM, 100 μM, and 100 μM, respectively. Solubility for albendazole at pH 7.4, 4.0, and 2.0 is 6.1 μM, 12 μM, and 100 μM, respectively.
Standards for the permeability assay include verapamil HCl (Pe = 1405 × 10–6 cm/s at pH 7.4 and 39 × 10–6 cm/s at pH 4.0), carbamazepine (Pe = 112 ×10–6 cm/s at pH 7.4 and 108 × 10–6 cm/s at pH 4.0), and ranitidine HCl (Pe = 0.5 × 10–6 cm/s at pH 7.4 and 0 × 10–6 cm/s at pH 4.0).
Standards for the log D7.4 assay include cinnarizine (log D7.4 = 5.68), hydrocortisone-21-acetate (log D7.4 = 2.19), ketoconazole (log D7.4 = 3.83), metronidazole (log D7.4 = −0.02), nadolol (log D7.4 = 0.68), pyrene (log D7.4 = 4.88), theophyline (log D7.4 = −0.05), and tolnaftate (log D7.4 = 5.40).
C.N.C. = could not be calculated. These compounds displayed little to no observable degradation throughout the assay duration (typically 250 min). Analogue 1 was used as a control for comparing half-lives of 4(1H)-quinolones. Compound 1 possesses half-lives of 7.9 min in mouse microsomes and 10.2 min in human microsomes.
In an effort to improve the results obtained with benzyloxyphenyl and phenoxybenzyl analogues 47 and 48, para-substituted analogues 63 and 64 were synthesized and tested. Analogue 64 showed a 2-fold improvement in activity against W2 while its activity against TM90-C2B remained the same. The solubility with the para-fluoro increased substantially while log D remained similar. Compound 65, possessing the fluoro substituent at the meta-position of the proximal ring rather than the para-position of the distal ring, showed a drastic decrease in activities but a moderate increase in solubility. Likewise, analogue 63, the para-trifluoromethoxy congener of 48, showed a decrease in activities while physicochemical data remained similar.
Based on the data generated from the compounds previously discussed, several 3-substituted 4(1H)-quinolones showed promising results. Biaryl 45 possessed excellent activities but poor solubility. The 3-pyridyl analogues 19–21 had enhanced solubility yet poor EC50 values, while the 3-para-substituted phenyl analogues 54 and 58 possessed good EC50 values coupled with excellent microsomal stability for 58. The new focus was to combine the desired properties of enhanced solubility and good microsomal stability while maintaining excellent activities against W2 and TM90-C2B strains. Analogues incorporating a pyridyl-based biaryl with fluoro substituents were envisioned; 66 and 67 were synthesized and tested. Both analogues showed excellent EC50 values against W2 and TM90-C2B and favorable RI values. Solubility testing rendered excellent data for 66, while 67 remained unchanged. Compound 67 also showed minimal degradation in microsomal stability testing.
In Vivo Efficacy Evaluation of Frontrunner Compounds
A modified Thompson test model was used for definitive in vivo efficacy evaluation of frontrunner compounds. Mice were infected with 1 × 106P. berghei-GFP parasites. Mice were treated once a day (days 3–5 PE) with 3, 10, or 50 mg/kg of test compound suspended in PEG400. On days 3, 6, 9, 13, 21, and 30 PE, parasitemia were monitored via blood smears. The primary evaluation was defined as the reduction in parasitemia on day 6 PE and the survival to day 30 PE, whereby a parasitemia level at day 30 of <1% (99% inhibition) was considered to be a cure. Animals were euthanized when >40% parasitemia was reached. The 4(1H)-quinolones assayed in these rigorous Thompson tests consisted of analogues that excelled in the in vivo efficacy screening assay, as well as optimized analogues showing promising in vitro activity and physicochemical properties.
3-Ethyl- and 3-phenyl-substituted 4(1H)-quinolones 3 and 4, previously regarded as adequate for in vivo efficacy studies, had no in vivo activity in the more vigorous challenge of the Thompson test assay, in which higher parasitemia is achieved prior to starting treatment.20 In comparison to reference 4, ortho-methylphenyl-substituted 4(1H)-quinolone 12, with a 3-fold increased solubility and a 4-fold improved in vitro antimalarial activity, produced a modest 17% parasitemia reduction at 10 mg/kg dosing (Table 7). A 41% parasitemia reduction was observed at an increased dose of 50 mg/kg for compound 12. Compounds 25 and 44, which were among the most soluble analogues, failed to demonstrate suppressive activity, whereas aminopyridine 23 showed 22% suppression of parasitemia albeit only at a dose of 50 mg/kg. Biphenyl and biaryl ether 4(1H)-quinolones 45 and 46, which were the most promising compounds in the preliminary in vivo efficacy screening, efficiently inhibited parasite development by day 6 at 10 and 50 mg/kg. Likewise, fluorinated compounds 49 and especially 52 were potent 4(1H)-quinolones with suppressive activities of up to 97%. These results supported our previous findings that the trifluoromethylphenyl analogue 52, which was superior in terms of antimalarial activity, solubility, and microsomal stability, was better suited for in vivo efficacy studies. Compounds 60 and 61, substituted analogues of 4(1H)-quinolone 12, were even more efficacious, increasing the survival period of mice to 11 and 13 days PE. These results are most likely due to the increased metabolic stability derived from the addition of the chloro (60) and trifluoromethyl substituents (61). Curative activities were obtained with 4(1H)-quinolones 7, 62, 66, and 67, which were structurally related to biphenyl 45 and biaryl ether 46, at doses as low as 3 mg/kg and 1 mg/kg. Based on results by GSK and insights gained from our own SAR studies and physicochemical evaluation, it was believed that 4(1H)-quinolones 7 and 62 would be among the most potent analogues in the 4(1H)-quinolone series.19 As expected, the biological activities of analogues 7 and 62 were excellent, with 98–99.5% inhibition on day 6 PE. Similar results were observed for biaryl ethers 45 and 46. Compound 67 cured all the mice at a dose of 10 mg/kg, whereas 62 was not as active, curing two out of five animals. There was no difference in the survival curves between the untreated group of mice and the one treated with 12 (Figure 4).
Table 7. Results of Thompson Test.
| efficacy |
||||
|---|---|---|---|---|
| compd | dose (mg/kg) | inhibition (%), day 6 PE | survival (daysa) | cure (%), n = 5 |
| 4 | 50 | <1 | 0 | 0 |
| 12 | 10 | 17 | 0 | 0 |
| 50 | 41 | 0 | 0 | |
| 23 | 10 | <1 | 0 | 0 |
| 50 | 22 | 0 | 0 | |
| 25 | 10 | <1 | 0 | 0 |
| 50 | <1 | 0 | 0 | |
| 44 | 10 | <1 | 0 | 0 |
| 50 | <1 | 0 | 0 | |
| 45 | 10 | 80 | 0 | 0 |
| 50 | 79 | 0 | 0 | |
| 46 | 10 | 87 | 4 | 0 |
| 50 | 70 | 4 | 0 | |
| 49 | 10 | 63 | 1 | 0 |
| 50 | 30 | 1 | 0 | |
| 52 | 10 | 92 | 3 | 0 |
| 50 | 97 | 4 | 0 | |
| 60 | 10 | 95 | 5 | 0 |
| 50 | 71 | 5 | 0 | |
| 61 | 3 | 84 | 6 | 0 |
| 10 | 80 | 6 | 0 | |
| 62 | 10 | 99 | 23 | 100 |
| 50 | 98 | 23 | 100 | |
| 7 | 1 | 99.3 | 23 | 100 |
| 3 | 99.3 | 23 | 100 | |
| 10 | 99.5 | 23 | 100 | |
| 50 | 99.2 | 23 | 100 | |
| 66 | 0.3 | 94.3 | 23 | 60 |
| 1 | 94.5 | 23 | 100 | |
| 3 | 96.3 | 23 | 100 | |
| 10 | 95.2 | 23 | 100 | |
| 67 | 0.3 | 93.9 | 23 | 80 |
| 1 | 95.8 | 23 | 100 | |
| 3 | 97.3 | 23 | 80 | |
| 10 | 97.3 | 23 | 100 | |
| amodiaquine | 30 | 99.8 | 23 | 100 |
| atovaquone | 50 | 99.1 | 23 | 100 |
Number of days animals survived beyond control, untreated animals.
Figure 4.
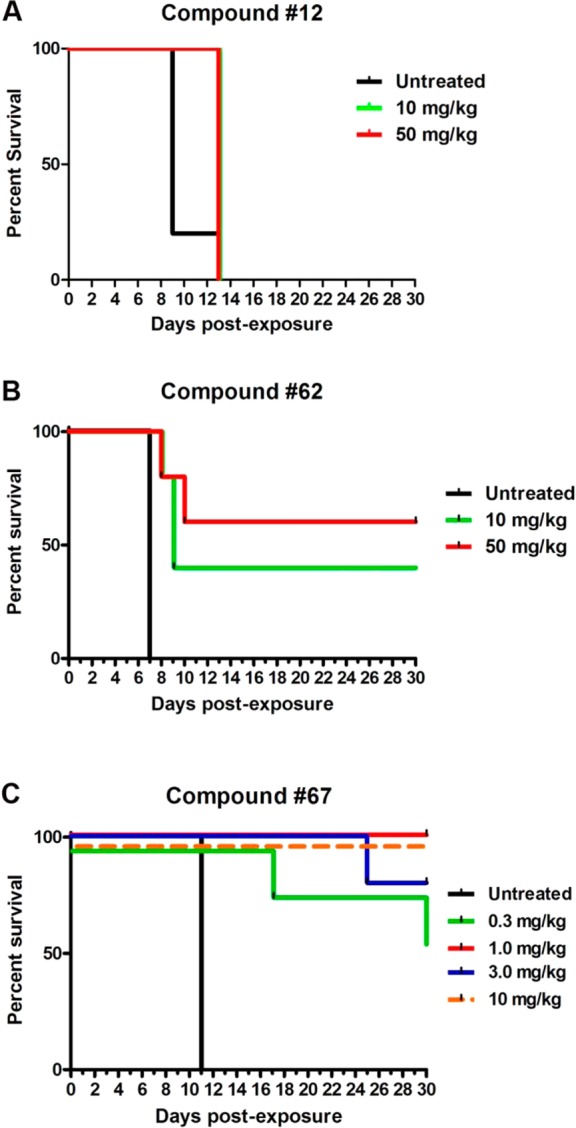
Representative survival curves for 4(1H)-quinolones 12 (A), 62 (B), and 67 (C) that demonstrate enhanced in vivo efficacy congruent with sequential improved physiological properties of the series.
In Vivo Exposure and Pharmacokinetics of Frontrunner Compounds
The in vivo exposure of four lead compounds (7, 52, 66 and 67) was assessed in Swiss mice following oral administration at a dose of 10 mg/kg in the same formulation used for efficacy studies (Figure 5). The highest exposure was observed for 7, and slightly lower exposure was observed for 66 and 67. Interestingly, exposure of 52 was substantially lower than those of the other three frontrunners, which may have partly contributed to its lower in vivo efficacy in the Thompson test.
Figure 5.

Abbreviated (three time-point) plasma exposure profiles for four frontrunner compounds in Swiss outbred mice following oral administration as PEG400 suspensions at 10 mg/kg: (●) 52, (△) 66, (■) 67, and (◇) 7.
Compound 7 was further examined for its pharmacokinetic properties in rats (Figure 6). Compound 7 exhibited a long in vivo half-life (32–42 h), low plasma clearance (0.3 mL/(min·kg)), and low volume of distribution (0.7 L/kg) following intravenous administration. These properties are likely to be influenced by the extensive binding to plasma proteins (>99%). The apparent oral bioavailability of 7 after dosing in PEG400 was approximately 14%. It is unlikely that bioavailability was limited by extensive first-pass metabolism or poor intestinal permeability, given that in vitro studies indicated that both of these were within acceptable ranges. Rather, the low aqueous solubility of 7, as described above, would suggest that absorption is likely to be limited by a combination of precipitation from the co-solvent vehicle and incomplete dissolution within the gastrointestinal tract.
Figure 6.

Plasma concentration versus time profiles for compound 7 in Sprague–Dawley rats (average of n = 2): (●) concentrations following IV administration at 0.15 mg/kg and (○) concentrations following oral administration of a PEG400 suspension at 10 mg/kg.
Conclusions
Inspired by encouraging results from previous SAR studies on the 6-chloro-7-methoxy-4(1H)-quinolone core, we synthesized a library of 58 analogues bearing a variety of aryl substituents at the 3-position of the core using various Suzuki–Miyaura cross-coupling conditions and tested for antimalarial activities against clinically relevant W2 and TM90-C2B strains. The library of compounds was also subjected to extensive physicochemical testing for solubility, permeability, log D, and microsomal stability in an effort to predict their potential for in vivo efficacy. This process was done in parallel with the synthetic chemistry to produce rapid SAR and SPR results, thus facilitating in vivo study timelines.
Nine of the 58 compounds gave positive results in some, but not all, of the physicochemical properties that were anticipated to predict effective in vivo activity. When the nine leads were subjected to in vivo testing, it was realized that microsomal stability was a key indicator for in vivo efficacy of the antimalarial 4(1H)-quinolones.
An optimized sub-series of 3-substituted-4(1H)-quinolones possessing structural features of the most active, most soluble, and most stable compounds in preliminary SAR studies were synthesized and subjected to a complete panel of in vitro and in vivo testing. All compounds possessed excellent activities against W2 and TM90-C2B strains along with desirable RI values and favorable cytotoxicity values. Most of the compounds showed low CLint values when tested for microsomal stability. Ultimately, six of the optimized compound sub-series were subjected to in vivo testing using a modified Thompson test. Four of the compounds, 7, 62, 66, and 67, resulted in >97% suppression of parasitemia 6 days PE, eliciting curative activities with as little as 1 mg/kg necessary for a curative dose. In conjunction with previous studies demonstrating potent exoerythrocytic activity and transmission blocking,22,39,40 3-substituted 4(1H)-quinolones have tremendous potential as antimalarial drug candidates.
Experimental Section
Synthetic Chemistry
All palladium catalysts and ligands were purchased from Strem. All boronic acids were purchased from Frontier Scientific. The identities of all title compounds were verified via 1H NMR, 13C NMR, and HPLC/HRMS. The chemical purity of the title compounds was determined using the following conditions: Agilent 1100 series LC/MSD with an Eclipse XDB-C18 (4.6 mm × 100 mm, 5 μm) reversed-phase column; 10% (v/v) acetonitrile (+0.05% TFA) in 90% (v/v) H2O (+0.05% TFA), ramped to 100% acetonitrile (+0.05% TFA) over 9 min, and holding at 100% acetonitrile for 4 min with a flow rate of 0.7 mL/min; UV detector, 254 nm. The purity of each compound was ≥95% in this analysis. NMR spectra were recorded at ambient temperature on a 400 or 500 MHz Varian NMR spectrometer in the solvent indicated. All 1H NMR experiments are reported in parts per million (ppm) downfield of TMS and were measured relative to the signals for chloroform (7.26 ppm) and dimethyl sulfoxide (2.50 ppm). All 13C NMR spectra are reported in ppm relative to the signals for chloroform (77 ppm) and dimethyl sulfoxide (39.5 ppm) with 1H decoupled observation. Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), integration, and coupling constant (Hz), whereas 13C NMR analyses were obtained at 101 MHz and reported in terms of chemical shift. NMR data were analyzed by using MestReNova software version 5.3.2-4936. High-resolution mass spectrometry (HRMS) was performed on an Agilent G3250AA LC/MSD TOF system. Isomers were separated by reversed-phase HPLC (Waters Prep LC 4000 system with Waters 996 photodiode array detector, Agilent Eclipse XDB-C18 column, 5 μM, 9.4 × 250 mm). Compounds were eluted using a gradient elution of 70/30 to 50/50 A/B over 30 min at a flow rate of 5.0 mL/min, where solvent A was water and solvent B was acetonitrile. Analytical thin-layer chromatography (TLC) was performed on silica gel 60 F254 pre-coated plates (0.25 mm) from EMD Chemical Inc., and components were visualized by UV light (254 nm). Silicycle silica gel 230–400 (particle size 40–63 μm) mesh was used for all flash column chromatography.
General Procedure A: Preparation of 3-Aryl-6-chloro-7-methoxy-4(1H)-quinolones via Suzuki–Miyaura Cross-Coupling
An oven-dried Schlenk tube was flame-dried and backfilled with argon (×3). The tube was then charged with quinolone (100 mg, 0.4 mmol), Pd2(dba)3 (14.5 mg, 0.016 mmol), SPHOS (13 mg, 0.032 mmol), boronic acid (0.6 mmol), and anhydrous powdered K3PO4 (168 mg, 0.79 mmol). The Schlenk tube was fitted with a rubber septum and then evacuated and backfilled with argon (this process was repeated three times). Dry solvent (toluene, DMF, or 2-butanol, 1 mL) was added through the septum via syringe, and the resulting solution was stirred for 1 min while purging with argon before replacing the rubber septum with the Teflon screwcap. The reaction was placed in an oil bath at 110 °C until completion was observed via HPLC analysis. The reaction was cooled to room temperature and then diluted with 20 mL of chloroform and 20 mL of methanol. The mixture was heated to boiling with a heat gun and then filtered over a pad of Celite. Then, 10–30 mL of boiling DMF was passed through the Celite pad. The eluent was concentrated under reduced pressure, and the residual oil was purified further via flash chromatography. Some 4(1H)-quinolones required two successive column purifications. The purified solids were then collected, placed in a 2-mL fritted funnel, and washed with ice-cold methanol, followed by diethyl ether; they were then dried in vacuo to obtain the NMR-pure samples. Some 4(1H)-quinolones could be triturated with cold ether upon in vacuo removal of the chloroform/methanol/DMF, placed in a 2-mL frit, and washed with ∼30 mL of diethyl ether and ∼30 mL of ice-cold methanol without any further purification.
General Procedure B: Preparation of 3-Aryl-6-chloro-7-methoxy-4(1H)-quinolones via Suzuki–Miyaura Cross-Coupling
An oven-dried Schlenk tube was flame-dried and backfilled with argon (×3). The tube was then charged with quinolone (100 mg, 0.4 mmol), Pd(PPh3)4 (14.5 mg, 0.016 mmol), boronic acid (0.6 mmol), and degassed 1–2 M Na2CO3 (1 mL). The Schlenk tube was fitted with a rubber septum and then evacuated and backfilled with argon (this process was repeated three times). DMF (5 mL) was added through the septum via syringe, and the resulting solution was stirred for 1 min while purging with argon before replacing the rubber septum with the Teflon screwcap. The reaction was placed in an oil bath at 110 °C until completion was observed via HPLC analysis. The reaction was cooled to room temperature and then diluted with 20 mL of chloroform and 20 mL of methanol. This mixture was heated to boiling with a heat gun and then filtered over a pad of Celite. Then, 10–30 mL of boiling DMF was passed through the Celite pad. The eluent was concentrated under reduced pressure, and the residual oil was purified further via flash chromatography. Some 4(1H)-quinolones required two successive column purifications. The purified solids were then collected, placed in a 2-mL fritted funnel, and washed with ice-cold methanol, followed by diethyl ether; they were then dried in vacuo to obtain the NMR-pure samples. Some 4(1H)-quinolones could be triturated with cold ether upon in vacuo removal of the chloroform/methanol/DMF, placed in a 2-mL fritted funnel, and washed with ∼30 mL of diethyl ether and ∼30 mL of ice-cold methanol without any further purification.
General Procedure C: Preparation of 3-Aryl-6-chloro-7-methoxy-4(1H)-quinolones via Suzuki–Miyaura Cross-Coupling
An oven-dried Schlenk tube was flame-dried and backfilled with argon (×3). The tube was then charged with quinolone (100 mg, 0.4 mmol), Pd(PPh3)4 (14.5 mg, 0.016 mmol), boronic acid (0.6 mmol), and K3PO4 (168 mg, 0.79 mmol). The Schlenk tube was fitted with a rubber septum and then evacuated and backfilled with argon (this process was repeated three times). A degassed mixture of toluene/H2O (9:1) was added through the septum via syringe, and the resulting solution was stirred for 1 min while purging with argon before replacing the rubber septum with the Teflon screwcap. The reaction was placed in an oil bath at 110 °C until completion was observed via HPLC analysis. The reaction was cooled to room temperature and then diluted with 20 mL of chloroform and 20 mL of methanol. This mixture was heated to boiling with a heat gun and then filtered over a pad of Celite. Then, 10–30 mL of boiling DMF was passed through the Celite pad. The eluent was concentrated under reduced pressure, and the residual oil was purified further via flash chromatography. Some 4(1H)-quinolones required two successive column purifications. The purifieded solids were then collected, placed in a 2-mL fritted funnel, and washed with ice-cold methanol, followed by diethyl ether; they were then dried in vacuo to obtain the NMR-pure samples. Some 4(1H)-quinolones could be triturated with cold ether upon in vacuo removal of the chloroform/methanol/DMF, placed in a 2-mL fritted funnel, and washed with ∼30 mL of diethyl ether and ∼30 mL of ice-cold methanol without any further purification.
General Procedure D: Preparation of 3-Aryl-6-chloro-7-methoxy-4(1H)-quinolones via Suzuki–Miyaura Cross-Coupling
An oven-dried Schlenk tube was flame-dried and backfilled with argon (×3). The tube was then charged with quinolone (100 mg, 0.4 mmol), Pd2(dba)3 (14.5 mg, 0.016 mmol), SPHOS (13 mg, 0.032 mmol), boronic acid (0.6 mmol), and anhydrous powdered K3PO4 (168 mg, 0.79 mmol). The Schlenk tube was fitted with a rubber septum and then evacuated and backfilled with argon (this process was repeated three times). A degassed mixture of toluene/H2O (9:1) was added through the septum via syringe, and the resulting solution was stirred for 1 min while purging with argon before replacing the rubber septum with the Teflon screwcap. The reaction was placed in an oil bath at 110 °C until completion was observed via HPLC analysis. The reaction was cooled to room temperature and then diluted with 20 mL of chloroform and 20 mL of methanol. This mixture was heated to boiling with a heat gun and then filtered over a pad of Celite. Then, 10–30 mL of boiling DMF was passed through the Celite pad. The eluent was concentrated under reduced pressure, and the residual oil was purified further via flash chromatography. Some 4(1H)-quinolones required two successive column purifications. The purified solids were then collected, placed in a 2-mL frit, and washed with ice-cold methanol, followed by diethyl ether; they were then dried in vacuo to obtain the NMR-pure samples. Some 4(1H)-quinolones could be triturated with cold ether upon in vacuo removal of the chloroform/methanol/DMF, placed in a 2-mL fritted funnel, and washed with ∼30 mL of diethyl ether and ∼30 mL of ice-cold methanol without any further purification.
General Procedure E: Preparation of 3-Aryl-6-chloro-7-methoxy-4(1H)-quinolones via Suzuki–Miyaura Cross-Coupling
An oven-dried Schlenk tube was flame-dried and backfilled with argon (×3). The tube was then charged with quinolone (100 mg, 0.4 mmol), Pd2(dba)3 (14.5 mg, 0.016 mmol), SPHOS (13 mg, 0.032 mmol), boronic acid (0.6 mmol), and degassed 1–2 M Na2CO3 (1 mL). The Schlenk tube was fitted with a rubber septum and then evacuated and backfilled with argon (this process was repeated three times). Dry solvent (toluene, DMF, or 2-butanol, 5 mL) was added through the septum via syringe, and the resulting solution was stirred for 1 min while purging with argon before replacing the rubber septum with the Teflon screwcap. The reaction was placed in an oil bath at 110 °C until completion was observed via HPLC analysis. The reaction was cooled to room temperature and then diluted with 20 mL of chloroform and 20 mL of methanol. This mixture was heated to boiling with a heat gun and then filtered over a pad of Celite. Then, 10–30 mL of boiling DMF was passed through the Celite pad. The eluent was concentrated under reduced pressure, and the residual oil was purified further via flash chromatography. Some 4(1H)-quinolones required two successive column purifications. The purified solids were then collected, placed in a 2-mL frit, and washed with ice-cold methanol, followed by diethyl ether; they were then dried in vacuo to obtain the NMR-pure samples. Some 4(1H)-quinolones could be triturated with cold ether upon in vacuo removal of the chloroform/methanol/DMF, placed in a 2-mL fritted funnel, and washed with ∼30 mL of diethyl ether and ∼30 mL of ice-cold methanol without any further purification.
General Procedure F: Preparation of 3-Aryl-6-chloro-7-methoxy-4(1H)-quinolones via Suzuki–Miyaura Cross-Coupling
An oven-dried Schlenk tube was flame-dried and backfilled with argon (×3). The tube was then charged with O-ethyl quinolone (100 mg, 0.39 mmol), Pd2(dba)3 (14.5 mg, 0.016 mmol), SPHOS (13 mg, 0.032 mmol), boronic acid (0.6 mmol), and anhydrous powdered K3PO4 (168 mg, 0.79 mmol). The Schlenk tube was fitted with a rubber septum and then evacuated and backfilled with argon (this process was repeated three times). Dry solvent (toluene, DMF, or 2-butanol, 1 mL) was added through the septum via syringe, and the resulting solution was stirred for 1 min while purging with argon before replacing the rubber septum with the Teflon screwcap. The reaction was placed in an oil bath at 110 °C until completion was observed via HPLC analysis. The reaction was cooled to room temperature and then diluted with 20 mL of chloroform and 20 mL of methanol. This mixture was heated to boiling with a heat gun and then filtered over a pad of Celite. Then, 10–30 mL of boiling DMF was passed through the Celite pad. The eluent was concentrated under reduced pressure, and the residual oil was purified further via flash chromatography. Some 4(1H)-quinolones required two successive column purifications. The purified solids were then collected, placed in a 2-mL fritted funnel, and washed with ice-cold methanol, followed by diethyl ether; they were then dried in vacuo to obtain the NMR-pure samples. Some 4(1H)-quinolones could be triturated with cold ether upon in vacuo removal of the chloroform/methanol/DMF, placed in a 2-mL frit, and washed with ∼30 mL of diethyl ether and ∼30 mL of ice-cold methanol without any further purification. The resulting Suzuki adduct was then dissolved in 1:1 AcOH/HBr and refluxed for 1.5 h. The solution was allowed to cool to room temperature and poured onto ice and water. Next, the crude compound was isolated via filtration and washed with 100 mL of water. The product was placed in a 60 °C oven, dried for 1 h, and then re-crystallized from DMF.
General Procedure G: Preparation of 3-Aryl-6-chloro-7-methoxy-4(1H)-quinolones via Suzuki–Miyaura Cross-Coupling
An oven-dried Schlenk tube was flame-dried and backfilled with argon (×3). The tube was then charged with O-ethyl quinolone (100 mg, 0.39 mmol), Pd2(dba)3 (14.5 mg, 0.016 mmol), SPHOS (13 mg, 0.032 mmol), boronic acid (0.6 mmol), and degassed 1–2 M Na2CO3 (1 mL). The Schlenk tube was fitted with a rubber septum and then evacuated and backfilled with argon (this process was repeated three times). DMF (5 mL) was added through the septum via syringe, and the resulting solution was stirred for 1 min while purging with argon before replacing the rubber septum with the Teflon screwcap. The reaction was placed in an oil bath at 110 °C until completion was observed via HPLC analysis. The reaction was cooled to room temperature and then diluted with 20 mL of chloroform and 20 mL of methanol. This mixture was heated to boiling with a heat gun and then filtered over a pad of Celite. Then, 10–30 mL of boiling DMF was passed through the Celite pad. The eluent was concentrated under reduced pressure, and the residual oil was purified further via flash chromatography. Some 4(1H)-quinolones required two successive column purifications. The purified solids were then collected, placed in a 2-mL fritted funnel, and washed with ice-cold methanol, followed by diethyl ether; they were then dried in vacuo to obtain the NMR-pure samples. Some 4(1H)-quinolones could be triturated with cold ether upon in vacuo removal of the chloroform/methanol/DMF, placed in a 2-mL fritted funnel, and washed with ∼30 mL of diethyl ether and ∼30 mL of ice-cold methanol without any further purification. The resulting Suzuki adduct was then dissolved in 1:1 AcOH/HBr and refluxed for 1.5 h. The solution was allowed to cool to room temperature and poured onto ice and water. Next, the crude compound was isolated via filtration and washed with 100 mL of water. The product was placed in a 60 °C oven, dried for 1 h, and then re-crystallized from DMF.
Preparation and Characterization of Compounds
6-Chloro-7-methoxy-2-methyl-3-(o-tolyl)quinolin-4(1H)-one (12)
Compound 12 was prepared using general procedure A in 31% yield as a white powder, mp = 337–339 °C. 1H NMR (400 MHz, DMSO): δ 11.66 (s, 1H), 7.99 (s, 1H), 7.22 (dt, J = 22.4, 7.1, 3H), 7.08 (s, 1H), 7.03 (d, J = 6.9, 1H), 3.96 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.16, 156.63, 146.50, 139.71, 137.35, 135.92, 130.81, 129.48, 127.04, 126.11, 125.46, 120.44, 118.58, 117.91, 99.44, 56.37, 19.30, 18.39. HRMS: calcd for C18H16ClNO2 (M+H)+ 314.09423, found 314.09470.
6-Chloro-7-methoxy-2-methyl-3-(p-tolyl)quinolin-4(1H)-one (13)
Compound 13 was prepared using general procedure A in 78% as a tan solid, mp = 336–341 °C. 1H NMR (400 MHz, DMSO): δ 11.59 (s, 1H), 7.99 (s, 1H), 7.18 (d, J = 7.7 Hz, 2H), 7.10 (d, J = 7.7 Hz, 2H), 7.03 (s, 1H), 3.94 (s, 3H), 2.33 (s, 3H), 2.18 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.57, 156.58, 146.33, 139.54, 135.51, 132.79, 130.75, 128.34, 126.16, 120.64, 118.74, 117.86, 99.35, 56.32, 20.80, 18.83. HRMS: calcd for C18H16ClNO2 (M+H)+ 314.09423, found 314.09456.
6-Chloro-3-(2,4-dimethylphenyl)-7-methoxy-2-methylquinolin-4(1H)-one (14)
Compound 14 was a prepared using general procedure A in 28%, mp = decomp at 346 °C. 1H NMR (400 MHz, DMSO): δ 11.63 (s, 1H), 7.98 (s, 1H), 7.06 (s, 2H), 6.99 (d, J = 7.6 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 3.95 (s, 3H), 2.29 (s, 3H), 2.04 (s, 3H), 1.97 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.98, 157.27, 147.26, 140.39, 137.77, 136.64, 133.58, 131.36, 130.91, 126.79, 121.02, 119.27, 118.54, 100.12, 57.04, 21.40, 19.95, 19.11. HRMS: calcd for C19H18ClNO2 (M+H)+ 328.1099, found 328.1102.
6-Chloro-3-mesityl-7-methoxy-2-methylquinolin-4(1H)-one (15)
Compound 15 was prepared using general procedure G in 10% as a tan solid, mp = 350–352 °C. 1H NMR (250 MHz, DMSO): δ 11.68 (s, 1H), 7.99 (s, 1H), 7.07 (s, 1H), 6.89 (s, 2H), 3.95 (s, 3H), 2.25 (s, 3H), 1.93 (d, J = 13.5 Hz, 9H). 13C NMR (126 MHz, DMSO): δ 173.2, 156.6, 146.4, 139.8, 136.6, 135.6, 132.5, 127.8, 126.1, 118.8, 118.5, 117.8, 99.5, 56.4, 20.7, 19.6, 17.8. HRMS: calcd for C20H20ClNO2 (M+H)+ 342.12553, found 342.12447.
6-Chloro-3-(2-ethylphenyl)-7-methoxy-2-methylquinolin-4(1H)-one (16)
Compound 16 was prepared following general procedure A in 8% yield, mp = decomp at 322 °C. 1H NMR (400 MHz, DMSO): δ 11.68 (s, 1H), 7.99 (s, 1H), 7.27 (d, J = 12.6 Hz, 2H), 7.21 (d, J = 5.2 Hz, 1H), 7.08 (s, 1H), 7.00 (d, J = 7.3 Hz, 1H), 3.96 (s, 3H), 2.35 (q, J = 7.3 Hz, 2H), 2.05 (s, 3H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO): δ 173.49, 156.63, 146.61, 143.23, 139.77, 135.35, 131.03, 127.86, 127.33, 126.13, 125.57, 120.42, 118.58, 117.93, 99.49, 56.38, 25.82, 18.59, 14.87.HRMS: calcd for C19H18ClNO2 (M+H)+ 328.1099, found 328.1107.
6-Chloro-3-(4-isopropylphenyl)-7-methoxy-2-methylquinolin-4(1H)-one (17)
Compound 17 was prepared using general procedure A in 66% as a white solid, mp = 359–360 °C. 1H NMR (400 MHz, DMSO): δ 11.60 (s, 1H), 7.99 (s, 1H), 7.24 (d, J = 7.8 Hz, 2H), 7.14 (d, J = 7.8 Hz, 2H), 7.05 (s, 1H), 3.95 (s, 3H), 2.91 (m, 1H), 2.19 (s, 3H), 1.24 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, DMSO): δ 173.61, 164.24, 156.60, 146.41, 139.54, 133.16, 130.80, 126.17, 125.64, 120.65, 118.73, 117.87, 99.35, 56.34, 33.17, 23.91, 18.89. HRMS: calcd for C20H20ClNO2 (M+H)+ 342.12553, found 342.12436.
3-(4-tert-Butylphenyl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (18)
Compound 18 was prepared using general procedure A in 35% as a white solid, mp = 360–362 °C. 1H NMR (400 MHz, DMSO): δ 11.60 (s, 1H), 7.99 (s, 1H), 7.40 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 7.06 (s, 1H), 3.96 (s, 3H), 2.20 (s, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO): δ 174.30, 157.31, 149.34, 147.07, 140.24, 133.48, 131.24, 126.86, 125.16, 121.26, 119.42, 118.57, 100.06, 57.05, 34.90, 31.88, 19.60. HRMS: calcd for C21H22ClNO2 (M+H)+ 356.14118, found 356.14311.
6-Chloro-7-methoxy-2-methyl-3-(pyridin-2-yl)quinolin-4(1H)-one (19)
Compound 19 was prepared using general procedure F in 29% over two steps, mp = decomp at 305 °C. 1H NMR (400 MHz, DMSO): δ 11.69 (s, 1H), 8.56 (s, 1H), 7.98 (s, 1H), 7.75 (td, J = 7.7, 1.7 Hz, 1H), 7.43 (d, J = 7.8 Hz, 1H), 7.24 (dd, J = 6.6, 5.1 Hz, 1H), 7.05 (s, 1H), 3.93 (s, 3H), 2.22 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.74, 157.23, 155.44, 149.10, 148.55, 139.97, 135.88, 127.29, 126.55, 122.03, 120.53, 119.50, 118.68, 100.05, 56.83, 19.03. HRMS: calcd for C16H13ClN2O2 (M+H)+ 301.0738, found 301.0747.
6-Chloro-7-methoxy-2-methyl-3-(pyridin-3-yl)quinolin-4(1H)-one (20)
Compound 20 was prepared using general procedure D in 12% as a purple solid, mp = 301–304 °C. 1H NMR (400 MHz, DMSO): δ 11.85 (s, 1H), 8.49 (s, 1H), 8.45 (s, 1H), 8.00 (s, 1H), 7.68 (d, J = 6.7, 1H), 7.43 (d, J = 4.5, 1H), 7.10 (s, 1H), 3.96 (s, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.40, 156.83, 151.34, 147.48, 147.24, 139.72, 138.44, 131.58, 126.10, 122.96, 118.62, 118.23, 117.24, 99.60, 56.42, 18.85. HRMS: calcd for C16H13ClN2O2 (M+H)+ 301.07383, found 314.07266.
6-Chloro-7-methoxy-2-methyl-3-(pyridin-4-yl)quinolin-4(1H)-one (21)
Compound 21 was prepared using general procedure B in 19% as a tan solid, mp = 307–309 °C. 1H NMR (400 MHz, DMSO): δ 11.80 (s, 1H), 8.58 (d, J = 5.8, 2H), 8.00 (s, 1H), 7.31–7.28 (m, 2H), 7.07 (s, 1H), 3.96 (s, 3H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO): δ 172.85, 156.90, 149.05, 146.95, 143.94, 139.60, 126.21, 126.11, 118.68, 118.34, 118.18, 99.55, 56.42, 18.78. HRMS: calcd for C16H13ClN2O2 (M+H)+ 301.07383, found 301.07303.
6-Chloro-7-methoxy-2-methyl-3-(pyrimidin-5-yl)quinolin-4(1H)-one (22)
Compound 22 was prepared using general procedure A in 12%. 1H NMR (400 MHz, DMSO): δ 11.90 (s, 1H), 9.11 (s, 1H), 8.74 (s, 2H), 8.01 (s, 1H), 7.09 (s, 1H), 3.97 (s, 3H), 2.30 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.88, 159.05, 157.73, 157.12, 148.56, 140.37, 130.39, 126.76, 119.27, 114.44, 108.95, 100.35, 57.15, 19.52. HRMS: calcd for C15H12ClN3O2 (M+H)+ 302.0691, found 302.0688.
6-Chloro-3-(6-(dimethylamino)pyridin-3-yl)-7-methoxy-2-methylquinolin-4(1H)-one (23)
Compound 23 was prepared using general procedure E in 16%, mp = decomp at 330 °C. 1H NMR (400 MHz, DMSO): δ 11.61 (s, 1H), 7.98 (s, 1H), 7.94 (d, J = 1.9 Hz, 1H), 7.39 (dd, J = 8.7, 2.2 Hz, 1H), 7.03 (s, 1H), 6.64 (d, J = 8.7 Hz, 1H), 3.95 (s, 3H), 3.04 (s, 6H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.90, 157.69, 156.58, 148.91, 146.65, 139.75, 139.49, 126.17, 118.68, 118.52, 117.88, 117.84, 104.83, 99.35, 56.33, 37.72, 18.93. HRMS: calcd for C18H18ClN3O2 (M+H)+ 344.11603, found 344.12216.
6-Chloro-7-methoxy-2-methyl-3-(6-(4-methylpiperazin-1-yl)pyridin-3-yl)quinolin-4(1H)-one (24)
Compound 24 was prepared using general procedure B in 10%, mp = decomp at 277 °C. 1H NMR (400 MHz, DMSO): δ 11.70 (s, 1H), 8.04 (s, 1H), 7.99 (d, J = 1.2 Hz, 1H), 7.52 (d, J = 8.6 Hz, 1H), 7.08 (s, 1H), 6.98 (d, J = 8.5 Hz, 1H), 3.97 (s, 3H), 2.80 (s, 3H), 2.26 (s, 3H). 13C NMR (101 MHz, DMSO): δ 174.20, 157.12, 156.91, 149.45, 147.35, 140.91, 139.97, 126.57, 122.05, 118.94, 118.47, 117.73, 107.18, 99.86, 56.85, 52.74, 42.87, 42.72, 19.35. HRMS: calcd for C21H23ClN4O2 (M+H)+ 399.1582, found 399.1583.
6-Chloro-3-(furan-2-yl)-7-methoxy-2-methylquinolin-4(1H)-one (25)
Compound 25 was prepared using general procedure A in 72% as a yellow solid, mp = 300–301 °C. 1H NMR (400 MHz, DMSO): δ 11.78 (s, 1H), 8.03 (s, 1H), 7.68 (s, 1H), 7.07 (s, 1H), 6.78 (d, J = 3.0 Hz, 1H), 6.55 (s, 1H), 3.96 (s, 3H), 2.45 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.17, 157.42, 149.37, 148.82, 141.91, 139.68, 126.81, 119.17, 119.09, 111.42, 111.27, 111.06, 100.24, 57.06, 20.11. HRMS: calcd for C15H12ClNO3 (M+H)+ 290.05785, found 290.05890.
6-Chloro-3-(furan-3-yl)-7-methoxy-2-methylquinolin-4(1H)-one (26)
Compound 26 was prepared using general procedure B in 45%, mp = 318–324 °C. 1H NMR (400 MHz, DMSO): δ 11.59 (s, 1H), 7.96 (s, 1H), 7.79 (s, 1H), 7.64 (d, J = 0.8 Hz, 1H), 6.99 (s, 1H), 6.61 (d, J = 0.8 Hz, 1H), 3.91 (s, 3H), 2.36 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.41, 156.74, 147.03, 142.01, 141.82, 139.25, 126.28, 118.37, 118.33, 118.23, 112.80, 111.63, 99.42, 56.48, 19.44. HRMS: calcd for C15H12ClNO3 (M+H)+ 290.0579, found 290.0574.
6-Chloro-3-(3,5-dimethylisoxazol-4-yl)-7-methoxy-2-methylquinolin-4(1H)-one (27)
Compound 27 was prepared using general procedure A in 16% as a red solid, mp = 274–275 °C. 1H NMR (400 MHz, DMSO): δ 11.82 (s, 1H), 7.99 (s, 1H), 7.08 (s, 1H), 3.96 (s, 3H), 2.17 (s, 6H), 1.98 (s, 3H). 13C NMR (101 MHz, DMSO): δ 174.14, 166.58, 160.84, 157.54, 149.51, 140.41, 126.74, 118.98, 118.87, 111.11, 108.96, 100.24, 57.12, 19.00, 11.94, 10.89. HRMS: calcd for C16H15ClN2O3 (M+H)+ 319.08440, found 319.08290.
3-(Benzo[c][1,2,5]oxadiazol-5-yl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (28)
Compound 28 was prepared using general procedure B in 22%. 1H NMR (400 MHz, DMSO): δ 11.87 (s, 1H), 8.03–7.94 (m, 2H), 7.90 (s, 1H), 7.50 (d, J = 9.3 Hz, 1H), 7.08 (s, 1H), 3.97 (s, 3H), 2.34 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.1, 157.0, 149.2, 148.1, 147.7, 140.8, 139.6, 137.4, 134.1, 126.0, 118.6, 118.5, 116.1, 114.3, 99.6, 56.5, 18.9. HRMS: calcd for C17H12ClN3O3 (M+H)+ 342.0640, found 342.0651.
3-(Benzofuran-2-yl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (29)
Compound 29 was prepared using general procedure A in 59%, mp = 322–323 °C. 1H NMR (400 MHz, DMSO): δ 11.94 (s, 1H), 8.06 (s, 1H), 7.67–7.62 (m, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.29–7.21 (m, 3H), 7.09 (s, 1H), 3.97 (s, 3H), 2.56 (s, 3H). 13C NMR (101 MHz, DMSO): δ 172.67, 156.95, 153.40, 151.64, 149.42, 139.08, 128.58, 126.15, 123.60, 122.59, 120.60, 118.73, 118.50, 110.67, 109.95, 106.91, 99.74, 56.44, 19.66. HRMS: calcd for C19H14ClNO3 (M+H)+ 340.0735, found 340.0734.
3-(Benzo[b]thiophen-2-yl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (30)
Compound 30 was prepared using general procedure C in 53%, mp = 345–346 °C. 1H NMR (500 MHz, DMSO): δ 11.91 (s, 1H), 8.04 (s, 1H), 7.93 (d, J = 7.7, 1H), 7.82 (d, J = 7.2, 1H), 7.38–7.30 (m, 3H), 7.09 (s, 1H), 3.98 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, DMSO): δ 173.60, 157.43, 149.08, 140.43, 139.77, 139.62, 138.12, 126.68, 124.70, 124.36, 124.29, 123.60, 122.33, 119.04, 118.83, 113.76, 100.10, 56.95, 20.08. HRMS: calcd for C19H14ClNO2S (M+H)+ 356.05065, found 356.05077.
6-Chloro-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-7-methoxy-2-methylquinolin-4(1H)-one (31)
Compound 31 was prepared using general procedure A in 61%, mp = 330–332 °C. 1H NMR (400 MHz, DMSO): δ 11.58 (s, 1H), 7.98 (s, 1H), 7.03 (s, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.74–6.59 (m, 2H), 4.25 (s, 4H), 3.94 (s, 3H), 2.20 (s, 3H). 13C NMR (101 MHz, DMSO): δ 174.29, 157.26, 147.21, 143.39, 142.77, 140.18, 129.37, 126.84, 124.46, 120.90, 120.20, 119.38, 118.56, 116.97, 100.02, 64.74, 64.69, 57.01, 19.54. HRMS: calcd for C19H16ClNO4 (M+H)+ 358.08406, found 358.08474.
6-Chloro-3-(dibenzo[b,d]furan-4-yl)-7-methoxy-2-methylquinolin-4(1H)-one (32)
Compound 32 was prepared using general procedure B in 24%, mp = decomp at 220 °C. 1H NMR (400 MHz, DMSO): δ 11.86 (s, 1H), 8.14 (dd, J = 18.0, 6.8 Hz, 2H), 8.02 (s, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.50–7.37 (m, 4H), 7.13 (s, 1H), 3.99 (s, 3H), 2.17 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.27, 156.84, 155.22, 154.07, 147.61, 139.81, 130.28, 127.35, 126.15, 123.86, 123.45, 122.95, 122.81, 121.04, 120.42, 119.95, 118.60, 118.25, 115.37, 111.63, 99.60, 56.44, 18.62. HRMS: calcd for C23H16ClNO3 (M+H)+ 390.0892, found 390.0892.
6-Chloro-7-methoxy-3-(4-methoxyphenyl)-2-methylquinolin-4(1H)-one (33)
Compound 33 was prepared using general procedure A in 68% as an off-white solid, mp = 297–298 °C. 1H NMR (400 MHz, DMSO): δ 11.59 (s, 1H), 7.99 (s, 1H), 7.14 (d, J = 7.8 Hz, 2H), 7.05 (s, 1H), 6.94 (d, J = 7.9 Hz, 2H), 3.95 (s, 3H), 3.78 (s, 3H), 2.20 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.73, 157.92, 156.61, 146.46, 139.55, 131.99, 127.82, 126.21, 120.39, 118.74, 117.88, 113.25, 99.38, 56.37, 55.01, 18.90. HRMS: calcd for C18H16ClNO3 (M+H)+ 330.08915, found 330.08983.
6-Chloro-3-(4-isopropoxyphenyl)-7-methoxy-2-methylquinolin-4(1H)-one (34)
Compound 34 was prepared using general procedure A in 62% as a brown solid, mp = 342–343 °C. 1H NMR (600 MHz, DMSO): δ 11.59 (s, 1H), 7.99 (s, 1H), 7.12 (d, J = 8.6 Hz, 2H), 7.05 (s, 1H), 6.90 (d, J = 8.6 Hz, 2H), 4.61 (m, 1H), 3.95 (s, 3H), 2.20 (s, 3H), 1.29 (d, J = 6.0 Hz, 6H). 13C NMR (126 MHz, DMSO): δ 173.70, 156.58, 156.10, 146.41, 139.53, 132.02, 127.51, 126.19, 120.41, 118.72, 117.83, 114.77, 99.35, 68.95, 56.35, 21.94, 18.92. HRMS: calcd for C20H20ClNO3 (M+H)+ 358.12045, found 358.11946.
4-(6-Chloro-7-methoxy-2-methyl-4-oxo-1,4-dihydroquinolin-3-yl)benzaldehyde (35)
Compound 35 was prepared using general procedure B in 15%, mp = decomp at 341 °C. 1H NMR (400 MHz, DMSO): δ 11.72 (s, 1H), 10.00 (s, 1H), 7.96 (s, 1H), 7.88 (d, J = 7.8 Hz, 2H), 7.47 (s, 2H), 7.03 (s, 1H), 3.92 (s, 3H), 2.20 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 197.97, 178.31, 162.03, 152.00, 147.81, 144.78, 139.73, 137.01, 134.08, 131.33, 124.86, 123.92, 123.44, 104.71, 61.60, 24.06. HRMS: calcd for C18H14ClNO3 (M+H)+ 328.0735, found 328.0740.
3-(4-Acetylphenyl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (36)
Compound 36 was prepared using general procedure A in 13% as a yellow solid, mp = 266–269 °C. 1H NMR (250 MHz, DMSO): δ 11.65 (s, 1H), 7.94–7.80 (m, 3H), 7.30 (d, J = 8.0 Hz, 2H), 6.96 (s, 1H), 3.85 (s, 3H), 2.39 (s, 3H), 2.11 (s, 3H). 13C NMR (101 MHz, DMSO): δ 198.28, 173.87, 157.48, 147.38, 141.81, 140.28, 135.75, 131.99, 128.32, 126.84, 120.46, 119.41, 118.86, 100.18, 57.09, 27.38, 19.54. HRMS: calcd for C19H16ClNO3 (M+H)+ 342.08915, found 342.08955.
4-(6-Chloro-7-methoxy-2-methyl-4-oxo-1,4-dihydroquinolin-3-yl)benzoic Acid (37)
Compound 37 was prepared using general procedure, mp = > 352 °C. 1H NMR (400 MHz, DMSO): δ 12.86 (s, 1H), 11.71 (s, 1H), 7.96 (dd, J = 13.4, 10.2 Hz, 3H), 7.37 (d, J = 8.1 Hz, 2H), 7.05 (s, 1H), 3.95 (s, 3H), 2.21 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.22, 167.32, 156.79, 146.70, 140.75, 139.60, 131.22, 128.92, 128.73, 126.15, 119.85, 118.73, 118.19, 99.49, 56.39, 18.85. HRMS: calcd for C18H14ClNO4 (M+H)+ 344.0684, found 344.0686.
Methyl 4-(6-Chloro-7-methoxy-2-methyl-4-oxo-1,4-dihydroquinolin-3-yl)benzoate (38)
Compound 38 was prepared using general procedure A in 32%. 1H NMR (400 MHz, DMSO): δ 11.73 (s, 1H), 7.98 (dd, J = 9.5, 1.8 Hz, 2H), 7.89 (d, J = 10.4 Hz, 1H), 7.43–7.34 (m, 2H), 7.05 (s, 1H), 3.95 (s, 3H), 3.87 (s, 3H), 2.21 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.16, 166.23, 156.81, 146.74, 141.23, 139.60, 131.41, 128.55, 127.75, 126.14, 119.68, 118.73, 118.22, 99.51, 95.86, 56.40, 52.07, 18.85. HRMS: calcd for C19H16ClNO4 (M+H)+ 358.0841, found 358.0847.
6-Chloro-3-(4-(dimethylamino)phenyl)-7-methoxy-2-methylquinolin-4(1H)-one (39)
Compound 39 was prepared using general procedure E in 49% as a tan solid, mp = 324–325 °C. 1H NMR (400 MHz, DMSO): δ 11.52 (s, 1H), 7.99 (s, 1H), 7.03 (s, 2H), 6.72 (d, J = 8.0 Hz, 2H), 3.94 (s, 3H), 2.91 (s, 6H), 2.20 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.86, 156.46, 149.07, 146.14, 139.45, 131.36, 126.21, 123.29, 120.83, 118.72, 117.65, 111.81, 99.29, 56.30, 40.19, 18.92. HRMS: calcd for C19H19ClN2O2 (M+H)+ 343.12078, found 343.12216.
4-(6-Chloro-7-methoxy-2-methyl-4-oxo-1,4-dihydroquinolin-3-yl)-N,N-dimethylbenzenesulfonamide (40)
Compound 40 was prepared using general procedure A in 34% as a brown solid, mp = 335–337 °C. 1H NMR (400 MHz, DMSO): δ 11.78 (s, 1H), 8.01 (s, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 7.08 (s, 1H), 3.97 (s, 3H), 2.66 (s, 6H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.06, 156.87, 146.98, 140.84, 139.60, 132.84, 131.88, 126.92, 126.09, 119.14, 118.69, 118.28, 99.54, 56.42, 37.60, 18.87. HRMS: calcd for C19H19ClN2O4S (M+H)+ 407.08268, found 407.08128.
6-Chloro-3-(4-hydroxyphenyl)-7-methoxy-2-methylquinolin-4(1H)-one (41)
Compound 41 was prepared using general procedure A in 44% as a gray solid, mp = 270–274 °C. 1H NMR (400 MHz, DMSO): δ 11.54 (s, 1H), 9.32 (s, 1H), 7.98 (s, 1H), 7.04 (s, 1H), 7.01 (d, J = 8.1 Hz, 2H), 6.76 (d, J = 8.0 Hz, 2H), 3.95 (s, 3H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO): δ 174.44, 157.22, 156.64, 146.98, 140.18, 132.55, 126.87, 126.74, 121.40, 119.40, 118.43, 115.28, 100.00, 57.02, 19.58. HRMS: calcd for C17H14ClNO3 (M+H)+ 316.07350, found 316.07254.
6-Chloro-3-(2-(hydroxymethyl)phenyl)-7-methoxy-2-methylquinolin-4(1H)-one (42)
Compound 42 was prepared using general procedure A in 32% as a white solid, mp = 267–269 °C. 1H NMR (400 MHz, DMSO): δ 11.74 (s, 1H), 7.99 (s, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.34 (t, J = 7.4 Hz, 1H), 7.26 (t, J = 7.3 Hz, 1H), 7.09 (s, 1H), 7.03 (d, J = 7.3 Hz, 1H), 4.90 (s, 1H), 4.28 (d, J = 13.4 Hz, 1H), 4.17 (d, J = 13.5 Hz, 1H), 3.96 (s, 3H), 2.06 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.50, 156.70, 147.16, 141.69, 139.74, 134.08, 130.70, 127.12, 126.80, 126.49, 126.09, 119.60, 118.49, 118.11, 99.51, 61.05, 56.41, 18.73. HRMS: calcd for C18H16ClNO3 (M+H)+ 330.08915, found 330.08830.
6-Chloro-3-(3-(hydroxymethyl)phenyl)-7-methoxy-2-methylquinolin-4(1H)-one (43)
Compound 43 was prepared using general procedure A in 23%, mp = 265–267 °C. 1H NMR (250 MHz, DMSO): δ 11.64 (s, 1H), 7.99 (s, 1H), 7.34 (t, J = 7.4 Hz, 1H), 7.24 (d, J = 7.6 Hz, 1H), 7.16 (s, 1H), 7.08 (d, J = 6.9 Hz, 2H), 5.19 (s, 1H), 4.51 (d, J = 4.2 Hz, 2H), 3.96 (s, 3H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO): δ 174.19, 157.30, 147.08, 142.64, 140.29, 136.30, 129.90, 129.69, 128.18, 126.83, 125.41, 121.60, 119.46, 118.57, 100.11, 63.62, 57.03, 19.53. HRMS: calcd for C18H16ClNO3 (M+H)+ 330.08915, found 330.09014.
6-Chloro-3-(4-(hydroxymethyl)phenyl)-7-methoxy-2-methylquinolin-4(1H)-one (44)
Compound 44 was prepared using general procedure A in 49% as a yellow solid, mp = 310–315 °C. 1H NMR (400 MHz, DMSO): δ 11.61 (s, 1H), 7.99 (s, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 7.05 (s, 1H), 5.17 (s, 1H), 4.52 (d, J = 3.7 Hz, 2H), 3.95 (s, 3H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.56, 156.62, 146.37, 140.71, 139.58, 134.15, 130.60, 126.17, 125.94, 120.70, 118.77, 117.89, 99.39, 62.87, 56.34, 18.85. HRMS: calcd for C18H16ClNO3 (M+H)+ 330.08915, found 330.09001.
3-([1,1′-Biphenyl]-4-yl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (45)
Compound 45 was prepared using general procedure A in 53% as a gray solid, mp = 352–353 °C. 1H NMR (400 MHz, DMSO): δ 11.63 (s, 1H), 7.99 (s, 1H), 7.65 (dd, J = 15.1, 7.6 Hz, 4H), 7.54–7.16 (m, 5H), 7.03 (s, 1H), 3.92 (s, 3H), 2.22 (s, 3H). 13C NMR (101 MHz, DMSO): δ 174.21, 157.36, 147.21, 140.75, 140.26, 138.98, 135.73, 132.22, 129.61, 127.97, 127.23, 126.87, 126.70, 120.95, 119.44, 118.68, 100.10, 57.05, 19.61. HRMS: calcd for C23H18ClNO2 (M+H)+ 376.10988, found 376.10541.
6-Chloro-7-methoxy-2-methyl-3-(4-phenoxyphenyl)quinolin-4(1H)-one (46)
Compound 46 was prepared using general procedure A in 65% as a white solid, mp = 303–305 °C. 1H NMR (400 MHz, DMSO): δ 11.64 (s, 1H), 8.00 (s, 1H), 7.41 (t, J = 7.9 Hz, 2H), 7.24 (d, J = 8.5 Hz, 2H), 7.15 (t, J = 7.4 Hz, 1H), 7.03 (dd, J = 22.4, 8.1 Hz, 5H), 3.95 (s, 3H), 2.23 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.53, 156.64, 155.31, 146.54, 139.55, 132.52, 130.80, 130.02, 126.15, 123.43, 120.00, 118.71, 117.94, 117.78, 99.38, 56.35, 18.90. HRMS: calcd for C23H18ClNO3 (M+H)+ 392.10480, found 392.10580.
3-(4-(Benzyloxy)phenyl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (47)
Compound 47 was prepared using general procedure A in 62%. 1H NMR (400 MHz, DMSO): δ 11.60 (s, 1H), 7.99 (s, 1H), 7.48 (d, J = 6.9 Hz, 2H), 7.41 (t, J = 6.9 Hz, 2H), 7.35 (d, J = 6.7 Hz, 1H), 7.15 (d, J = 7.7 Hz, 2H), 7.05 (s, 1H), 7.02 (d, J = 7.9 Hz, 2H), 5.13 (s, 2H), 3.95 (s, 3H), 2.20 (s, 3H). 13C NMR (101 MHz, DMSO): δ 173.64, 157.03, 156.59, 146.38, 139.53, 137.23, 131.98, 128.41, 128.04, 127.76, 127.64, 126.18, 120.32, 118.72, 117.83, 114.06, 99.36, 69.15, 56.35, 18.89. HRMS: calcd for C24H20ClNO3 (M+H)+ 406.12045, found 406.12204.
6-Chloro-7-methoxy-2-methyl-3-(4-(phenoxymethyl)phenyl)quinolin-4(1H)-one (48)
Compound 48 was prepared using general procedure A in 57% as a beige solid, mp = 301–302 °C. 1H NMR (400 MHz, DMSO): δ 11.60 (s, 1H), 7.96 (s, 1H), 7.42 (d, J = 7.6 Hz, 2H), 7.35–7.20 (m, 4H), 7.06–6.87 (m, 4H), 5.08 (s, 2H), 3.92 (s, 3H), 2.17 (s, 3H). 13C NMR (101 MHz, DMSO): δ 174.17, 159.11, 157.35, 147.15, 140.25, 136.07, 135.95, 131.67, 130.16, 127.90, 126.85, 121.34, 121.09, 119.43, 118.65, 115.39, 100.09, 69.74, 57.04, 19.55. HRMS: calcd for C24H20ClNO3 (M+H)+ 406.12045, found 406.12223.
6-Chloro-3-(4-fluorophenyl)-7-methoxy-2-methylquinolin-4(1H)-one (49)
Compound 49 was prepared using general procedure B in 52% as a gray solid, mp = 344–345 °C. 1H NMR (400 MHz, DMSO): δ 11.66 (s, 1H), 7.99 (s, 1H), 7.27 (dd, J = 8.5, 5.9, 2H), 7.20 (t, J = 8.9, 2H), 7.07 (s, 1H), 3.96 (s, 3H), 2.20 (s, 3H). 19F NMR (376 MHz, DMSO): δ −116.51. 13C NMR (101 MHz, DMSO): δ 171.50, 160.26 (d, J = 242.4), 154.66, 152.60, 145.84, 135.60, 132.845 (d, J = 7.07), 125.55, 120.67, 118.38, 115.66, 113.93 (d, J = 21.21), 104.47, 55.83, 22.45. HRMS: calcd for C17H13ClFNO2 (M+H)+ 318.06916, found 318.06988.
6-Chloro-3-(2,4-difluorophenyl)-7-methoxy-2-methylquinolin-4(1H)-one (50)
Compound 50 was prepared using general procedure A in 20% as a tan solid, mp = 318–323 °C. 1H NMR (400 MHz, DMSO): δ 11.79 (s, 1H), 7.98 (s, 1H), 7.32–7.26 (m, 2H), 7.12–7.07 (m, 2H), 3.97 (s, 3H), 2.16 (s, 3H). 19F NMR (376 MHz, DMSO): δ −108.97, −111.77. 13C NMR (101 MHz, DMSO): δ 173.73, 162.32 (dd, J = 246.44, 12.12), 160.81 (dd, J = 247.45, 13.13), 157.53, 148.29, 140.40, 134.95, 126.72, 120.37 (d, J = 17.17), 118.99 (d, J = 7.07), 114.51, 111.78 (d, J = 202.20), 104.52 (d, J = 26.26), 104.26 (d, J = 26.26), 100.24, 57.10, 19.12. HRMS: calcd for C17H12ClF2NO2 (M+H)+ 336.05974, found 336.06018.
6-Chloro-3-(2,6-difluorophenyl)-7-methoxy-2-methylquinolin-4(1H)-one (51)
mp = decomp at 278 °C. 1H NMR (400 MHz, DMSO): δ 11.95 (s, 1H), 7.99 (s, 1H), 7.41–7.52 (m, 1H), 7.15 (t, J = 7.69 Hz, 2H), 7.10 (s, 1H), 3.97 (s, 3H), 2.17 (s, 3H). 19F NMR (376 MHz, DMSO): δ −111.5. 13C NMR (101 MHz, DMSO): δ 172.67, 160.55 (dd, J = 245.2, 7.8 Hz), 156.98, 148.32, 139.83, 129.97 (t, J = 10.2 Hz), 126.20, 125.99, 118.32 (d, J = 39.6 Hz), 112.47, 111.40 (d, J = 25.6 Hz), 108.41, 99.70, 56.44, 18.25. HRMS: calcd for C17H12ClF2NO2 (M+H)+ 336.0597, found 336.0598.
6-Chloro-7-methoxy-2-methyl-3-(4-(trifluoromethyl)phenyl)quinolin-4(1H)-one (52)
Compound 52 was prepared using general procedure A in 31% as a yellow solid, mp = 322–323 °C. 1H NMR (400 MHz, DMSO): δ 11.79 (s, 1H), 8.00 (s, 1H), 7.74 (d, J = 8.1, 2H), 7.49 (d, J = 8.0, 2H), 7.09 (s, 1H), 3.97 (s, 3H), 2.22 (s, 3H). 19F NMR (376 MHz, DMSO): δ −61.24 13C NMR (126 MHz, DMSO): δ 173.20, 156.85, 146.87, 140.39, 139.64, 131.87, 127.11 (q, J = 31.6 Hz), 126.13, 124.56 (q, J = 3.6 Hz), 124.47 (q, J = 271.9 Hz), 119.37, 118.71, 118.26, 99.52, 56.43, 18.85. HRMS: calcd for C18H13ClF3NO2 (M+H)+ 368.06597, found 368.06445.
3-(3,5-Bis(trifluoromethyl)phenyl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (53)
Compound 53 was prepared using general procedure A in 7% as a yellow solid, mp = 305–307 °C. 1H NMR (400 MHz, DMSO): δ 11.88 (s, 1H), 8.12–7.81 (m, 4H), 7.10 (s, 1H), 3.98 (s, 3H), 2.28 (s, 3H). 19F NMR (376 MHz, DMSO): δ −61.54. 19F NMR (376 MHz, DMSO): δ −61.16. 13C NMR (101 MHz, DMSO): δ 172.99, 156.99, 147.62, 139.63, 138.52, 131.83, 129.73 (q, J = 32.5 Hz), 126.08, 123.49 (q, J = 271.0 Hz), 120.24, 118.54 (d, J = 12.5 Hz), 117.67, 99.60, 56.43, 18.84. HRMS: calcd for C19H12ClF6NO2 (M+H)+ 356.14118, found 356.14311.
6-Chloro-3-(2-fluoro-4-(trifluoromethyl)phenyl)-7-methoxy-2-methylquinolin-4(1H)-one (54)
Compound 54 was prepared using general procedure F in 75% over two steps, mp = decomp at 342 °C. 1H NMR (400 MHz, DMSO): δ 11.88 (s, 1H), 7.99 (d, J = 0.8 Hz, 1H), 7.70 (d, J = 9.5 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.09 (s, 1H), 3.97 (s, 3H), 2.19 (s, 3H). 19F NMR (376 MHz, DMSO): δ −61.03, −110.61. 13C NMR (101 MHz, DMSO): δ 173.20, 160.35 (d, J = 246.3 Hz), 157.41, 148.12, 140.19, 136.29–134.27 (m), 130.43–129.90 (m), 129.06–128.16 (m), 126.44, 122.03–120.77 (m), 118.93, 118.74, 113.88, 113.55–112.85 (m), 100.08, 56.88, 18.87. HRMS: calcd for C18H12ClF4NO2 (M+H)+ 386.0565, found 386.0576.
6-Chloro-3-(3-ethoxy-4-fluorophenyl)-7-methoxy-2-methylquinolin-4(1H)-one (55)
Compound 55 was prepared using general procedure A in 77% as a tan solid, mp = 328–329 °C. 1H NMR (400 MHz, DMSO): δ 11.64 (s, 1H), 7.98 (s, 1H), 7.16–7.03 (m, 3H), 6.97 (d, J = 8.0 Hz, 1H), 4.13 (q, J = 6.6 Hz, 2H), 3.95 (s, 3H), 2.21 (s, 3H), 1.37 (t, J = 6.7 Hz, 3H). 19F NMR (376 MHz, DMSO): δ −136.21. 13C NMR (101 MHz, DMSO): δ 174.12, 157.34, 151.76 (d, J = 243.41), 147.38, 145.62 (d, J = 11.11), 140.19, 129.15, 127.81, 126.82, 120.04, 119.28 (d, J = 15.15), 119.03, 118.66, 114.75, 100.06, 64.90, 57.03, 19.54, 15.33. HRMS: calcd for C19H17ClFNO3 (M+H)+ 362.09538, found 362.09390.
6-Chloro-3-(2,3-difluoro-4-methoxyphenyl)-7-methoxy-2-methylquinolin-4(1H)-one (56)
mp = decomp at 328 °C. 1H NMR (400 MHz, DMSO): δ 11.79 (s, 1H), 7.98 (s, 1H), 6.97–7.11 (m, 3H), 3.97 (s, 3H), 3.91 (s, 3H), 2.19 (s, 3H). 19F NMR (376 MHz, DMSO): δ −137.30, −161.36 13C NMR (101 MHz, DMSO): δ 173.07, 156.86, 148.60 (dd, J = 243.7, 10.1 Hz), 147.82, 147.72–147.56 (m), 140.29 (dd, J = 244.3, 16.2 Hz), 139.70, 126.76 (t, J = 4.0 Hz), 126.06, 118.31 (d, J = 4.4 Hz), 117.00, 116.86, 113.68, 108.54, 99.56, 56.51, 56.42, 18.49. HRMS: calcd for C18H14ClF2NO3 (M+H)+ 366.0703, found 366.0707.
6-Chloro-7-methoxy-2-methyl-3-(2-(trifluoromethoxy)phenyl)quinolin-4(1H)-one (57)
Compound 57 was prepared using general procedure A in 25% as a gray solid, mp = 312–316 °C. 1H NMR (400 MHz, DMSO): δ 11.87–11.79 (s, 1H), 7.99 (s, 1H), 7.46 (dt, J = 18.2, 8.8 Hz, 3H), 7.34 (d, J = 7.3 Hz, 1H), 7.10 (s, 1H), 3.97 (s, 3H), 2.12 (s, 3H). 19F NMR (376 MHz, DMSO): δ −56.13. 13C NMR (101 MHz, DMSO): δ 172.83, 156.83, 147.15, 139.74, 133.53, 129.49, 129.12, 128.13, 127.14, 126.05, 120.67, 120.02 (q, J = 256.0 Hz), 118.28, 118.24, 115.78, 99.65, 56.40, 18.28. HRMS: calcd for C18H13ClF3NO3 (M+H)+ 384.06088, found 384.06162.
6-Chloro-7-methoxy-2-methyl-3-(4-(trifluoromethoxy)phenyl)quinolin-4(1H)-one (58)
Compound 58 was prepared using general procedure B in 47% as a white solid, mp = 321–322 °C. 1H NMR (400 MHz, DMSO): δ 11.69 (s, 1H), 7.99 (s, 1H), 7.37 (s, 4H), 7.05 (s, 1H), 3.96 (s, 3H), 2.21 (s, 3H). 19F NMR (376 MHz, DMSO): δ −56.71. 13C NMR (101 MHz, DMSO): δ 173.31, 156.75, 146.97, 146.71, 139.57, 135.15, 132.83, 126.10, 120.30, 119.29, 118.65, 118.12, 99.43, 56.37, 18.83. HRMS: calcd for C18H13ClF3NO3 (M+H)+ 384.06088, found 384.06184.
6-Chloro-3-(4-fluoro-2-methylphenyl)-7-methoxy-2-methylquinolin-4(1H)-one (59)
Compound 59 was prepared using general procedure B in 34% yield, mp = decomp at 336 °C. 1H NMR (400 MHz, DMSO): δ 7.95 (s, 1H), 7.07 (d, J = 2.6 Hz, 1H), 7.04–7.00 (m, 3H), 3.92 (s, 3H), 2.00 (d, J = 12.1 Hz, 6H). 19F NMR (376 MHz, DMSO): δ −116.63. 13C NMR (101 MHz, DMSO): δ 173.19, 161.26 (d, J = 242.2 Hz), 156.67, 146.88, 140.26 (d, J = 8.0 Hz), 139.73, 132.56, 132.09, 126.09, 119.33, 118.53, 118.00, 115.97 (d, J = 20.9 Hz), 112.11 (d, J = 20.8 Hz), 99.47, 56.38, 19.34 (d, J = 1.3 Hz), 18.41. HRMS: calcd for C18H15ClFNO2 (M+H)+ 332.0848, found 332.0846.
6-Chloro-3-(4-chloro-2-methylphenyl)-7-methoxy-2-methylquinolin-4(1H)-one (60)
Compound 60 was prepared using general procedure A in 17% as a tan solid, mp = 340–341 °C. 1H NMR (400 MHz, DMSO): δ 11.68 (s, 1H), 7.95 (s, 1H), 7.32 (s, 1H), 7.22 (d, J = 7.9 Hz, 1H), 7.03 (d, J = 10.3 Hz, 2H), 3.93 (s, 3H), 2.01 (d, J = 15.7 Hz, 6H). 13C NMR (101 MHz, DMSO): δ 173.71, 157.40, 147.46, 140.82, 140.42, 135.62, 133.30, 132.18, 129.84, 126.76, 126.09, 119.83, 119.19, 118.75, 100.18, 57.08, 19.80, 19.08. HRMS: calcd for C18H15Cl2NO2 (M+H)+ 348.05526, found 348.05562.
6-Chloro-7-methoxy-2-methyl-3-(2-methyl-4-(trifluoromethyl)phenyl)quinolin-4(1H)-one (61)
Compound 61 was prepared using general procedure B in 10% yield, mp = 326–332 °C. 1H NMR (400 MHz, DMSO): δ 11.73 (s, 1H), 7.96 (d, J = 2.4 Hz, 1H), 7.61 (s, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.25 (d, J = 8.1 Hz, 1H), 7.05 (d, J = 2.1 Hz, 1H), 3.93 (d, J = 2.2 Hz, 3H), 2.08 (s, 3H), 2.03 (d, J = 2.2 Hz, 3H). 19F NMR (376 MHz, DMSO): δ −60.88. 13C NMR (101 MHz, DMSO): δ 172.86, 156.79, 146.72, 140.62, 139.77, 139.18, 131.82, 127.74 (q, J = 31.2 Hz), 126.04, 125.99 (q, J = 3.4 Hz), 124.45 (q, J = 271.9 Hz), 122.20 (q, J = 3.3 Hz), 119.14, 118.49, 118.20, 99.54, 56.42, 19.16, 18.38. HRMS: calcd for C19H15ClF3NO2 (M+H)+ 382.0816, found 382.0824.
6-Chloro-7-methoxy-2-methyl-3-(2-methyl-4-(4-(trifluoromethoxy)phenoxy)phenyl)-quinolin-4(1H)-one (62)
Compound 62 was prepared using general procedure B in 20%, mp = decomp at 311 °C. 1H NMR (400 MHz, DMSO): δ 7.98 (s, 1H), 7.40 (d, J = 9.0 Hz, 2H), 7.14 (d, J = 9.0 Hz, 2H), 7.06 (d, J = 10.5 Hz, 2H), 6.98 (s, 1H), 6.88 (s, 1H), 3.96 (s, 3H), 2.08 (s, 3H), 2.01 (s, 3H). 19F NMR (376 MHz, DMSO): δ −57.20. 13C NMR (126 MHz, DMSO): δ 173.25, 156.67, 155.92, 155.01, 146.91, 143.49, 139.90, 139.74, 132.50, 131.83, 126.12, 122.97, 120.12 (q, J = 255.0 Hz), 119.98, 119.62, 118.56, 117.98, 116.02, 99.47, 56.41, 19.45, 18.51. HRMS: calcd for C25H19ClF3NO4 (M+H)+ 490.1028, found 490.1042.
6-Chloro-7-methoxy-2-methyl-3-(4-((4-(trifluoromethoxy)phenoxy)methyl)phenyl)-quinolin-4(1H)-one (63)
Compound 63 was prepared using general procedure B in 3%. 1H NMR (400 MHz, DMSO): δ 11.66 (s, 1H), 8.01 (s, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 9.1 Hz, 2H), 7.28 (d, J = 7.9 Hz, 2H), 7.16 (d, J = 9.1 Hz, 2H), 7.08 (s, 1H), 5.17 (s, 2H), 3.98 (s, 3H), 2.23 (s, 3H). 19F NMR (376 MHz, DMSO): δ −57.30. 13C NMR (126 MHz, DMSO): δ 173.48, 157.33, 156.69, 146.54, 141.81, 139.63, 135.59, 134.81, 131.06, 127.36, 126.18, 122.56, 120.38, 120.19 (q, J = 255.1 Hz), 118.77, 117.98, 115.95, 99.46, 69.71, 56.39, 18.91. HRMS: calcd for C25H19ClF3NO4 (M+H)+ 490.1028, found 490.1043.
6-Chloro-3-(4-(4-fluorobenzyloxy)phenyl)-7-methoxy-2-methylquinolin-4(1H)-one (64)
Compound 64 was prepared using general procedure B in 25%, mp = decomp at 304 °C. 1H NMR (400 MHz, DMSO): δ 11.59 (s, 1H), 7.99 (s, 1H), 7.53 (dd, J = 8.3, 5.7 Hz, 2H), 7.24 (t, J = 8.7 Hz, 2H), 7.15 (d, J = 8.3 Hz, 2H), 7.06–7.00 (m, 3H), 5.11 (s, 2H), 3.96 (s, 3H), 2.20 (s, 3H). 19F NMR (376 MHz, DMSO): δ −114.60. 13C NMR (126 MHz, DMSO): δ 173.61, 161.72 (d, J = 243.2 Hz), 156.93, 156.60, 146.43, 139.52, 133.42, 131.99, 129.88 (d, J = 8.2 Hz), 128.09, 126.17, 120.30, 118.69, 117.87, 115.22 (d, J = 21.4 Hz), 114.07, 99.36, 68.43, 56.35, 18.89. HRMS: calcd for C24H19ClFNO3 (M+H)+ 424.1110, found 424.1123.
3-(4-(Benzyloxy)-3-fluorophenyl)-6-chloro-7-methoxy-2-methylquinolin-4(1H)-one (65)
Compound 65 was prepared using general procedure A in 41%, mp = 302–303 °C. 1H NMR (400 MHz, DMSO): δ 11.65 (s, 1H), 7.98 (s, 1H), 7.42 (ddd, J = 33.2, 19.1, 7.1 Hz, 5H), 7.24 (t, J = 8.7 Hz, 1H), 7.13–6.94 (m, 3H), 5.21 (s, 1H), 3.96 (s, 3H), 2.22 (s, 3H). 19F NMR (376 MHz, DMSO): δ −136.41. 13C NMR (101 MHz, DMSO): δ 174.09, 157.37, 151.87 (d, J = 244.42), 147.40, 145.50, 140.20, 137.36, 129.57, 129.16, 128.69, 128.47, 127.79, 126.83, 119.98, 119.35, 119.24 (d, J = 17.17), 118.68, 115.29, 100.08, 70.88, 57.05, 19.56. HRMS: calcd for C24H19ClFNO3 (M+H)+ 424.11103, found 424.10894.
6-Chloro-3-(6-(2-fluoro-4-(trifluoromethyl)phenyl)pyridin-3-yl)-7-methoxy-2-methylquinolin-4(1H)-one (66)
1H NMR (400 MHz, DMSO): δ 11.83 (s, 1H), 8.64–8.67 (m, 1H), 8.24 (t, J = 8.00 Hz, 1H), 8.02 (s, 1H), 7.81–7.95 (m, 3H), 7.74 (d, J = 8.20 Hz, 1H), 7.08 (s, 1H), 3.97 (s, 3H), 2.31 (s, 3H). 19F NMR (376 MHz, DMSO): δ −61.21, −114.51. 13C NMR (101 MHz, DMSO): δ 173.38, 159.56 (d, J = 251.0 Hz), 156.91, 151.79, 148.72, 147.44, 139.63, 139.36, 132.04 (d, J = 3.2 Hz), 131.57, 131.32–130.19 (m), 126.12, 123.61 (d, J = 8.5 Hz), 123.29 (q, J = 273.9 Hz), 121.57 (dd, J = 7.1, 2.9 Hz), 118.56, 118.38, 116.60, 114.00 (d, J = 26.8 Hz), 99.57, 56.42, 18.93. HRMS: calcd for C23H15ClF4N2O2 (M+H)+ 463.0831, found 463.0850.
6-Chloro-7-methoxy-2-methyl-3-(6-(4-(trifluoromethoxy)phenyl)pyridin-3-yl)quinolin-4(1H)-one (67)
1H NMR (400 MHz, DMSO): δ 11.80 (br. s., 1H), 8.57 (s, 1H), 8.26 (d, J = 7.81 Hz, 2H), 7.99–8.08 (m, 2H), 7.81 (d, J = 8.20 Hz, 1H), 7.49 (d, J = 8.20 Hz, 2H), 7.09 (s, 1H), 3.97 (s, 3H), 2.30 (s, 3H). 19F NMR (376 MHz, DMSO): δ −56.70. 13C NMR (101 MHz, DMSO): δ 173.4, 156.9, 152.4, 151.4, 148.8, 147.3, 139.6, 137.8, 130.7, 128.3, 126.1, 121.1, 119.5, 118.6, 118.3, 116.8, 99.5, 56.4, 18.9. HRMS: calcd for C23H16ClF3N2O3 (M+H)+ 461.0874, found 461.0880.
Determination of Physicochemical Properties
Aqueous Solubility, Permeability Pe, and Partition Coefficient log D
Aqueous solubility, permeability Pe and partition coefficient have been determined as previously reported.20,25,28
Microsomal Stability
The microsomal stability of 4(1H)-quinolones was determined as previously reported.20 Briefly, compounds were incubated at a concentration of 1 μM with human microsomes at 37 °C and 0.4 mg/mL of microsomal protein. The metabolic reaction was initiated with the addition of an NADPH-regenerating system (i.e., NADPH is the cofactor required for CYP450-mediated metabolism) and quenched at various time points over the incubation period by the addition of acetonitrile. Controls were included in the absence of NADPH to assess the non-CYP-mediated degradation. The relative loss of compound was monitored by LC-MS using a Waters/Micromass ZQ mass spectrometer.
Test compound concentration versus time data were fitted to an exponential decay function to determine the first-order rate constant for substrate degradation and the in vitro intrinsic clearance (CLint, μL/(min·mg protein)). CLint values of <7 μL/(min·mg) indicate little to no observable degradation throughout the assay duration (typically 250 min), whereas N.D. entries indicate molecules that were not assayed.
Evaluation for Antimalarial Activity and Cytotoxicity
In Vitro Antimalarial Activity and Cytotoxicity
In vitro antimalarial activity and cytotoxicity were determined as previously reported.20,22,25,28
Screening for in Vivo Efficacy
For the in vivo studies, female Balb/c mice (average weight 18 g) were obtained from Harlan (Fredrick, MD). An initial assessment of the in vivo antimalarial activity of selected compounds was performed using a rapid screening assay. Briefly, 38 compounds with potent activity against P. falciparum in vitro were selected. Experimental mice (n = 2 per compound) were infected with 1 × 106P. berghei-GFP ANKA parasites. On day 1 PE, mice were treated with a single dose of the test compound or control drug atovaquone (ATOV) at a concentration of 50 mg/kg. All compounds were reconstituted in poly(etheylene glycol)400 (PEG400). Parasitemia were determined from blood collected from the tail vein on days 3 and 6 PE. Compounds with >50% inhibition of parasitemia on days 3 and 6 PE were selected for more in-depth study using the Thompson test.
In Vivo Thomson Test
The in vivo efficacies of the new compounds were determined by a modified Thompson test. This test measures the survivability of mice and parasite clearance following administration of the drug on days 3–5 post-exposure. In brief, 1 × 106P. berghei-infected erythrocytes (ANKA-GFP) were inoculated into the intraperitoneal cavity of female mice that weighed 18–20 g. By day 3 PE, parasitemia ranged from 0.1 to 1.0%. There were five mice per dosage group, and each drug, suspended in PEG400, was administered orally (p.o.) once daily from days 3 to 5 PE. Blood smears were prepared on days 6, 9, 13, 21, and 30 PE. Compounds were considered active if suppression of parasitemia was ≥80% on day 6 PE. Mice that were blood-film negative on day 30 PE were considered cured. Mice developing ≥40% parasitemia or more were sacrificed. All animal studies for antimalarial efficacy were approved by the University of South Florida Institutional Animal Care and Use Committee.
Plasma Exposure of Frontrunner Compounds in Swiss Mice
The plasma exposure of four frontrunner compounds (7, 52, 66 and 67) was studied in non-fasted male Swiss outbred mice (5–7 weeks) according to procedures approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee (protocol no. VCPA.2010.41). Mice had access to food and water ad libitum throughout the pre- and post-dose sampling period.
Each compound was administered orally via gavage (0.2 mL of a 1–1.5 mg/mL suspension in PEG400), and blood samples were collected via sub-mandibular bleed or cardiac puncture at three sample time points (0.5, 7.5/8, and 24 h). Blood was collected directly into polypropylene Eppendorf tubes containing heparin as anticoagulant and stabilization cocktail [containing Complete (a protease inhibitor cocktail), potassium fluoride, and EDTA] to minimize potential for ex vivo compound degradation. Blood samples were centrifuged, and supernatant plasma was removed and stored frozen until analysis by LC-MS.
In Vivo Pharmacokinetics in Rats
The pharmacokinetics of the most promising frontrunner (compound 7) were studied in overnight-fasted male Sprague–Dawley rats (6–8 weeks; weighing 291–299 g) according to procedures approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee (protocol no. VCPA.2010.38). Rats had access to water ad libitum throughout the pre- and post-dose sampling period, and access to food was re-instated 4 h post-dose. Compound 7 was administered intravenously as a 10 min constant rate infusion (1 mL of a 0.044 mg/mL solution in 80:20 (v/v) propylene glycol/ethanol; n = 2 rats) and orally by gavage (1 mL of a 3 mg/mL solution in PEG400, followed by 1 mL of water; n = 2 rats). Samples of arterial blood were collected up to 48 h post-dose into borosilicate vials (at 4 °C) containing heparin, Complete (a protease inhibitor cocktail), potassium fluoride, and EDTA to minimize potential for ex vivo compound degradation. Blood samples were centrifuged, and supernatant plasma was removed and stored frozen (−20 °C) until plasma concentrations were determined by LC-MS. The analytical limit of quantitation (LLQ) was 0.0020 μM. Pharmacokinetic parameters were determined via standard non-compartmental methods using WinNonLin software (version 5.2.1).
Plasma Protein Binding
Plasma protein binding was determined via an ultracentrifugation method based on that described by Nakai et al.41 Briefly, frozen plasma was thawed and spiked with a DMSO/acetonitrile/water solution of test compound, with the final DMSO and acetonitrile concentrations being 0.2% (v/v) and 0.4% (v/v), respectively. Plasma was incubated at 37 °C for 60 min to equilibrate, and aliquots were transferred into polyallomer ultracentrifuge tubes and subjected to ultracentrifugation (Beckman Rotor type 42.2 Ti; 223000g and 37 °C) for 4.2 h, resulting in removal of >99% of the plasma proteins (and bound compound) from the supernate. Following ultracentrifugation, aliquots of supernate (containing the unbound fraction) were taken and stored frozen together with non-centrifuged samples at −20 °C until analysis by LC-MS. Based on the concentrations observed in non-centrifuged plasma maintained at 37 °C (Cplasma) and in the protein-free supernate (Cplasma-water), the percentage of compound bound to plasma proteins (% bound) was calculated according to the following equation:
Acknowledgments
We thank the Medicines for Malaria Venture (MMV 08/0068) and the National Institutes of Health (R01 GM097118) for financial support. This project was established as a collaboration by the MMV with the Portland Veterans Affairs Medical Center and Drexel University, and all of the authors would like to recognize the significant contributions to the overall 4(1H)-quinolone optimization project provided by Michael K. Riscoe, Rolf W. Winter, and Akhil B. Vaidya and members of their respective laboratories. We thank the Genshaft Family Doctoral Fellowship from the University of South Florida for financial support of J.R.M. We thank the Florida Center of Excellence for Drug Discovery and Innovation for providing the Biomolecular Identification of Targeted Therapeutics Fellowship (FCoE-BITT) for R.M.C. We thank Drs. R. Kiplin Guy and Fangyi Zhu for the discussions and guidance at implementing various structureproperty relationship assays in the Manetsch lab.
Glossary
Abbreviations Used
- ACT
artemisinin combination therapy
- SPR
structure–property relationship
- SPHOS
dicyclohexyl(2′,6′-dimethoxybiphenyl-2-yl)phosphine
- RPMI
Roswell Park Memorial Institute
- RI
resistance index
Supporting Information Available
Additional experimental details and data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊗ F.E.S.: Centro de Investigación en Enfermedades Infecciosas, Escuela de Ciencias Biológicas, Pontificia Universidad Católica del Ecuador, Quito, Ecuador.
Author Present Address
¶ R.M.: Department of Chemistry and Chemical Biology and Department of Pharmaceutical Sciences, Northeastern University, 102 Hurtig Hall, 360 Huntington Avenue, Boston, MA 02115, United States.
Author Contributions
# R.M.C. and D.L.F. contributed equally.
The authors declare no competing financial interest.
Author Status
∇ I.B.: Deceased June 26, 2011.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Greenwood B. M.; Fidock D. A.; Kyle D. E.; Kappe S. H.; Alonso P. L.; Collins F. H.; Duffy P. E. Malaria: progress, perils, and prospects for eradication. J. Clin. Invest. 2008, 118, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow R. W.; Guerra C. A.; Noor A. M.; Myint H. Y.; Hay S. I. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 2005, 434, 214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Malaria Report 2012; World Health Organization, 2013.
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P.; Lindegardh N.; Socheat D.; White N. J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White N. J.; Olliaro P. Artemisinin and derivatives in the treatment of uncomplicated malaria. Med. Trop. (Mars.) 1998, 58, 54–56. [PubMed] [Google Scholar]
- Noedl H.; Se Y.; Schaecher K.; Smith B. L.; Socheat D.; Fukuda M. M. Artemisinin Resistance in Cambodia 1 Study, C. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. [DOI] [PubMed] [Google Scholar]
- Plowe C. V. Malaria: Resistance nailed. Nature 2014, 505, 30–31. [DOI] [PubMed] [Google Scholar]
- Ariey F.; Witkowski B.; Amaratunga C.; Beghain J.; Langlois A. C.; Khim N.; Kim S.; Duru V.; Bouchier C.; Ma L.; Lim P.; Leang R.; Duong S.; Sreng S.; Suon S.; Chuor C. M.; Bout D. M.; Menard S.; Rogers W. O.; Genton B.; Fandeur T.; Miotto O.; Ringwald P.; Le Bras J.; Berry A.; Barale J. C.; Fairhurst R. M.; Benoit-Vical F.; Mercereau-Puijalon O.; Menard D. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill P. M.; Ward S. A.; Berry N. G.; Jeyadevan J. P.; Biagini G. A.; Asadollaly E.; Park B. K.; Bray P. G. A medicinal chemistry perspective on 4-aminoquinoline antimalarial drugs. Curr. Top. Med. Chem. 2006, 6, 479–507. [DOI] [PubMed] [Google Scholar]
- Stocks P.; Raynes K.; Ward S.. Novel Quinoline Antimalarials. In Antimalarial Chemotherapy; Rosenthal P., Ed.; Humana Press: New Jersey, 2001; pp 235–253. [Google Scholar]
- Ray S.; Madrid P. B.; Catz P.; LeValley S. E.; Furniss M. J.; Rausch L. L.; Guy R. K.; DeRisi J. L.; Iyer L. V.; Green C. E.; Mirsalis J. C. Development of a new generation of 4-aminoquinoline antimalarial compounds using predictive pharmacokinetic and toxicology models. J. Med. Chem. 2010, 53, 3685–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennerstrom J. L.; Arbe-Barnes S.; Brun R.; Charman S. A.; Chiu F. C.; Chollet J.; Dong Y.; Dorn A.; Hunziker D.; Matile H.; McIntosh K.; Padmanilayam M.; Santo Tomas J.; Scheurer C.; Scorneaux B.; Tang Y.; Urwyler H.; Wittlin S.; Charman W. N. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 2004, 430, 900–904. [DOI] [PubMed] [Google Scholar]
- Salzer W.; Timmler H.; Andersag H. Über einen neuen, gegen Vogelmalaria wirksamen Verbindungstypus. Chem. Ber. 1948, 81, 12–19. [Google Scholar]
- Casey A. C. 4(1H)-quinolones. 2. Antimalarial effect of some 2-methyl-3-(1′-alkenyl)-or-3-alkyl-4(1H)-quinolones. J. Med. Chem. 1974, 17, 255–256. [DOI] [PubMed] [Google Scholar]
- Casey A. C.; Reynolds S.; Neubeck R. Synthesis of Some 2-Methyl-3-(2′-Alkenyl)-4(1h)Quinolones. J. Heterocycl. Chem. 1972, 9, 415–418. [Google Scholar]
- Puri S. K.; Dutta G. P. Quinoline esters as potential antimalarial drugs: effect on relapses of Plasmodium cynomolgi infections in monkeys. Trans. R. Soc. Trop. Med. Hyg. 1990, 84, 759–760. [DOI] [PubMed] [Google Scholar]
- Ryley J. F.; Peters W. The antimalarial activity of some quinolone esters. Ann. Trop. Med. Parasitol. 1970, 64, 209–222. [DOI] [PubMed] [Google Scholar]
- Bueno J. M.; Manzano P.; Garcia M. C.; Chicharro J.; Puente M.; Lorenzo M.; Garcia A.; Ferrer S.; Gomez R. M.; Fraile M. T.; Lavandera J. L.; Fiandor J. M.; Vidal J.; Herreros E.; Gargallo-Viola D. Potent antimalarial 4-pyridones with improved physico-chemical properties. Bioorg. Med. Chem. Lett. 2011, 21, 5214–5218. [DOI] [PubMed] [Google Scholar]
- Yeates C. L.; Batchelor J. F.; Capon E. C.; Cheesman N. J.; Fry M.; Hudson A. T.; Pudney M.; Trimming H.; Woolven J.; Bueno J. M.; Chicharro J.; Fernandez E.; Fiandor J. M.; Gargallo-Viola D.; Gomez de las Heras F.; Herreros E.; Leon M. L. Synthesis and structure-activity relationships of 4-pyridones as potential antimalarials. J. Med. Chem. 2008, 51, 2845–2852. [DOI] [PubMed] [Google Scholar]
- Cross R. M.; Monastyrskyi A.; Mutka T. S.; Burrows J. N.; Kyle D. E.; Manetsch R. Endochin optimization: structure-activity and structure-property relationship studies of 3-substituted 2-methyl-4(1H)-quinolones with antimalarial activity. J. Med. Chem. 2010, 53, 7076–7094. [DOI] [PubMed] [Google Scholar]
- Winter R.; Kelly J. X.; Smilkstein M. J.; Hinrichs D.; Koop D. R.; Riscoe M. K. Optimization of endochin-like quinolones for antimalarial activity. Exp. Parasitol. 2011, 127, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen A.; LaCrue A. N.; White K. L.; Forquer I. P.; Cross R. M.; Marfurt J.; Mather M. W.; Delves M. J.; Shackleford D. M.; Saenz F. E.; Morrisey J. M.; Steuten J.; Mutka T.; Li Y.; Wirjanata G.; Ryan E.; Duffy S.; Kelly J. X.; Sebayang B. F.; Zeeman A. M.; Noviyanti R.; Sinden R. E.; Kocken C. H.; Price R. N.; Avery V. M.; Angulo-Barturen I.; Jimenez-Diaz M. B.; Ferrer S.; Herreros E.; Sanz L. M.; Gamo F. J.; Bathurst I.; Burrows J. N.; Siegl P.; Guy R. K.; Winter R. W.; Vaidya A. B.; Charman S. A.; Kyle D. E.; Manetsch R.; Riscoe M. K. Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med. 2013, 5, 177ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidathala C.; Amewu R.; Pacorel B.; Nixon G. L.; Gibbons P.; Hong W. D.; Leung S. C.; Berry N. G.; Sharma R.; Stocks P. A.; Srivastava A.; Shone A. E.; Charoensutthivarakul S.; Taylor L.; Berger O.; Mbekeani A.; Hill A.; Fisher N. E.; Warman A. J.; Biagini G. A.; Ward S. A.; O’Neill P. M. Identification, design and biological evaluation of bisaryl quinolones targeting Plasmodium falciparum type II NADH:quinone oxidoreductase (PfNDH2). J. Med. Chem. 2012, 55, 1831–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagini G. A.; Fisher N.; Shone A. E.; Mubaraki M. A.; Srivastava A.; Hill A.; Antoine T.; Warman A. J.; Davies J.; Pidathala C.; Amewu R. K.; Leung S. C.; Sharma R.; Gibbons P.; Hong D. W.; Pacorel B.; Lawrenson A. S.; Charoensutthivarakul S.; Taylor L.; Berger O.; Mbekeani A.; Stocks P. A.; Nixon G. L.; Chadwick J.; Hemingway J.; Delves M. J.; Sinden R. E.; Zeeman A. M.; Kocken C. H.; Berry N. G.; O’Neill P. M.; Ward S. A. Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 8298–8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross R. M.; Namelikonda N. K.; Mutka T. S.; Luong L.; Kyle D. E.; Manetsch R. Synthesis, antimalarial activity, and structure-activity relationship of 7-(2-phenoxyethoxy)-4(1H)-quinolones. J. Med. Chem. 2011, 54, 8321–8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley R.; Leung S.; Fisher N.; Al-Helal M.; Berry N. G.; Lawrenson A. S.; Sharma R.; Shone A. E.; Ward S. A.; Biagini G. A.; O’Neill P. M. The development of quinolone esters as novel antimalarial agents targeting the Plasmodium falciparum bc(1) protein complex. MedChemComm 2012, 3, 39–44. [Google Scholar]
- Zhang Y.; Clark J. A.; Connelly M. C.; Zhu F.; Min J.; Guiguemde W. A.; Pradhan A.; Iyer L.; Furimsky A.; Gow J.; Parman T.; El Mazouni F.; Phillips M. A.; Kyle D. E.; Mirsalis J.; Guy R. K. Lead optimization of 3-carboxyl-4(1H)-quinolones to deliver orally bioavailable antimalarials. J. Med. Chem. 2012, 55, 4205–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross R. M.; Maignan J. R.; Mutka T. S.; Luong L.; Sargent J.; Kyle D. E.; Manetsch R. Optimization of 1,2,3,4-tetrahydroacridin-9(10H)-ones as antimalarials utilizing structure-activity and structure-property relationships. J. Med. Chem. 2011, 54, 4399–4426. [DOI] [PubMed] [Google Scholar]
- Kelly J. X.; Smilkstein M. J.; Brun R.; Wittlin S.; Cooper R. A.; Lane K. D.; Janowsky A.; Johnson R. A.; Dodean R. A.; Winter R.; Hinrichs D. J.; Riscoe M. K. Discovery of dual function acridones as a new antimalarial chemotype. Nature 2009, 459, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen A.; Miley G. P.; Forquer I. P.; Mather M. W.; Katneni K.; Li Y. X.; Pou S.; Pershing A. M.; Stickles A. M.; Ryan E.; Kelly J. X.; Doggett J. S.; White K. L.; Hinrichs D. J.; Winter R. W.; Charman S. A.; Zakharov L. N.; Bathurst I.; Burrows J. N.; Vaidya A. B.; Riscoe M. K. Discovery, Synthesis, and Optimization of Antimalarial 4(1H)-Quinolone-3-Diarylethers. J. Med. Chem. 2014, 57, 3818–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross R. M.; Manetsch R. Divergent route to access structurally diverse 4-quinolones via mono or sequential cross-couplings. J. Org. Chem. 2010, 75, 8654–8657. [DOI] [PubMed] [Google Scholar]
- Desjardins R. E.; Canfield C. J.; Haynes J. D.; Chulay J. D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looareesuwan S.; Viravan C.; Webster H. K.; Kyle D. E.; Hutchinson D. B.; Canfield C. J. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am. J. Trop. Med. Hyg. 1996, 54, 62–66. [DOI] [PubMed] [Google Scholar]
- Milhous W. K.; Gerena L.; Kyle D. E.; Oduola A. M. In vitro strategies for circumventing antimalarial drug resistance. Prog. Clin. Biol. Res. 1989, 313, 61–72. [PubMed] [Google Scholar]
- Bueno J. M.; Herreros E.; Angulo-Barturen I.; Ferrer S.; Fiandor J. M.; Gamo F. J.; Gargallo-Viola D.; Derimanov G. Exploration of 4(IH)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochrome bcl. Future Med. Chem. 2012, 4, 2311–2323. [DOI] [PubMed] [Google Scholar]
- LaPlante S. R.; Edwards P. J.; Fader L. D.; Jakalian A.; Hucke O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 2011, 6, 505–513. [DOI] [PubMed] [Google Scholar]
- LaPlante S. R.; L D. F.; Fandrick K. R.; Fandrick D. R.; Hucke O.; Kemper R.; Miller S. P.; Edwards P. J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 2011, 54, 7005–7022. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. [DOI] [PubMed] [Google Scholar]
- Lacrue A. N.; Saenz F. E.; Cross R. M.; Udenze K. O.; Monastyrskyi A.; Stein S.; Mutka T. S.; Manetsch R.; Kyle D. E. 4(1H)-Quinolones with liver stage activity against Plasmodium berghei. Antimicrob. Agents Chemother. 2013, 57, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz F. E.; Lacrue A. N.; Cross R. M.; Maignan J. R.; Udenze K.; Manetsch R.; Kyle D. E. 4-(1H)-Quinolones and 1,2,3,4-tetrahydroacridin-9(10H)-ones prevent the transmission of Plasmodium falciparum to Anopheles freeborni. Antimicrob. Agents Chemother. 2013, 57, 6187–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai D.; Kumamoto K.; Sakikawa C.; Kosaka T.; Tokui T. Evaluation of the protein binding ratio of drugs by a micro-scale ultracentrifugation method. J. Pharm. Sci. 2004, 93, 847–854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.