

Abstract

Neuropeptide FF1 and FF2 receptors (NPFF1-R and NPFF2-R), and their endogenous ligand NPFF, are one of only several systems responsible for mediating opioid-induced hyperalgesia, tolerance, and dependence. Currently, no small molecules displaying good affinity or selectivity for either subtype have been reported, to decipher the role of NPFF2-R as it relates to opioid-mediated analgesia, for further exploration of NPFF1-R, or for medication development for either subtype. We report the first nonpeptide small molecule scaffold for NPFF1,2-R, the guanidino-piperidines, and SAR studies resulting in the discovery of a NPFF1 agonist (7b, Ki = 487 ± 117 nM), a NPFF1 antagonist (46, Ki = 81 ± 17 nM), and a NPFF2 partial antagonist (53a, Ki = 30 ± 5 nM), which serve as leads for the development of pharmacological probes and potential therapeutic agents. Testing of 46 alone was without effect in the mouse 48 °C warm-water tail-withdrawal test, but pretreatment with 46 prevented NPFF-induced hyperalgesia.
Introduction
The neuropeptide FF (NPFF) system comprises two receptor subtypes, NPFF1 and NPFF2 receptors (NPFF1-R and NPFF2-R), which are Gi/o coupled G-protein coupled receptors (GPCRs) with 30–35% homology to neuropeptide Y and orexin receptors,1 and four endogenous ligands belonging to the Arg-Phe-NH2 (RFa) peptide family.2 These peptides include hNPFF (SQAFLFQPQRFa), hNPSF (SLNFEELKDWGPKNVIKMSTPAVNKMPHSFANLPLRFa), and hNPAF (AGEGLSSPFWSLAAPQRFa) (10.7-, 1.2-, and 10.1-fold selective in radioligand competition binding assays for the hNPFF2 subtype, respectively), and hNPVF (VPNLPQRFa) (27.8-fold selective for the hNPFF1 subtype).2 Both NPFF1-R and NPFF2-R are present in the rat brain; however; only NPFF2-R is found in the spinal cord.3 Conversely, in humans, the distribution of NPFF1-R is not restricted to the CNS, since NPFF1 mRNA4 is found in the spinal cord. Talmont and colleagues noted species-related differences in the pharmacological profiles between mouse and human NPFF2-R, further highlighting the consideration that must be taken in the interpretation of preliminary in vitro and in vivo results in mouse models as they translate to human targets.5
The opioid-modulating properties (among others) of the NPFF peptide have been chronicled throughout the literature since its discovery in 1985,6 described as producing either a pro- or antinociceptive effect depending on the route of administration. For example, when administered in rats by intracerebroventricular (icv) injection alone or in combination with morphine or other opioid agonists, NPFF or its stable analog 1DMe (D.YL(N-Me)FQPQRFa) produces an antiopioid effect,6−9 however; when given intrathecally (it.), NPFF yields an opioid-like effect and potentiates morphine-induced analgesia in various pain models.10−13 Thus, a precise role for NPFF1-R and NPFF2-R as anti- or pronociceptive systems in preliminary in vivo models has not been delineated. In rats, the lack of NPFF1-R at the spinal level has formed the basis of the hypothesis that the antinociceptive activity of NPFF observed upon it. administration is a result of its interaction with NPFF2-R, while its pronociceptive activity at the supraspinal (i.e., brain) level is a result of its interaction with NPFF1-R.2,14 However, the pronociceptive activity of NPFF in the rat brain cannot be exclusively attributed to its activity at the NPFF1-R, because the activity of the rNPFF peptide (NPAFLFQPQRFa) at rNPFF1,2-R and hence its selectivity has not been reported. Failing the availability of any truly selective ligands for NPFF receptors, gene knockout studies would prove valuable in elucidating the pharmacology of these subtypes.
A limited number of dipeptides and small molecules have been used to try to define the pharmacology of these subtypes, with mixed results. Although a pronociceptive/antiopioid role has been assigned to the NPFF system as a whole based on the in vivo activity of a nonselective dipeptide competitive antagonist RF9 (administered subcutaneously (sc) or intraperitoneally (ip)) (Chart 1)15,16 in opioid-induced hyperalgesia (OIH), tolerance, and dependence assays, conflicting reports exist as to the nociceptive- or opioid-modulating properties of each subtype. NPFF1-R has been classified as a pronociceptive/antiopioid system using a selective (1843-fold) dipeptide antagonist 1 (administered sc) (Chart 1)17 and a modestly selective (19.5-fold) small molecule antagonist AC-262620 (administered ip)14 from Acadia Pharmaceuticals (hNPFF1 Ki = 16.4 ± 10.1 nM, similar in structure to AC-099 and 2) based on their activities within in vivo models of pain. Further confirmation of NPFF1-R’s pronociceptive role in terms of opioid analgesia could be realized with NPFF1-selective agonists. While being similarly classified as a pronociceptive/antiopioid construct due to its antiopioid activity at the cellular level using the stable peptide agonist 1DMe,18 NPFF2-R is also contrarily described as a antinociceptive/pro-opioid system using small molecules of modest affinity, putative full agonist AC-099 (Chart 1) and partial agonist 2 (Chart 1) (both administered ip) reported by Acadia Pharmaceuticals.14,19,20 However, it must be noted that 1DMe is not highly selective, exhibiting full agonist activity on both NPFF1-R (EC50 = 71 ± 14 nM) and NPFF2-R (EC50 = 2.7 ± 0.5 nM),21 and thus both receptors are likely to contribute to its pharmacological response. Moreover, the modest activity and selectivity of aryliminoguanidines AC-099 and 2 displayed in unconventional in vitro assays limits their utility in defining NPFF2-R with respect to opioid-mediated analgesia (Chart 1). Additionally, no off-target opioid affinity is reported for these aryliminoguanidines, which calls into question the interpretation of their in vivo analgesic activity.
Chart 1. Reported Ligands Used for Pharmacological Characterization of NPFF-1 and NPFF-2 Receptorsa.

a The Ki and EC50 values associated with the ligands described are included for discussion purposes only and cannot be directly compared.
b Ki values were determined by radioligand displacement assays.
c EC50 values were calculated from pEC50 values determined in RSAT assay.20
d EC50 values were calculated from pEC50 values determined in cAMP assay.20
e EC50 values were taken directly, as determined in the IP accumulation assay.22
f EC50 values were calculated from pEC50 values determined in RSAT assay,19 using hNPFF1-R and hNPFF2-R.
A single core scaffold, on which modifications can be made to yield a selective agonist or antagonist functional profile, would be of use to help define the pharmacology of NPFF2-R. The functional profiles of the compounds summarized in Chart 1 underscore the need for compounds with NPFF1 agonist or NPFF2 agonist and antagonist efficacy to aid in the exploration of the pharmacology of these subtypes. Other small molecule ligands for NPFF1-R and NPFF2-R, containing a wide range of scaffolds and thus no definitive SAR or pharmacophore, have been reported mainly in the patent literature.23 However; to our knowledge, no pharmacologically pure and high affinity NPFF1 and NPFF2 agonists and antagonists have been reported (apart from dipeptide NPFF1 antagonist 1). Moreover, the in vivo activities of these compounds as they pertain to pain modulation, if any, have not been reported.
As summarized above, the majority of these compounds are proprietary and limited in terms of binding affinity, selectivity, and functional profiles necessary to define the pharmacology of these subtypes. Due to the lack of availability of these compounds to the scientific community, a tremendous design space exists to develop novel, selective NPFF1 and NPFF2 ligands of varying functional profiles to aid in the study of these receptors.
SAR of the residues conserved among mouse, rat, bovine and human NPFF (FLFQPQRFa) (Ki 0.21/0.34 nM in the membrane of rat spinal cord or CHO hNPFF2-R)24,25 indicate that the peptide adheres to a message–address concept,26 with the C-terminal amide, guanidine of arginine,7 and aromatic ring of phenylalanine8 serving as the message component conferring the majority of the peptide’s affinity and functional activity. To date, a good number of NPFF ligands reported in the literature contain at least phenyl and guanidine moieties, including dipeptides RF9 and 1, and aryliminoguanidines AC-099 and 2. Taking these functionalities into consideration, we desired a scaffold on which to accommodate these pharmacophoric elements while at the same time maintaining chemical feasibility. The 4-(phenylamino)piperidine-4-carbonitrile template (Figure 1) seemed to be a reasonable starting point, because the 4-anilido piperidine portion is known to be a privileged scaffold for GPCRs, particularly in the field of opioid analgesics (i.e., fentanyl). Moreover, the piperidine nitrogen portion allows for lipophilic substitutions to be made, and the nitrile moiety could be converted to the corresponding amine, to allow for the introduction of guanidine-containing groups. We report herein a series of designed guanidino-piperidines based on this scaffold, which led to the first class of ligands to demonstrate a tractable SAR for modulation of affinity, efficacy, and selectivity at NPFF1-R and NPFF2-R.
Figure 1.
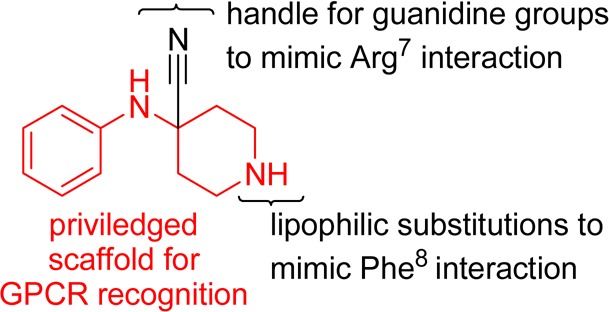
4-(Phenylamino)piperidine-4-carbonitrile scaffold for designed NPFF ligands.
Results and Discussion
Chemistry
Piperidones 3a–c and 12d (Scheme 1) bearing various lipophilic substitutions at the nitrogen (either commercially available or prepared via the ketal 10) were subjected to standard Strecker conditions27 to generate N-substituted carbonitriles 4a–d in 70–94% yield. Reduction of the nitriles using either LiAlH4 or hydrogenation afforded the corresponding amines 5a–d in 35–90% yield, followed by coupling with Boc-Arg-(Boc)2-OH using EDCI/HOBt to give Boc-protected arginine intermediates 6a–d or HgCl2-assisted guanidation to give protected guanidine intermediates 8a–c in 30–70% yield. Deprotection with HCl/dioxane afforded guanidines 7a–d and 9a–c in excellent yield (95%).
Scheme 1. Synthesis of 7a–d and 9a–c.

Reagents and conditions: (a) aniline, TMSCN, AcOH, 0 °C, 1 h, 70–94%; (b) LiAlH4, diethyl ether (ah), 0 °C, 35–40% (for 5a,5c) or NH3/MeOH, Raney Ni, 3 atm H2(g), 4 h, rt, 82–90% (for 5b, 5d); (c) Boc-Arg-(Boc)2-OH, EDCI, HOBt, Et3N, DCM, rt, 24 h, 50–70%; (d) HCl/dioxane, 3 days, rt, 95%; (e) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, HgCl2, Et3N, DMF, 0 °C → rt, 10 h, 30%; (f) HCl/dioxane, 4 days, rt, quantitative; (g) 2-bromomethylnaphthalene, K2CO3, KI, 4-methyl-2-pentanone, reflux, 5 h; (h) AcOH, conc. HCl (aq), reflux, 18 h, 40%.
Des-aniline 15 (Scheme 2) was prepared starting with (1-benzyl-4-piperidinyl)methylamine (13) and using analogous chemistry to that used for 6a–d (Scheme 1) in 70% yield, followed by deprotection with HCl/dioxane in excellent yield (95–99%).
Scheme 2. Synthesis of 15.

Reagents and conditions: (a) Boc-Arg-(Boc)2-OH, EDCI, HOBt, Et3N, DCM, rt, 24 h, 70%; (b) 4 M HCl/dioxane, rt, 10 h to 4 days (95–99%).
Guanidated glycine 21 (1-carbon aliphatic linker) was synthesized by coupling Boc-Gly-OH with 4-(aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (5b) using EDCI/HOBt to give intermediate 16b in 65% yield, followed by Boc removal with TFA to yield amine 17b (92%) (Scheme 3). Boc-protected guanidation of amine 17b with 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea afforded intermediate 20 in 84% yield, and deprotection with TFA afforded one-carbon linked guanidine 21 in 90% yield. Two carbon homologue 17a was prepared in a similar manner via EDCI/HOBt coupling of cyanoacetic acid with amine 5b (Scheme 1) to yield intermediate 16a in 50% yield, then reduction of the cyano moiety with H2(g)/Raney Ni to amine 18a (76%). Boc-protected guanidation of amine 18a afforded intermediate 19a in 87% yield; subsequent deprotection with TFA yielded two-carbon linked guanidine 17a (77%). Three and four carbon homologues 17c,d were prepared in a similar manner by first converting 4-aminobutanoic acid and 5-aminopentanoic acid to their corresponding Boc-protected guanidines as described above (17–26%), followed by EDCI/HOBt coupling with amine 5b to yield intermediates 16c,d (77–80%). Boc-removal from intermediates 16c,d with TFA afforded analogs 17c,d. Phenyl linked guanidines 17e–g were prepared by converting 3-aminobenzoic acid, 4-aminobenzoic acid, and 4-aminophenylacetic acid to the Boc-protected guanidines as described above (16-31%), followed by EDCI/HOBt coupling with amine 5b to yield intermediates 16e–g (66–84%). Boc removal from intermediates 16e–g with TFA afforded analogs 17e–g in quantitative yield.
Scheme 3. Synthesis of 17a–g and 21.

Reagents and conditions: (a) carboxylic acid, EDCI, HOBt, Et3N, DCM, 0 °C → rt, 15 h, 50–84%; (b) TFA, DCM, rt, 2 h, 77–100%; (c) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, HgCl2, Et3N, THF/MeOH, rt, 15 h, 16-31%; (d) NH3/MeOH, Raney Ni, 2.4 atm H2(g), 15 h, rt, 76%; (e) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, Et3N, DMF(ah), rt, 18 h, 87%; (f) TFA, DCM(ah), rt, 3 h, 77%; (g) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, Et3N, DMF(ah), rt, 2 days, 84%; (h) TFA, DCM(ah), rt, 3 h, 90%.
One, three-substituted analog 26 was obtained (Scheme 4) using similar chemistry as shown in Schemes 1 and 2. 1-Benzyl-3-piperidone 22 was coupled to aniline under Strecker conditions to give nitrile 23 in 74% yield, followed by catalytic hydrogenation to amine 24 (78% yield). Boc-protected guanidated glycine 25 was accessed by EDCI/HOBt coupling of amine 24 with 1,3-di-Boc-2-(carboxymethyl)guanidine in 37% yield; subsequent TFA deprotection afforded 1,3-substituted piperidine 26 in 65% yield (Scheme 4).
Scheme 4. Synthesis of 26.

Reagents and conditions: (a) aniline, Ti(OiPr)4, diethylaluminum cyanide, DCE, rt, 24 h, 74%; (b) NH3/MeOH, Raney Ni, 2.7 atm H2(g), 24 h, 78%; (c) 1,3-di-Boc-2-(carboxymethyl)guanidine, HOBt, EDCI, Et3N, DCM, 0 °C → rt, 20 h, 37%; (d) TFA, DCM, rt, 6 h, 65%.
1-Naphthyl, 3,5-dimethoxyphenyl, 3-methoxyphenyl, and 3,4-dichlorophenyl analogs were prepared by reacting the corresponding substituted aniline with N-benzyl piperidone under Strecker conditions to give nitriles 27a–d in 55–77% yield (Scheme 5). Subsequent reduction of the nitriles using catalytic hydrogenation afforded amines 28a–d in excellent yields (89–95%), followed by EDCI/HOBt coupling with 1,3-di-Boc-2-(carboxymethyl)guanidine to give Boc protected guanidines 29a–d in moderate yields (33–67%). Deprotection with TFA afforded guanidines 30a–d in good yield (53–83%).
Scheme 5. Phenyl (Ring 1) Modification of Lead 21: Synthesis of 30a–d.

Reagents and conditions: (a) acetic acid, substituted aniline, KCN(aq) or TMSCN, 0 °C → rt, 4 h, 55–77%; (b) NH3/MeOH, Raney Ni, 2.7 atm H2(g), 16–24 h, 89–95%; (c) 1,3-di-Boc-2-(carboxymethyl)guanidine, HOBt, EDCI, Et3N, DCM, 0 °C → rt, 20 h, 33–67%; (d) TFA, DCM, rt, 2–4 h, 53–83%.
Analog 34 was synthesized by first coupling commercially available 1-(1-benzyl-4-phenylpiperidin-4-yl)methanamine 31 with Boc-gly-OH using EDCI/HOBt in 78% yield to give intermediate 32, deprotection with TFA and HgCl2-assisted guanidation (33, 89% overall yield), and finally deprotection with TFA (Scheme 6). Methylene 42 and ethylene 43 analogs were prepared by initially generating the carbanion at the 4-position of commercially available 1-benzylpiperidine-4-carbonitrile (35) with LDA, then displacing benzyl bromide (to intermediate 36) or (2-bromoethyl)benzene (to intermediate 37) in 86–89% yield. Catalytic hydrogenation of the cyano moieties of intermediates 36 and 37 afforded the corresponding amines 38 and 39 in 61–90% yield. The Boc-protected guanidated glycines of amines 38 and 39 (intermediates 40 and 41, respectively) were prepared by either coupling with 1,3-di-Boc-2-(carboxymethyl)guanidine using EDCI/HOBt or employing the sequence above (coupling with Boc-gly-OH, deprotection, HgCl2-assisted Boc guanidation) in reasonable yields. Deprotection with TFA afforded methylene and ethylene analogs 42–43 in 71–85% yield (Scheme 6).
Scheme 6. Aniline Amine Modification of Lead 21: Synthesis of 34, 42, and 43.

Reagents and conditions: (a) Boc-gly-OH, HOBt, EDCI, Et3N, DCM, 0 °C → rt, 24 h, 78%; (b) TFA, DCM, rt, 1 h then 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, HgCl2, Et3N, DMF, rt, 1 h, 89%; (c) TFA, DCM, rt, 2 h; (d) benzyl bromide (for 36) or (2-bromoethyl)benzene (for 37), LDA, THF(ah), −70 °C → rt, 2–3 h, 86–89%; (e) NH3/MeOH, Raney Ni, 2.4–2.7 atm H2(g), 24 h, 61–90%; (f) 1,3-di-Boc-2-(carboxymethyl)guanidine, HOBt, EDCI, Et3N, DCM, 0 °C → rt, 22 h, 48% (for 40); for 41 (i) Boc-gly-OH, HOBt, EDCI, Et3N, DCM, 0 °C → rt, 20 h, 91%, (ii) TFA, DCM, rt, 1 h, (iii) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, HgCl2, Et3N, DMF, rt, 2 h, 76%; (g) TFA, DCM, rt, 2–3 h, 71–85%.
Reverse amide 46 was prepared by coupling commercially available 1-benzyl-4-phenylamino-4-carboxy-piperidine 44 (Scheme 7) with 2-(2-aminoethyl)-1,3-di-Boc-guanidine using EDCI/HOBt to give intermediate 45 in 70% yield, followed by TFA deprotection in 54% yield.
Scheme 7. Methylene Amide Modification of Lead 21: Synthesis of 46.

Reagents and conditions: (a) 2-(2-aminoethyl)-1,3-di-Boc-guanidine, HOBt, EDCI, Et3N, 1-methyl-2-pyrrolidone, rt, 24 h, 70%; (b) TFA, DCM, rt, 4 h, 54%.
Various aromatic and nonaromatic groups were explored at the piperidine nitrogen (49b–g, Scheme 8) by initial debenzylation of tert-butyl 1-(1-benzyl-4-(phenylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate, 20 (Scheme 8), using catalytic hydrogenation to give intermediate 47 in 68% yield and subsequent displacement of the corresponding substituted bromide or chloride in 27–96% yield to afford intermediates 48b–d,f,g. Intermediate 48e was prepared by coupling 4-(aminomethyl)-1-(naphthalen-2-ylmethyl)-N-phenylpiperidin-4-amine, 5d (Scheme 1), with 1,3-di-Boc-2-(carboxymethyl)guanidine using EDCI/HOBt in 48% yield. TFA deprotection of Boc-protected guanidated glycines 48a–g afforded N-substituted analogs 49a–g in 54–85% yield.
Scheme 8. Phenyl (Ring 2) Modification of Lead 21: Synthesis of 49a–g.

Reagents and conditions: (a) Pd/C 10%, 4 atm H2(g), MeOH, 7 days, 68%; (b) RCH2Br(or Cl), DMF or NMP, 0 °C → rt, 33–44 h, 27–96%; (c) TFA, DCM, rt 3–15 h, 54–85%; (d) 1,3-di-Boc-2-(carboxymethyl)guanidine, HOBt, EDCI, Et3N, DMF, 0 °C → rt, 15 h, 48%.
1-Naphthyl (53a), 3,4-dichlorophenyl (53b), 2-naphthyl (53c), and biphenyl (53d) replacements were prepared using the same synthetic sequence as that of benzyl 42 and phenethyl 43 using 1-benzylpiperidine-4-carbonitrile, 35 (Scheme 9). Briefly, displacement of phenyl-substituted halides with the LDA-generated carbanion of carbonitrile 35 gave intermediates 50a–d in 59–91% yield, followed by catalytic hydrogenation of the cyano moieties to yield amines 51a–d (38–93%). The Boc-protected guanidated glycines of amines 51a–d (intermediates 52a–d) were prepared by either coupling with 1,3-di-Boc-2-(carboxymethyl)guanidine using EDCI/HOBt or employing the sequence above (coupling with Boc-gly-OH, deprotection, HgCl2-assisted Boc guanidation) (Scheme 6) in 41–77% yield. Deprotection with TFA afforded 1-naphthyl (53a), 3,4-dichlorophenyl (53b), 2-naphthyl (53c), and biphenyl analogs (53c) in 54–76% yield.
Scheme 9. Phenyl (Ring 1) Modification of NPFF2-Preferring Ligand 42: Synthesis of 53a–d.

Reagents and conditions: (a) RCH2Br(or Cl), LDA, THF(ah), −70 °C → rt, 2–3 h, 59–91%; (b) NH3/MeOH, Raney Ni, 2.4 atm H2(g), 6–20 h, 38–93%; (c) 1,3-di-Boc-2-(carboxymethyl)guanidine, HOBt, EDCI, Et3N, DCM, 0 °C → rt, 15 h, 41–72% (for 52a–c), for 52d: (i) Boc-gly-OH, HOBt, EDCI, Et3N, DCM, 0 °C → rt, 15 h, 77%, (ii) TFA, DCM, rt, 1 h, (iii) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, HgCl2, Et3N, DMF, rt, 15 h, 80%; (d) TFA, DCM, rt, 3 h, 54–76%.
Biology
Binding Affinity and Functional Efficacy at hNPFF1-R and hNPFF2-R
The affinities of 7a–d, 9a–c, 15, 17a–g, 21, 26, 30a–d, 34, 42–43, 46, 49a–g, and 53a–d at hNPFF1-R and hNPFF2-R were evaluated by competition experiments using the selective NPFF1 and NPFF2 radioligands [125I]YVP (YVPNLPQRFa) and [125I]EYF (EYWSLAAPQRFa), or [3H]-NPVF and [3H]-EYF, respectively, in membranes of CHO cells stably expressing each receptor (Tables 1–4). Tested compounds were compared with previously reported high affinity peptides 1DMe and NPVF.21 Selected compounds were tested for off-target affinity at μ-, δ-, and κ-opioid receptors (MOP, DOP, and KOP; Table 5) in competition experiments using the selective radioligands [3H]DAMGO, [3H]DPDPE, and [3H]U-69,593, respectively, in membranes of HEK 293 cells stably expressing each receptor (Table 5) and compared to the standard naloxone. The functional activity of 7b was tested by measuring its capacity to increase the binding of [35S]GTPγS in the membranes of CHO cells stably expressing each receptor (Figure 2). The functional activity of 21, 30a–c, 42, 46, and 53a was assessed by testing each compound’s capacity to inhibit forskolin-induced intracellular cAMP accumulation in CHO cells stably expressing NPFF1-R and NPFF2-R (Figures 3A,B, 6A,B, 7, and 9A–D). The antagonist properties of 21, 46, and 53a were evaluated by measuring its ability to reverse the NPVF- and 1DMe-induced inhibition of cAMP production in NPFF1-R and NPFF2-R, respectively (Figures 3A,B, 6B, and 9C,D), and in the case of 53a to right-shift the dose–response curve of the aforementioned NPFF agonists in their respective subtypes (Figures 10A,B).
Table 1. Binding Affinities of Guanidino-piperidines 7a–d, 9a–c, and 15a,b at NPFF1-R and NPFF2-Ra.
| cmpd | hNPFF1-R (Ki, nM) | hNPFF2-R (Ki, nM) | ratio |
|---|---|---|---|
| 1DMe | 4 ± 0.5 | 0.8 ± 0.1 | |
| 7a | >5K | >5K | |
| 7b | 487 ± 117 | 1052 ± 32 | 2.2-fold (NPFF1) |
| 7c | 2906 ± 576 | 1694 ± 183 | |
| 7d | 479 ± 39 | 304 ± 14 | 1.6-fold (NPFF2) |
| 9a | >5K | >5K | |
| 9b | 1880 ± 53 | 6616 ± 377 | |
| 9c | 2965 ± 112 | 2773 ± 470 | |
| 15 | >5K | 7579 ± 54 |
Ki values were determined by using [125I]YVP and [125I]EYF for hNPFF1-R and hNPFF2-R, respectively, and are the mean ± SEM from two to four experiments performed in duplicate.
Table 4. Binding Affinities of Guanidino-piperidines 53a–d at NPFF1-R and NPFF2-Ra.
| cmpd | hNPFF1-R (Ki, nM) | hNPFF2-R (Ki, nM) | ratio |
|---|---|---|---|
| NPVF | 3.66 ± 0.95 | ||
| 1DMe | 3.85 ± 0.42 | 0.47 ± 0.08 | |
| 42 | 417 ± 74 | 84 ± 12 | 5-fold (NPFF2) |
| 53a | 112 ± 19 | 30 ± 5 | 3.7-fold (NPFF2) |
| 53b | 432 ± 14 | 82 ± 8 | 5.3-fold (NPFF2) |
| 53c | 316 ± 11 | 205 ± 8 | 1.5-fold (NPFF2) |
| 53d | 296 ± 14 | 910 ± 107 | 3-fold (NPFF1) |
Ki values were determined by using [3H]NPVF and [3H]EYF for hNPFF1-R and hNPFF2-R, respectively, and are the mean ± SEM from two-four experiments performed in duplicate.
Table 5. Binding Affinities of Selected Guanidino-piperidines at μ-, δ-, and κ-Opioid Receptorsa.
| cmpd | hNPFF1-R (Ki, nM) | hNPFF2-R (Ki, nM) | MOP (Ki, nM) | DOP (Ki, nM) | KOP (Ki, nM) |
|---|---|---|---|---|---|
| naloxone | 3.39 ± 0.08 | 37.2 ± 1.3 | 4.83 ± 0.08 | ||
| RF9 | 58 ± 5b | 75 ± 9b | >5Kb | >5Kb | >5Kb |
| 7b | 487 ± 117 | 1052 ± 32 | >5K | >5K | 1233 ± 30 |
| 21 | 319 ± 19 | 405 ± 56 | 2022 ± 170 | >5K | 705 ± 80 |
| 30a | 94 ± 24 | 309 ± 55 | 688 ± 60 | >5K | 3678 ± 530 |
| 30b | 114 ± 17 | 987 ± 65 | >5K | >5K | 4247 ± 580 |
| 42 | 417 ± 74 | 84 ± 12 | 201 ± 10 | 2486 ± 200 | 224 ± 20 |
| 46 | 81 ± 17 | 1426 ± 49 | >5K | 2607 ± 650 | 1470 ± 190 |
| 53a | 112 ± 19 | 30 ± 5 | 354 ± 90 | 1516 ± 330 | 362 ± 30 |
Ki values were determined using radioligands [3H]DAMGO, [3H]DPDPE, and [3H]U-69,593 for MOP, DOP, and KOP receptors, respectively, and are the mean ± SEM from three experiments.
Data taken from ref (15).
Figure 2.

NPFF1-preferring ligand 7b in the [35S]GTPγS assay in cloned NPFF1-R and NPFF2-R. Compound 7b (10 μM) increased by 36% and 184% the binding of [35S]GTPγS on membranes of NPFF1-R and NPFF2-R expressing CHO cells, respectively, compared with a 71% and 231% increase by the agonist 1DMe (10 μM). Data represents the mean ± SEM from two-four experiments performed in duplicate.
Figure 3.

Ability of the nonselective, low efficacy NPFF1 antagonist 21 to inhibit the effect of NPFF agonists in the forskolin-induced cAMP assay in CHO cells expressing human NPFF1-R (A) or NPFF2-R (B). Increasing doses of 21 were tested either alone or in the presence of 0.1 μM NPVF in CHO hNPFF1 cells or of 0.01 μM 1DMe in CHO hNPFF2 cells. ***, p < 0.001; **, p < 0.01; *, p < 0.05; different from NPVF alone (white bar) (one-way ANOVA followed by Dunnett’s multiple comparison test). Data represents the mean ± SEM from two to four experiments performed in duplicate.
Figure 6.

(A) NPFF1-preferring ligands 30a–c and NPFF1-selective ligand 46 in the forksolin-induced cAMP assay in cloned NPFF1-R, compared with the NPFF1 selective agonist NPVF. (B) Ability of the selective NPFF1 antagonist 46 to reverse the effect of 0.1 μM NPVF in the forskolin-induced cAMP assay in CHO cells expressing hNPFF1-R. Increasing doses of 46 were tested either alone or in the presence of NPVF. ***, p < 0.001; **, p < 0.01; *, p < 0.05; different from NPVF alone (white bar) (one-way ANOVA followed by Dunnett’s multiple comparison test). Data represents the mean ± SEM from two-four experiments performed in duplicate.
Figure 7.

NPFF2-preferring ligand 42 in the forksolin-induced cAMP assay in cloned NPFF2-R, compared with the NPFF agonist 1DMe. Data represents the mean ± SEM from two to four experiments performed in duplicate.
Figure 9.

NPFF2-preferring ligand 53a in the forskolin-induced cAMP assay in cloned NPFF1-R (A) and NPFF2-R (B) compared with the NPFF1 and NPFF2 selective agonists NPVF and 1DMe, respectively. Ability of NPFF2-preferring partial antagonist 53a to reverse the effect of 0.1 μM NPVF (C) or 0.01 μM 1DMe (D) in the forskolin-induced cAMP assay in CHO cells expressing hNPFF1-R or hNPFF2-R, respectively. Increasing doses of 53a were tested either alone or in the presence of NPVF or 1DMe. ***, p < 0.001; **, p < 0.01; *, p < 0.05; different from NPVF or 1DMe alone (white bar) (one-way ANOVA followed by Dunnett’s multiple comparison test). Data represents the mean ± SEM from two to four experiments performed in duplicate.
Figure 10.

Ability of NPFF2-preferring partial antagonist 53a to right-shift the dose–response curve of NPVF (A) or 1DMe (B) in the forskolin-induced cAMP assay in CHO cells expressing hNPFF1-R or hNPFF2-R, respectively. Data represents the mean ± SEM from two to four experiments performed in duplicate.
Among compounds bearing either an arginine or guanidine moiety at the 4-position of the piperidine ring and various lipophilic substitutions at the piperidine nitrogen (methyl, benzyl, phenethyl, and 2-naphthalenylmethyl) (7a–d, 9a–c), arginines 7b and 7d with benzyl and 2-naphthalenylmethyl substitutions (Scheme 1) yielded affinity below 500 nM, indicating that both arginine and compact aromatic substitutions on the 4-(phenylamino)piperidine scaffold are detrimental for NPFF1,2 affinity (Table 1). While 2-naphthalenylmethyl 7d gave nonselective affinity at NPFF1 and NPFF2, Ki = 479 ± 39 and 304 ± 14 nM (1.6-fold preference for NPFF2), respectively, we chose to utilize 7b as the lead compound due to the N-benzyl being less bulky and for facile analog synthesis.
This benzyl analog (7b) bound to the NPFF1 subtype at Ki = 487 ± 117 nM with 2.2-fold preference over NPFF2. Indeed, the 2-naphthalenylmethyl substitution improved affinity at NPFF2 and can be incorporated into future analog development if necessary. To further investigate the moieties of arginine 7b responsible for NPFF1,2-binding, the aniline was removed (15, Scheme 2). This modification resulted in a complete loss of NPFF1 affinity and a 7-fold loss of affinity at NPFF2-R (Table 1), emphasizing the importance of this group on our core scaffold for generating NPFF ligands, as shown in Figure 1.
In the GTPγS assay for functional activity, 7b exhibits agonist activity at both subtypes when tested at a high concentration of 10 μM (36% stimulation at NPFF1-R and 184% stimulation at NPFF2-R), albeit lower than the effect of full NPFF agonist 1DMe at 10 μM (71% stimulation at NPFF1-R and 231% stimulation at NPFF2-R; Figure 2). Although 7b has modest affinity and preference for NPFF1-R, this analog serves as a good starting point to develop small molecule NPFF1-selective agonists, a pharmacological profile that is currently lacking in the literature (as shown in Chart 1). From the limited SAR above, we note that preference for either subtype for these arginine-bearing analogs can be modulated in part by substitution at the piperidine nitrogen (7b vs 7d) with 1-C linked aromatic groups. Further substitutions, including 1-C linked cycloalkyl, bicyclic and tricyclic aromatic rings, and heterocyclic rings could be explored to potentially afford NPFF1 or NPFF2 agonists.
SAR of NPFF1 Agonist 7b
From the SAR above, it was determined that disubstitution at the 4-position of the piperidine ring with aniline and a C-terminal linked arginine (linked via a methylene amide) were necessary for affinity at NPFF1,2-R, in addition to compact aromatic substitution at the 1-position. To evaluate the importance of the two positively charged moieties of 7b, the α-amino and guanidinium groups, the α-amino group was removed in favor of retaining the guanidine moiety present in most reported NPFF1,2 ligands (17d) (Scheme 3, Table 2). Additionally, the linker length between the guanidinium and amide groups was explored with aliphatic (one to three carbons, 17a, 17c, 21) and rigidified aromatic linkers (three to five carbons, 17e–g) (Scheme 3, Table 2).
Table 2. Binding Affinities of Guanidino-piperidines 7b, 17a–g and 21 at NPFF1-R and NPFF2-Ra.
| cmpd | hNPFF1-R (Ki, nM) | hNPFF2-R (Ki, nM) | ratio |
|---|---|---|---|
| NPVF | 3.66 ± 0.95 | ||
| 1DMe | 3.85 ± 0.42 | 0.47 ± 0.08 | |
| 7bb | 487 ± 117 | 1052 ± 32 | 2.2-fold (NPFF1) |
| 17a | 441 ± 14 | 2907 ± 216 | 6-fold (NPFF1) |
| 17b | 3429 ± 377 | 3220 ± 53 | |
| 17cb | 2692 ± 121 | 2682 ± 570 | |
| 17db | 2454 ± 338 | 2223 ± 388 | |
| 17eb | 1623 ± 630 | 993 ± 87 | |
| 17fb | 1966 ± 324 | 1620 ± 179 | |
| 17gb | 2348 ± 413 | 2398 ± 288 | |
| 21 | 319 ± 19 | 405 ± 56 |
Ki values were determined by using [3H]NPVF and [3H]EYF for hNPFF1-R and hNPFF2-R, respectively, and are the mean ± SEM from two-four experiments performed in duplicate.
Ki values were determined by using [125I]YVP and [125I]EYF for hNPFF1-R and hNPFF2-R, respectively, and are the mean ± SEM from two to four experiments performed in duplicate.
Removal of the α-amino group of 7b (17d) yields a 5- and 2-fold loss of affinity at NPFF1-R and NPFF2-R, respectively, indicating the importance of a positively charged moiety near the core piperidine structure. The distance between the positively charged guanidine and the core structure was then investigated by deleting one to three aliphatic linkers (17c, 17a, 21). Shortening the linker length by one carbon (17c) affords similar affinity at both subtypes as 17d, however; two carbon deletion (17a) yields similar affinity at NPFF1 as 7b and 6-fold selectivity. Three carbon deletion (21) affords a 1.5- and 2.6-fold increase in affinity at NPFF1-R and NPFF2-R, respectively, compared with lead 7b, with Ki values of 319 ± 19 and 405 ± 56 nM, greatly simplifying our lead for optimization. Thus, considering 7b vs 21, only one positively charged group is needed on this scaffold, projected a short distance from the core piperidine. Substituting an amine (17b) in place of the guanidine moiety of 21 results in an 11- and 8-fold loss of affinity at NPFF1-R and NPFF2-R, respectively, suggesting a potential bidentate interaction. Exploring spacers between the guanidinium group and the amide of 7b with aromatic-linked guanidines (three to five carbons), 17e–g afford a 3.3–4.8-fold loss of affinity at NPFF1 relative to 7b, and a 1.5–2.3-fold loss of affinity at NPFF2-R. Such a result indicates that only one aromatic group is tolerable at the 4-position of the piperidine ring, in addition, as seen with the aliphatic linked analogs above, the placement of the guanidine group relative to the core structure is of paramount importance.
When evaluated alone in the forskolin-induced cAMP assay at both subtypes, 21 displays no agonist activity at concentrations up to 1 μM and weak partial agonist activity at concentrations >1 μM (data not shown), in contrast to the effect observed for peptide agonists NPVF and 1DMe at NPFF1-R and NPFF2-R, respectively. In the same assay, guanidated glycine 21 partially reverses the inhibitory effect of 0.1 μM NPVF at NPFF1-R, with an EC50 of 21.98 ± 5.08 μM and corresponding Emax of 83 ± 2% (Figure 3A). At NPFF2-R, this analog exhibits low agonist activity (20–25% inhibition of cyclase at high concentrations of 10 and 30 μM) and less potently inhibits the effect of 0.01 μM 1DMe compared with its effect on NPVF at NPFF1-R (Figure 3B). We considered this simplified structure (21) as our lead for optimization to generate NPFF2 selective small molecule ligands, since compounds of this profile are not available and would aid in the pharmacological classification of this subtype.
Collectively, based on these compounds born from our proposed general scaffold for designed NPFF1,2 ligands (Figure 1), we have noted a 2D pharmacophore for both affinity and modulation of functional activity at both subtypes (Figure 4). Both subtypes favor substitution at the piperidine nitrogen with methylene-linked aromatic groups such as a phenyl and 2-naphthyl.
Figure 4.

Two-dimensional pharmacophore for affinity and modulation of functional activity at NPFF1-R and NPFF2-R.
Disubstitution at the 4-position of the piperidine ring with an aniline moiety and a methylene amide linked to a positively charged moiety (or moieties) is also necessary for recognition at both subtypes. The nature of the positively charged moiety is crucial for modulation of functional efficacy: while an arginine side chain (7b) yields a modestly preferring NPFF1 agonist, a guanidated glycine (21) affords a nonselective compound that weakly antagonizes NPFF1-R.
SAR of Low Efficacy NPFF1 Antagonist 21
In our quest to identify NPFF2 selective ligands, we performed SAR analysis of low efficacy NPFF1 antagonist 21 (Ki NPFF1-R = 319 ± 19 nM; Ki NPFF2-R = 405 ± 56 nM), focusing on four areas: (1) the aniline phenyl ring (ring 1), (2) the aniline amine, (3) the methylene amide, and (4) the benzyl ring (ring 2) at the N-1 position (Figure 5).
Figure 5.

Sites of modification (1–4) for low efficacy NPFF1 antagonist 21.
To first validate the 4,4-substitution pattern of 21 about the piperidine ring before exploring modifications 1–4 above, the corresponding 3,3-substituted analog was investigated (26) (Scheme 4), resulting in a 2.8- and 4.7-fold loss of affinity at NPFF1-R and NPFF2-R (Table 3). Although disubstitution of these pharmacophoric elements at the two-position of the piperidine ring was not explored, we felt a 4,4-substitution pattern was optimal for our novel scaffold.
Table 3. Binding Affinities of Guanidino-piperidines 26, 30a–d, 34, 42−43, 46, and 49a–g at NPFF1-R and NPFF2-Ra.
| cmpd | hNPFF1-R (Ki, nM) | hNPFF2-R (Ki, nM) | ratio |
|---|---|---|---|
| NPVF | 3.66 ± 0.95 | ||
| 1DMe | 3.85 ± 0.42 | 0.47 ± 0.08 | |
| 21 | 319 ± 19 | 405 ± 56 | |
| 26 | 904 ± 237 | 1893 ± 490 | |
| 30a | 94 ± 24 | 309 ± 55 | 3.3-fold (NPFF1) |
| 30b | 114 ± 17 | 987 ± 65 | 8.7-fold (NPFF1) |
| 30c | 201 ± 35 | 884 ± 177 | 4.4-fold (NPFF1) |
| 30d | 191 ± 31 | 409 ± 39 | 2.1-fold (NPFF1) |
| 34 | 511 ± 79 | 3505 ± 847 | 6.9-fold (NPFF1) |
| 42 | 417 ± 74 | 84 ± 12 | 5-fold (NPFF2) |
| 43 | 151 ± 15 | 465 ± 108 | 3-fold (NPFF1) |
| 46 | 81 ± 17 | 1426 ± 49 | 17.6-fold (NPFF1) |
| 49a | 15085 ± 355 | 2757 ± 81 | |
| 49b | 1638 ± 499 | 298 ± 14 | 5.5-fold (NPFF2) |
| 49c | 1057 ± 150 | 330 ± 28 | 3.2-fold (NPFF2) |
| 49d | 538 ± 40 | 129 ± 26 | 4.2-fold (NPFF2) |
| 49e | 141 ± 34 | 504 ± 95 | 3.6-fold (NPFF1) |
| 49f | 112 ± 11 | 454 ± 4 | 4-fold (NPFF1) |
| 49g | 340 ± 91 | 1513 ± 33 | 4.5-fold (NPFF1) |
Ki values were determined by using [3H]NPVF and [3H]EYF for hNPFF1-R and hNPFF2-R, respectively, and are the mean ± SEM from two to four experiments performed in duplicate.
We next turned our attention to substituting the aniline phenyl ring of lead 21 (modification 1, Scheme 5) with bulky, hydrophobic 1-naphthyl 30a and 3,4-dichlorophenyl 30d groups, as well as groups containing H-bond acceptor moieties 3,5-dimethoxyphenyl 30b and 3-methoxyphenyl 30c (Scheme 5), because the aniline moiety as a whole affects NPFF1,2 binding (7b vs 15). These modifications afforded a 1.6–3.4-fold gain of affinity for NPFF1-R compared with lead 21 (Ki = 94 ± 24 to 201 ± 35 nM), while showing comparable or less affinity at NPFF2-R (Table 3). In particular, 1-naphthyl 30a and 3,5-dimethoxyphenyl 30b substitutions were favored at NPFF1-R (Ki = 94 ± 24 and 114 ± 17 nM, respectively), while 3-methoxyphenyl 30c and 3,4-dichlorophenyl 30d proved to be half as active at this subtype (Ki = 191 ± 31 to 201 ± 35 nM), indicating that additional hydrophobic interaction(s) accessed by 30a and 30b in this region of the scaffold are beneficial for NPFF1 binding only.
Despite showing modest (3.3–8.7-fold) preference for NPFF1-R binding, analogs 30a–c were assayed alone for functional activity in the forskolin-induced cAMP assay (Figure 6A). No activity was observed at concentrations up to 10 μM; thus, a lack of agonist activity was retained on our scaffold.
Continuing to explore the aniline moiety, the aniline NH (modification 2, Scheme 6) was removed (34) and replaced with methylene (42) and ethylene (43) groups. Removal of the amine (34) results in only a modest loss of affinity at NPFF1-R compared with lead 21 (1.6-fold) but a significant loss of affinity at NPFF2-R for this scaffold (8.7-fold) (Table 3), suggesting the necessity of a spacer atom between the phenyl and piperidine rings for binding at NPFF2-R only. Substitution with a methylene moiety (42) again yields similar affinity at NPFF1-R, and a 5-fold increase in affinity at NPFF2-R compared with lead 21 (Ki = 84 ± 12 nM), indicating a greater degree of lipophilicity required for NPFF2 binding. Additionally, the spatial arrangement of the phenyl moiety could be important for NPFF2 preference, since switching from a trigonal pyramidal NH (21) to an sp3 C (42) yields 5-fold preference for NPFF2-R. In the forskolin-induced cAMP assay at NPFF2-R, benzyl 42 alone is inactive at concentrations up to 1 μM (Figure 7), again indicating this scaffold’s lack of agonist activity. Extending the phenyl ring by an additional carbon (phenethyl 43) is not tolerated at NPFF2-R, resulting in a 5.5-fold loss of affinity relative to methylene 42 (Ki = 465 ± 108 nM), instead binding to NPFF1-R with 151 nM affinity. Collectively, mostly NPFF2 binding is affected by substitution of the aniline NH, while NPFF1 binding is affected by substitution of the aniline phenyl ring.
The importance of the methylene amide at the four-position of the piperidine ring of lead 21 was explored by simultaneous removal of the methylene moiety and replacement with a reverse amide (46; modification 3, Scheme 7). Additionally, the linker length between the amide and guanidine moieties was extended by one carbon, in order to retain the same distance between the guanidine and piperidine moieties as lead 21. This change allowed for a 4-fold improvement at NPFF1 relative to lead 21 (Ki = 81 ± 17 nM) and 17.6-fold selectivity vs NPFF2 (Table 3). Thus, selectivity for NPFF1 is controlled by positioning of the amine and carbonyl groups; in addition, the spatial orientation of the guanidine in relation to the core piperidine scaffold may also contribute to selectivity (connected via a planar carbonyl group (46) vs a flexible methylene group (21)). In the forskolin-induced cAMP assay at NPFF1-R, reverse amide 46 alone is inactive at concentrations up to 10 μM (Figure 6A,B) and dose-dependently antagonizes the effect of 0.1 μM NPVF at an EC50 = 3.52 ± 1.42 μM and Emax = 68 ± 16% (Figure 6B). In terms of its good affinity and selectivity, reverse amide 46 is the first small molecule, nonpeptide NPFF1-R antagonist known, in contrast to dipeptide NPFF1-R antagonist 1. Considering its reduced polar surface area and number of metabolically labile amide bonds compared with dipeptide 1, reverse amide 46 represents a small molecule lead for drug development targeting the NPFF1-R subtype, since dipeptide 1 was recently shown to strongly reduce OIH induced by the opioid agonist fentanyl.17
Various methylene-linked aromatic and nonaromatic substitutions of phenyl ring 2, located off of the 1-position of the core piperidine ring, were also explored (49b–g; modification 4, Scheme 8). As suggested by N-methyl analogs 7a and 9a (Scheme 1, Table 1), removal of the N-substituent (49a) resulted in a significant loss of affinity at both subtypes (Table 3). Cyclohexyl 49b, benzyl 49c, and 1-naphthyl 49d provided modest improvement (1.2–3.1-fold) in binding to NPFF2-R compared with lead 21 (Ki = 298 ± 14, 330 ± 28, and 129 ± 26 nM, respectively), with 3.2–5.5-fold preference vs NPFF1-R. Interestingly, replacement of the benzyl group of lead 21 with a cyclohexyl moiety shows the importance of this aromatic ring for NPFF1-R affinity only, since 49b yields a 5-fold drop in NPFF1-R affinity, while retaining similar affinity at NPFF2-R. Conversely, 2-naphthyl 49e and 4-bromophenyl 49f are more active at NPFF1-R, providing 2.3–2.8-fold improvement in binding to NPFF1-R compared with lead 21 (Ki = 112 ± 11 to 141 ± 34 nM), with 3.6–4.5-fold preference vs NPFF2-R. 4-Methyl benzoate analog 49g yields similar affinity at NPFF1-R as lead 21, with 4.5-fold preference. In summary, on this scaffold, selectivity for either subtype can be controlled by N-1 substitution: both cycloalkyl (49b) and unsubstituted aromatic moieties (49c) afford NPFF2-R-preferring compounds, whereas a broad range of substituted aromatic groups favor the NPFF1-R subtype, including both bromo (49f) and methyl ester (49g) groups. Additionally, selectivity for NPFF2-R vs NPFF1-R can be achieved with a 1- vs 2-naphthyl group (49d vs 49e), indicating that the NPFF2-R pocket is of a more discriminate volume.
In Vivo Testing of NPFF1-Selective Ligand 46
Mice exposed to a 48 °C warm-water bath demonstrated a 7.85 ± 0.51 s latency to tail-withdrawal. NPFF (30 nmol, icv) significantly reduced tail-withdrawal latency in the mouse 48 °C warm-water tail-withdrawal assay over time (F(9,118) = 5.56, p < 0.0001, one-way ANOVA),6,8 whereas vehicle (icv) was without effect (F(9,74) = 0.19, p = 0.99, one-way ANOVA; Figure 8A,B).
Figure 8.
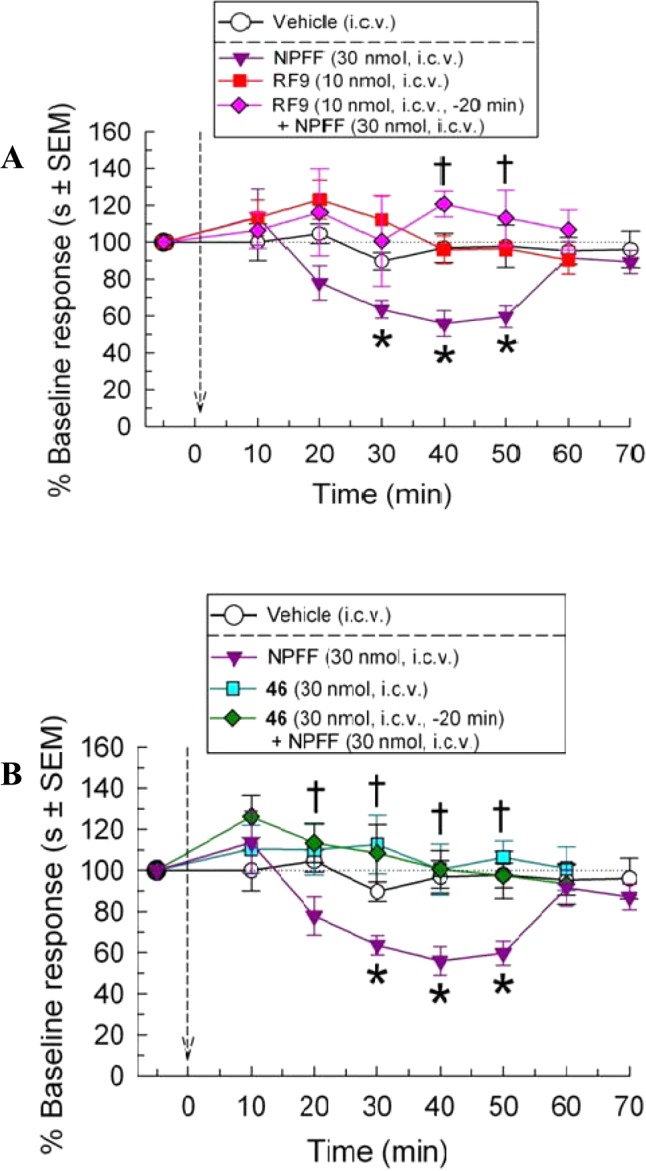
NPFF-induced hyperalgesia is prevented by pretreatment with NPFF-receptor antagonists. Mouse latencies to withdraw their tail from a 48 °C warm-water stimulus were measured repeatedly over 80 min after administration of test compounds. After collection of baseline responses (left of arrow), arrow denotes single administration of vehicle (50% DMSO, icv; white circles), NPFF (30 nmol, icv; purple triangles), RF9 (10 nmol icv, squares in part A) or 46 (30 nmol icv; squares in part B). Additional mice were pretreated 20 min with RF9 (A) or 46 (B) prior to administration of NPFF (diamonds). Points represent n = 7–12 mice, with average % baseline response ± SEM plotted. *p < 0.05 compared with baseline response with one-way ANOVA with Tukey HSD post hoc test; †p < 0.05 compared with NPFF response with two-way ANOVA.
Administration of the nonselective NPFF1,2-R antagonist RF9 (10 nmol, icv) was without effect on the tail-withdrawal latency (F(9,77) = 1.74, p = 0.09, one-way ANOVA), but a 20 min pretreatment significantly reversed NPFF-mediated hyperalgesia (F(3,188) = 8.92, p < 0.0001, two-way ANOVA; Figure 8A). Similarly, pretreatment with the NPFF1-R selective antagonist 46 (30 nmol, icv) also significantly prevented the NPFF-induced hyperalgesic effects (F(3,233) = 9.58, p < 0.0001, two-way ANOVA; Figure 8B), without demonstrating significant differences from either baseline or vehicle-treated responses.
SAR of NPFF2-Preferring Ligand 42
Since substitution at the aniline NH (modification 2) with a methylene group (benzyl 42) yielded a high affinity NPFF2 ligand (Ki = 84 ± 12 nM) with 5-fold preference, we considered this as our new lead compound to generate NPFF2 ligands and explored the effect of replacing the proximal phenyl ring 1 of this scaffold with bulky aromatic rings as shown in Scheme 9.
Substitution of phenyl ring 1 of lead 42 with a 1-naphthyl group (53a) afforded a 2.8-fold increase in affinity at NPFF2-R (Ki = 30 ± 5 nM) relative to lead 42, with similar preference (Table 4). Substitution with a 3,4-dichlorophenyl moiety (53b) decreased affinity by 2.7-fold at NPFF2 relative to 1-naphthyl 53a, suggesting a potential π–π stacking interaction in the NPFF2 pocket. Interestingly, substitution with 2-naphthyl (53c) or biphenyl groups (53d) results in 6.8- and 30-fold losses of affinity at NPFF2-R relative to 1-naphthyl 53a, indicating that correct placement of the additional aromatic group on this scaffold is important for maintaining affinity at NPFF2-R.
We note that substitution of phenyl ring 1 on this scaffold improves NPFF2 binding and maintains preference for this subtype, while the same substitutions on the aniline-containing scaffold (30a, 30d, Scheme 5, Table 3) afford compounds with preference toward the NPFF1 subtype. This result suggests different binding conformations for the aniline- vs benzyl-containing scaffolds at NPFF1-R and NPFF2-R, respectively. This observation could be further investigated by installing NPFF2-preferring N-1 substituents (as in 49b–d) onto the benzyl scaffold.
In the forskolin-induced cAMP assay at NPFF1-R and NPFF2-R, 1-naphthyl 53a alone exhibits low activity at concentrations up to 10 μM (Figures 9A–D). To evaluate the antagonistic property of naphthyl 53a, various concentrations were tested in the presence of a fixed concentration of NPVF (0.1 μM, at NPFF1-R) and the stable NPFF peptide agonist 1DMe (0.01 μM, at NPFF2-R) in the cAMP assay (Figures 9C,D). 1-Naphthyl 53a dose-dependently reversed both NPVF- and 1DMe-inhibition of forskolin-induced cAMP accumulation with EC50 values of 2.6 ± 0.69 μM (Figure 9C) and 645 ± 143 nM (Figure 9D), respectively. Corresponding Emax values at NPFF1-R and NPFF2-R were 56.2 ± 2.6% and 22 ± 1.5%, respectively.
In addition, a dose–response curve of NPVF and 1DMe in the same assay was performed in the absence or the presence of 10 μM 53a (Figures 10A,B) at each subtype. 1-Naphthyl 53a (10 μM) induces a 12-fold loss of functional activity for NPVF at NPFF1-R (EC50 = 181 ± 38 nM in the presence of 53a compared with 15 ± 0.6 nM alone; Figure 10A); similarly, this compound induces a 4-fold loss of functional activity for 1DMe at NPFF2-R (EC50 = 11.4 ± 3.2 nM in the presence of 53a compared with 2.8 ± 0.3 nM alone; Figure 10B). As a result, 1-naphthyl 53a represents the first small molecule NPFF2 partial antagonist reported in the literature. Although exhibiting modest preference toward the NPFF2 subtype (3.7-fold), this compound’s unprecedented high affinity (30 nM) at NPFF2-R bears further SAR investigation, serving as an excellent lead to develop selective small molecule NPFF2 antagonists as pharmacological probes.
Binding Affinity of Selected Compounds at μ-, δ-, and κ-Opioid Receptors
Because no selective, pharmacologically pure, and high affinity NPFF1 and NPFF2 agonists and antagonists are available to the scientific community (save dipeptide NPFF1 antagonist 1) to investigate the opioid-modulating properties of the NPFF1 and NPFF2 subtypes, we tested compounds displaying reasonable affinities and a variety of pharmacological profiles (summarized in Table 6) at both subtypes for off-target affinity at the three opioid subtypes (Table 5).
Table 6. Summary of Functional Activities of Selected Guanidino-piperidines on hNPFF1-R and hNPFF2-R Receptors Expressed in CHO Cells.
| cAMP hNPFF1-R |
cAMP hNPFF2-R |
|||
|---|---|---|---|---|
| cmpd | antagonist activity EC50,a nM | Emax, % | antagonist activity EC50,a nM | Emax, % |
| RF9 | 4700 ± 1200b | 79 ± 5c | h | h |
| 7b | 36d | 184d | ||
| 21 | 21980 ± 5080 | 83 ± 2 | g | 20–25e |
| 30a | >10Kf | g | g | g |
| 30b | >10Kf | g | g | g |
| 42 | g | g | 1Kf | g |
| 46 | 3520 ± 1420 | 68 ± 16 | g | g |
| 53a | 2600 ± 690 | 56.2 ± 3 | 645 ± 143 | 22 ± 1.5 |
EC50 is the concentration that produces a 50% reversion of the NPVF (0.1 μM) or 1DMe (0.01 μM)-induced inhibition of forksolin-stimulated cAMP accumulation in CHO hNPFF1 and hNPFF2 cells, respectively.
Data taken from ref (15).
Unpublished observation.
% Maximal activation in the [35S]GTPγS assay at 10 μM.
% Inhibition of forskolin-stimulated cAMP accumulation at concentrations of 10 and 30 μM.
Highest concentration showing no agonist activity when tested alone in the forskolin-induced cAMP assay.
Not determined.
Not reported.
In general, most compounds possessed modest preference for NPFF1 or NPFF2 vs MOP and KOP (2–12-fold), and most ligands did not bind at DOP with any appreciable affinity (>5K) (Table 5). Notably, the NPFF1-selective antagonist 46 afforded ≥18-fold selectivity at NPFF1 vs MOP/DOP/KOP, while NPFF2-preferring ligands 42 and 53a yielded 2- and 12-fold selectivity, respectively, for NPFF2-R vs MOP and KOP. Similar to compound 46, NPFF1-preferring ligands 30a and 30b were also reasonably selective vs MOP and KOP (7–43-fold).
Such modest preference for NPFF1,2-R vs MOP and KOP is not surprising, given that most of these ligands contain a 4-anilido piperidine, a known scaffold for MOP analgesia (i.e., fentanyl).28 Additionally, the seemingly requisite guanidine moiety on our scaffold is reminiscent of 5′-guanidinonaltrindole (GNTI),29 a potent and selective KOP antagonist. These moieties will be taken into account during further SAR exploration of our lead compounds, namely, 7b, 46, and 53a, in order to generate selective NPFF1 and NPFF2 ligands. Additionally, docking of 7b, 46, and 53a into the active state homology models and crystal structures of MOP and KOP (depending on their functional activities at each subtype) could also shed light on further modifications to be made to this scaffold in order to minimize off-target opioid affinity. While the opioid affinity of these leads is of primary concern, we also recognize the need to assay these compounds for any off-target affinity at neuropeptide Y and orexin receptors due to 30–35% homology with NPFF1,2-R, other receptors within the RFa peptide family (GPR10, GPR54, and GPR103), and the nociceptin opioid receptor (NOPr, an opioid-modulating receptor), as previously done for RF9, the most well characterized NPFF ligand to date.15
Collectively, SAR analysis of four regions of nonselective NPFF1 antagonist 21 led to the discovery of three small molecule, NPFF2-preferring ligands, benzyl 42, 3,4-dichlorophenyl 53b, and partial antagonist 1-naphthyl 53a. All analogues displayed good affinity at NPFF2-R, 84 ± 12, 82 ± 8, and 30 ± 5 nM, respectively, and 3.7–5-fold preference vs NPFF1-R. Additionally, SAR analysis of 21 allowed us to generate a 2D pharmacophore for NPFF2-preferring ligands (Figure 11).
Figure 11.

Two-dimensional pharmacophore for NPFF2-preferring ligands from SAR analysis of low efficacy NPFF1 antagonist 21.
We found that substitution of the aniline moiety of nonselective antagonist 21 with compact aromatic groups such as benzyl (42), 1-naphthalenylmethyl (53a), and 3,4-dichlorophenylmethylene (53b) improved affinity and preference for NPFF2, particularly for 53a (14-fold improvement in binding affinity, 3.7-fold preference vs NPFF1). The methylene amide at the four-position of the piperidine ring proved crucial for maintaining recognition at NPFF2 (46). Additionally, lipophilic groups substituted at N-1, including both cycloalkyl (49b) and aromatic (49c,d) moieties, were preferred at NPFF2, indicating that substitution at the 4- rather than 1-position of the ring may mimic the Phe8 interaction of the endogenous ligand NPFF. Further SAR studies of NPFF2-preferring lead 53a, to be done by substituting NPFF2-preferring N-1 substituents (as in 49b–d), are planned to potentially increase affinity and preference for this subtype.
Conclusion
Considering the structures of a limited number of NPFF1,2 dipeptides and low affinity small molecules, as well as the endogenous ligand NPFF, ligand-based rational design was employed to construct the first small molecule scaffold for NPFF1,2-R displaying significant improvements in affinity relative to the aryliminoguanidines (Chart 1), as well as a tractable SAR for determining functional activity and preference for either subtype, as summarized in our 2D pharmacophore models (Figures 4 and 11). Specifically, the guanidino-piperidine class of NPFF ligands represents the first disclosure of a small molecule scaffold whose SAR results in a wide range of pharmacological profiles at NPFF1,2-R to serve as lead compounds to develop pharmacological tools, namely, a NPFF1 agonist (7b, Ki = 487 ± 117 nM), a NPFF1 antagonist (46, Ki = 81 ± 17 nM), and a NPFF2 partial antagonist (53a, Ki = 30 ± 5 nM). In vivo testing of the novel NPFF1-R antagonist 46 demonstrated antagonism of NPFF-mediated hyperalgesia without significant effect alone on tail-withdrawal latencies (Figure 8B), similar to the effects seen with dipeptide NPFF1,2 antagonist RF9 (Figure 8A),15,16 the most established NPFF ligand to date, and dipeptide NPFF1 antagonist 1.17 SAR studies of reverse amide 46, including exploration of this scaffold with NPFF1-preferring moieties (such as in those of 30a,b, 43, and 49e,f), could afford compounds of an improved NPFF1 profile to combat opioid-induced hyperalgesia. The SAR of arginine 7b, reverse amide 46, and 1-naphthyl 53a will be further explored, using the approaches described herein, in order to generate NPFF1 and NPFF2 selective agonists and antagonists to further explore and characterize the pharmacology of these subtypes as they relate to opioid-mediated analgesia.
Experimental Section
Chemistry
All the solvents and reagents were obtained commercially and used as received unless noted otherwise. Anhydrous diethyl ether was prepared by refluxing with sodium; anhydrous THF was prepared by refluxing with sodium and benzophenone. Flash chromatography was performed with Merck silica gel 60 (230 × 400 mesh). NMR spectra were recorded on either a Bruker AVIII 400 spectrometer or Bruker DRX500. For 7a–7e, 9a–9c and 15a,b, HPLC/MS analyses were recorded on a Waters Alliance LC/MS System, consisting of a Waters ZQ mass detector, photodiode array detector, and an Alliance HPLC system, equipped with an XTerra column (C-18, 2.1 mm × 5 mm). HPLC analysis was done using gradient of ACN/H2O/10% HCl (aq). For all remaining final compounds, mass spectra were obtained on a Waters micromass ZQ detector or a Micromass Q-TOF micro hybrid quadrupole/orthogonal high resolution time of flight MS. HPLC analysis was performed on a reverse phase XBridge C18 column (4.6 mm × 75 mm) column, using 40% H2O/50% ACN/10% of 0.2% TFA(aq) as the mobile phase, in gradient conditions at a flow rate of 1 mL/min. Eluted peaks were monitored at 254 nm with a Waters 996 PDA detector. All final compounds tested were confirmed to be of ≥95% purity by the HPLC methods described above.
General Procedure 1: Strecker Synthesis To Generate Carbonitriles
To a solution of N-substituted piperidone (15.9 mmol) in acetic acid (25 mL) was slowly added substituted aniline (17.6 mmol). The mixture was cooled to 5 °C with an ice bath, and a solution of KCN or TMSCN (17.6 mmol) in water (5 mL) was added dropwise. The solution was stirred for 1 h at 0 °C and for 3 h at room temperature and then poured into water. The pH was adjusted to 10 by adding NH4OH, and the aqueous layer was extracted with methylene chloride (3 × 50 mL). The combined organic extracts were washed with water and brine and dried. The solvent was removed in vacuo, and the residue was purified using one of the following methods: flash chromatography using hexanes/EtOAc, trituration with cold ether, or recrystallization.
General Procedure 2: Nitrile Reduction
Method A: Anhydrous ether (500 mL) was added to the nitrile intermediate (0.5 mmol), which was then added dropwise to a solution of LiAlH4 (1.0 mmol) in 1 L of anhydrous ether under nitrogen. The solution was quenched with a few drops of water, decanted, and evaporated. The resulting oil was purified by silica gel chromatography (49:50:1 chloroform–methanol–NH4OH) affording the amine as a pale yellow solid in 35–40% yield. Method B: The nitrile intermediate (7.21 mmol) was hydrogenated in a solution of methanol saturated with ammonia (210 mL) and Raney Ni (0.4 g) under pressure (2.4–3 atm) for 4–24 h. The catalyst was removed by filtration, and the solvent was evaporated in vacuo. The residue was chromatographed on a silica gel column using methylene chloride/methanol as the eluent to afford the amine.
General Procedure 3: Introduction of Arginine Side Chain
A solution of Boc-protected arginine (0.4 mmol) in 10 mL of methylene chloride was stirred at −10 °C for 20 min. Then, triethylamine (0.9 mmol), HOBt (0.4 mmol), and EDCI (0.4 mmol) were added, and the mixture was stirred at −10 °C for 30 min. A solution of amine (0.4 mmol) in 10 mL of methylene chloride was cooled at −10 °C and added dropwise. After stirring overnight at room temperature, water was added to the reaction mixture, followed by 10% NaOH solution to pH 12, and the mixture was extracted 3 times. The organic layer was dried using MgSO4 and evaporated affording the Boc-protected arginine intermediate (50–60% yield).
General Procedure 4: Conversion of Various Amines to Boc-Protected Guanidines
Method A: To a stirred mixture of Boc-protected guanidino carboxylic acid (1.11 mmol), 1-hydroxy-benzotriazole hydrate (1.11 mmol), and triethylamine (1.20 mmol) in methylene chloride (20 mL) at −10 °C was added N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (1.11 mmol). After 2 h of stirring at −10 °C, a solution of 4-(aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (5b) (0.85 mmol) in methylene chloride (4 mL) was slowly added, and the reaction mixture was stirred for 1 h at −10 °C and for 15 h at room temperature. The solvent was removed in vacuo, and the residue was chromatographed on a silica gel column using a gradient of petroleum ether/ethyl acetate as the eluent to give the desired material. Method B: A mixture of amine (0.57 mmol), 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (0.68 mmol), and triethylamine (1.42 mmol) in anhydrous DMF (4 mL) was stirred at room temperature for 2 days. The reaction mixture was poured into water and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by chromatography on a silica gel column using methylene chloride/methanol as the eluent to give the desired material. Method C: To a stirred mixture of 1,3-di-Boc-2-(carboxymethyl)guanidine (1.15 mmol), 1-hydroxy-benzotriazole hydrate (1.12 mmol), and triethylamine (2.87 mmol) in methylene chloride (10 mL) under argon at 0 °C was added N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (1.18 mmol). The reaction mixture was kept at 0 °C for 2 h, and a solution of amine (1.02 mmol) in methylene chloride (10 mL) was slowly added. The reaction mixture was stirred 1 h at 0 °C and then allowed to warm to room temperature. After 15 h of stirring, the solvent was evaporated. Ethyl acetate (50 mL) and water were added. The layers were separated, and the organic layer was washed with brine. The solvent was dried and evaporated. The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol to give the desired material. Method D: (Step I) To a stirred mixture of amine (1.21 mmol), 1-hydroxy-benzotriazole hydrate (1.45 mmol), Boc-gly-OH (1.33 mmol), and triethylamine (3.64 mmol) in methylene chloride (15 mL) at 0 °C under argon was added N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (1.33 mmol). The reaction mixture was then allowed to warm to room temperature. After 20 h of stirring, methylene (80 mL) and water (30 mL) were added. The layers were separated, and the organic layer was extracted again with methylene chloride (30 mL). The combined organic layers were washed with brine. The solvent was dried and evaporated. The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol to yield the Boc-Gly-coupled amine. (Step II) To a solution of the Boc-Gly-coupled amine (0.558 mmol) in anhydrous methylene chloride (6 mL) was added trifluoroacetic acid (3 mL). The reaction mixture was stirred at room temperature under argon for 1 h and evaporated. Methylene chloride (80 mL) and a 10% solution of NaOH (40 mL) were added. The layers were separated, and the organic layer was washed with water and brine. The solvent was dried and evaporated. The residue was dissolved in anhydrous DMF (7 mL). Triethylamine (1.40 mmol), mercury(II) chloride (0.558 mmol), and 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (0.670 mmol) were added, and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was poured into water and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol as the eluent to provide the title material.
General Procedure 5: Boc Deprotection To Give Final Products
Method A: The Boc-protected guanidine (0.1 mmol) was dissolved in an excess of HCl/dioxane under nitrogen and stirred for 3–4 days after which it was concentrated under reduce pressure to afford the hydrochloride salt of guanidine as colorless solid. Method B: To a solution of Boc-protected guanidine (0.3 mmol) in anhydrous methylene chloride (4 mL) was added trifluoroacetic acid (4 mL). The reaction mixture was stirred at room temperature under argon for 3 h. The solvent was removed in vacuo, and the residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol as the eluent to the title material as a tritriflate salt.
General Procedure 6: Synthesis of Various Aromatic Substituted Carbonitriles
1-Benzylpiperidine-4-carbonitrile (5 mmol) was added to a dry three-necked flash equipped with a low temperature thermometer. Dry THF (60 mL) was added, and the solution was cooled to −70 °C in a dry ice–acetone bath under positive argon pressure. Lithium diisopropylamide (2.0 M solution in heptane/tetrahydrofuran/ethylbenzene, 6.54 mmol) was slowly added while maintaining the temperature below −65 °C, and the resulting mixture was stirred for 30 min at −70 °C. Substituted bromide or chloride (6.54 mmol) was quickly added, and the resulting mixture was then allowed to warm to room temperature, stirred for another 2 h, poured into 40 mL of NH4Cl saturated solution, and extracted with ethyl acetate (3 × 40 mL). The combined organic extracts were washed with brine and dried. The solvent was removed in vacuo, and the residue was purified by chromatography on a silica gel column using ethyl acetate and hexanes as the eluent to afford the title material.
General Procedure 7: Synthesis of N-Substituted Analogs
A mixture of tert-butyl 10,10-dimethyl-3,8-dioxo-1-(4-(phenylamino)piperidin-4-yl)-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (47, 0.257 mmol) and potassium carbonate (0.773 mmol) in anhydrous DMF (3 mL) was stirred at 0 °C for 30 min under argon. A solution of substituted bromide or chloride (0.231 mmol) in DMF (1 mL) was added dropwise, and the mixture was stirred at 0 °C for 1 h and then allowed to warm to room temperature. After 3 h of stirring, the reaction mixture was poured into brine (50 mL) and extracted with ethyl acetate (3 × 30 mL). The organic layers were combined, washed with brine, dried, and evaporated. The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol as the eluent to give the title material.
1-Methyl-4-(phenylamino)piperidine-4-carbonitrile (4a)
Prepared according to general procedure 1 to afford the title material in 90% yield. 1H NMR (600 MHz, CD3OD): δ 6.72–6.79 (m, 3H), 7.13–7.17 (m, 2H), 3.642 (s, 3H), 2.37–2.40 (m, 4H), 1.92–1.96 (m, 4H). MS (ESI) 216.29 [M + H]+.
1-Benzyl-4-(phenylamino)piperidine-4-carbonitrile (4b)
Prepared according to general procedure 1 to afford the title material in 94% yield. 1H NMR (CD3OD): δ 7.32–7.20 (m, 7H), 6.90–6.87 (m, 3H), 3.67 (s, 1H), 3.53 (s, 2H), 2.79–2.76 (m, 2H), 2.43 (t, J = 7.5 Hz, 2H), 2.30–2.27 (m, 2H), 1.89 (t, J = 7.8 Hz, 2H). MS (ESI) 292 [M + H]+.
1-Phenethyl-4-(phenylamino)piperidine-4-carbonitrile (4c)
Prepared according to general procedure 1 to afford the title material in 90% yield. 1H NMR (400 MHz, CD3OD): δ 7.31–7.19 (m, 7H), 6.94–6.92 (m, 3H), 3.72 (d, 2H), 3.01–2.42 (m, 4H), 2.42–2.39 (m, 2H), 2.08–2.05 (m, 4H). MS (ESI) 306.2 [M + H]+.
1-(Naphthalen-2-ylmethyl)-4-(phenylamino)piperidine-4-carbonitrile (4d)
Prepared according to general procedure 1 to afford the title material in 70% yield. 1H NMR (600 MHz, CDCl3): δ 7.82–7.80 (m, 4H), 7.52–7.24 (m, 3H), 7.23–7.22 (m, 3H), 6.92–6.90 (m, 2H), 3.70 (s, 2H), 3.69 (s, 1H), 2.93–2.59 (m, 4H), 2.37–2.35 (m, 4H). MS (ESI) 342.5 [M + H]+.
4-(Aminomethyl)-1-methyl-N-phenylpiperidin-4-amine (5a)
Prepared according to general procedure 2, method A, to afford the title material in 35–40% yield. 1H NMR (600 MHz, CD3OD): δ 6.72–6.79 (m, 3H), 7.13–7.17 (m, 2H), 3.642 (s, 3H), 2.86 (s, 2H), 2.37–2.40 (m, 4H), 1.92–1.96 (m, 4H). MS (ESI) 220.2 [M + H]+.
4-(Aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (5b)
Prepared according to general procedure 2, method B, to afford the title material in 82% yield. 1H NMR (CDCl3): δ 7.34–7.16 (m, 7H), 6.80–6.75 (m, 3H), 3.53 (s, 2H), 2.89 (s, 2H), 2.65–2.62 (m, 2H), 2.33 (t, J = 8.1 Hz, 2H), 2.00–1.62 (m, 6H). MS (ESI) m/z 296 [M + H]+.
4-(Aminomethyl)-1-phenethyl-N-phenylpiperidin-4-amine (5c)
Prepared according to general procedure 2, method A, to afford the title material in 35–40% yield. 1H NMR (600 MHz, CD3OD): δ 7.30–7.14 (m, 7H), 6.83–6.78 (m, 3H), 3.68–3.67 (d, J = 4, 2H), 2.95 (s, 2H), 2.88–2.42 (m, 4H), 2.05–2.02 (m, 2H), 1.82–1.76 (m, 4H). MS (ESI) 310.5 [M + H]+.
4-(Aminomethyl)-1-(naphthalen-2-ylmethyl)-N-phenylpiperidin-4-amine (5d)
Prepared according to general procedure 2, method B, to afford the title material in 90% yield. 1H NMR (CDCl3): δ 7.81–7.71 (m, 4H), 7.48–7.44 (m, 3H), 7.14 (t, J = 6.2 Hz, 2H), 6.76–6.71 (m, 3H), 3.64 (s, 2H), 2.86 (s, 2H), 2.64 (d, J = 9.1 Hz, 2H), 2.34 (t, J = 8.7 Hz, 2H), 1.95 (d, J = 10.6 Hz, 2H), 1.75 (br s, 2H), 1.65–1.60 (m, 2H). MS (ESI) m/z 346 [M + H]+.
2-Amino-5-guanidino-N-((1-methyl-4-(phenylamino)piperidin-4-yl)methyl)pentanamide Hydrochloride (7a)
Prepared according to general procedure 3, starting with intermediate 5a, to afford the corresponding Boc-protected arginine intermediate 6a in 50–60% yield, which was deprotected using general procedure 5, method A, to afford the title material in 95% yield. 1H NMR (600 MHz, CD3OD): δ 7.17–7.14 (m, 3H), 6.95–6.93 (m, 2H), 3.95–3.72 (m, 2H), 3.66–3.55 (m, 1H), 3.41–3.33 (m, 6H), 2.85 (s, 3H), 2.31–1.97 (m, 6H), 1.66–1.64 (2H). 13C NMR (600 MHz, CD3OD): δ 170.8, 169.7, 157.4, 130, 129.5, 126, 72.3, 71.2, 67, 61, 53.2, 53.09, 42.4, 40.7, 24.5. MS (ESI) 376.51 [M + H]+.
2-Amino-N-((1-benzyl-4-(phenylamino)piperidin-4-yl)methyl)-5-guanidinopentanamide Hydrochloride (7b)
Prepared according to general procedure 3, starting with intermediate 5b, to afford the corresponding Boc-protected arginine intermediate 6b in 50–60% yield, which was deprotected using general procedure 5, method A, to afford the title material in 95% yield. 1H NMR (600 MHz, CD3OD): δ 7.58–7.47 (m, 7H), 6.97–6.95 (m, 3H), 3.73–3.34 (m, 11 H), 2.30–3.31 (m, 2H), 2.12–2.03 (m, 4H), 1.31–1.28 (s, 2H). 13C NMR (600 MHz, CD3OD): δ 169, 157, 149, 132.5, 132.3, 130.3, 130.2, 124.9, 120, 118.9, 73.4, 72.3, 62.0, 61.4, 54.1, 41.7, 30.7, 25.5. MS (ESI) 452.5 [M + H]+.
2-Amino-5-guanidino-N-((1-phenethyl-4-(phenylamino)piperidin-4-yl)methyl)pentanamide Hydrochloride (7c)
Prepared according to general procedure 3, starting with intermediate 5c, to afford the corresponding Boc-protected arginine intermediate 6c in 50–60% yield, which was deprotected using general procedure 5, method A, to afford the title material in 95% yield. 1H NMR (600 MHz, CD3OD): δ 7.63–7.75 (m, 3H), 7.30–7.23 (m, 7H), 3.72–3.69 (m, 4H), 3.66–3.64 (m, 1H), 3.63–3.57 (m, 2H), 3.56–3.37 (4H), 2.35–2.97 (m, 2H), 2.20–2.00 (m, 4H), 2.0–1.8 (m, 2H), 1.30–1.26 (2H). 13C NMR (600 MHz, CD3OD): δ 170.9, 169.9, 157.4, 130.8, 130.2, 130.06, 129.2, 127.4, 126.6, 72.6, 71.6, 61.3, 58.2, 53.4, 43.6, 41.1, 30.8, 24.8, 24.5. MS (ESI) 466.60 [M + H]+.
2-Amino-5-guanidino-N-((1-(naphthalen-2-ylmethyl)-4-(phenylamino)piperidin-4-yl)methyl)pentanamide (7d)
Prepared according to general procedure 3, starting with intermediate 5d, to afford the corresponding Boc-protected arginine intermediate 6d in 70% yield. This material was deprotected using general procedure 5, method A, to afford the title material in 99% yield. 1H NMR (600 MHz, CD3OD), δ 7.81–7.71 (m, 4H), 7.48–7.43 (m, 3H), 7.16–7.13 (m, 3H), 6.79–6.66 (m, 2H), 4.09–4.06 (m, 1H), 3.85–3.83 (m, 2H), 3.66 (s, 2H), 3.63–3.47 (m, 2H), 2.64–2.62 (m, 4H), 1.49 (s, 9H). 13C NMR (400 MHz, CD3OD): δ 172.5, 163.6, 160.9, 155.1, 133.5, 133.0, 129.5, 128.1, 127.9, 127.8, 127.5, 126.1, 125.8, 119.3, 117.5, 63.2, 54.9, 49.3, 44.3, 33.9, 33.5, 25.3.
tert-Butyl (tert-Butoxycarbonylamino)((1-methyl-4-(phenylamino)piperidin-4-yl)methylamino)methylenecarbamate (8a)
Prepared according to general procedure 4, method B, also using HgCl2 (1 equiv) to afford the title material in 30% yield. 1H NMR (400 MHz, CD3OD): δ 6.72–6.79 (m, 3H), 7.13–7.17 (m, 2H), 3.642 (s, 2H), 2.27 (s, 3H), 1.9–2.2(m, 4H), 1.657–1.8 (m, 4H), 1.45–1.50 (db, J = 20 Hz, 18H). MS (ESI) 462.80 [M + H]+.
tert-Butyl ((1-Benzyl-4-(phenylamino)piperidin-4-yl)methylamino)(tert-butoxycarbonylamino)methylenecarbamate (8b)
Prepared according to general procedure 4, method B, also using HgCl2 (1 equiv) to afford the title material in 30% yield. 1H NMR (400 MHz, CD3OD): δ 7.33–7.16 (m, 7H), 6.83–6.78 (m, 3H), 3.68 (s, 2H), 3.52 (s, 2H), 2.64–2.61 (m, 2H), 2.32–2.30 (m, 2H), 1.99–1.97 (m, 2H), 1.75–1.70 (m, 2H), 1.53–1.48 (d, J = 20 Hz, 18H). MS (ESI) 538.50 [M + H]+.
tert-Butyl (tert-Butoxycarbonylamino)((1-phenethyl-4-(phenylamino)piperidin-4-yl)methylamino)methylenecarbamate (8c)
Prepared according to general procedure 4, method B, also using HgCl2 (1 equiv) to afford the title material in 30% yield. 1H NMR (600 MHz, CD3OD): δ 7.30–7.14 (m, 7H), 6.83–6.78 (m, 3H), 3.68–3.67 (d, J = 4, 2H), 2.95 (s, 2H), 2.88–2.42 (m, 4H), 2.05–2.02 (m, 2H), 1.82–1.76 (m, 4H), 1.51–1.47 (d, J = 16 Hz, 18H). MS (ESI) 552.50 [M + H]+.
1-((1-Methyl-4-(phenylamino)piperidin-4-yl)methyl)guanidine Hydrochloride (9a)
Prepared according to general procedure 5, method A, from intermediate 8a to afford the title material in quantitative yield. 1H NMR (600 MHz, CD3OD): δ 6.72–6.79 (m, 3H), 7.13–7.17 (m, 2H), 3.642 (s, 2H), 2.86 (s, 3H), 2.37–2.40 (m, 4H), 1.92–1.96 (m, 4H). 13C NMR (600 MHz, CD3OD): δ 157.9, 144.5, 129.3, 119.7, 117.6, 52.6, 50.2, 48.9, 48.5, 30.7. MS (ESI) 262.5 [M + H]+.
1-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methyl)guanidine Hydrochloride (9b)
Prepared according to general procedure 5, method A, from intermediate 8b to afford the title material in quantitative yield. 1H NMR (600 MHz, CD3OD): δ 7.59–7.13 (m, 7H), 6.89–6.75 (m, 3H), 3.47 (s, 2H), 3.34 (m, 2H), 2.41–2.38 (m, 4H), 2.06–2.01 (m, 4H). 13C NMR (600 MHz, CD3OD): δ 157.9, 144.5, 134, 131.4, 130.0, 129.1, 129.11, 119.3, 117.1, 60.3, 52.9, 48.4, 48.2, 30.2. MS (ESI) 338.0 [M + H]+.
1-((1-Phenethyl-4-(phenylamino)piperidin-4-yl)methyl)guanidine Hydrochloride (9c)
Prepared according to general procedure 5, method A, from intermediate 8c to afford the title material in quantitative yield. 1H NMR (600 MHz, CD3OD): δ 7.30–7.14 (m, 7H), 6.83–6.78 (m, 3H), 3.68–3.67 (d, J = 4, 2H), 2.95 (s, 2H), 2.88–2.42 (m, 4H), 2.05–2.02 (m, 2H), 1.82–1.76 (m, 4H). 13C NMR (600 MHz, CD3OD): δ 163.7, 145.2, 139.1, 129.35, 128.93, 128.65, 126.35, 119.8, 118.4, 60.6, 54.0, 49.3, 33.6. MS (ESI) 352.5 [M + H]+.
1-(Naphthalen-2-ylmethyl)piperidin-4-one (12d)
A solution of 2-bromomethylnaphthalene (5 g, 22.6 mmol), 4-piperidone ethylene ketal (3.23 g, 22.6 mmol), K2CO3 (9.12 g, 55.25 mmol), and KI (117 mg, 0.7 mmol) in methyl isobutyl ketone (300 mL) was heated at reflux for 5 h, cooled, and filtered. The residue was purified using silica gel chromatography (30:70 ethyl acetate–hexane) to afford 1-naphthalen-2-ylmethyl-piperidin-4-one ethylene ketal (11d). 1H NMR (600 MHz, CDCl3): δ 7.81–7.75 (m, 4H), 7.52–7.43 (m, 3H), 3.93 (s, 4H), 3.72 (s, 2H), 2.81–2.61 (m, 4H), 1.79–1.77 (m, 4H). MS (ESI) 284.36 [M + H]+. This material was hydrolyzed directly in a mixture of concentrated HCl (40 mL) and acetic acid (210 mL) at reflux for 18 h. The mixture was poured onto ice water, neutralized with 32% NaOH to pH 8, extracted with ethyl acetate, and dried using MgSO4. Evaporation afforded the title material (5g, 40%). 1H NMR (600 MHz, CDCl3): δ 7.83–7.75 (m, 4H), 7.54–7.45 (m, 3H), 3.77 (s, 2H), 2.80–2.78 (m, 4H), 2.48–2.46 (m, 4H). MS (ESI) 240.31 [M + H]+.
2-Amino-N-((1-benzylpiperidin-4-yl)methyl)-5-guanidinopentanamide Hydrochloride (15)
Prepared according to general procedure 3 starting with intermediate 13, to afford the corresponding Boc-protected arginine intermediate 14 in 70% yield. This material was deprotected using general procedure 5, method A, to afford the title material in 95–99% yield. 1H NMR (600 MHz, CD3OD): δ 7.62–7.61 (m, 2H), 7.40–7.48 (m, 3H), 4.34 (s, 4H), 3.66 (s, 2H), 3.50–3.47 (m, 2H), 3.31–3.24 (m, 3H), 3.19–3.07 (m, 4H), 1.99–1.91 (6H), 1.73–1.63 (m, 3H). 13C NMR (600 MHz, CD3OD): δ 169, 157.4, 131.4, 130, 129.3, 129.1, 66.9, 60.5, 53, 52.2, 40.64, 34, 28.6, 27, 24.5. MS (ESI) 361.5 [M + H]+.
N-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methyl)-2-cyanoacetamide (16a)
Prepared according to general procedure 4, method A, using cyanoacetic acid (1.3 equiv) to afford 1.26 g (50%) of the title material as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 7.32–7.27 (m, 5H), 7.21 (t, J = 7.7 Hz, 2H), 6.86 (t, J = 7.2 Hz, 1H), 6.75 (d, J = 7.8 Hz, 2H), 6.68–6.66 (m, 1H), 3.57–3.53 (m, 4H), 3.30 (s, 2H), 2.62–2.60 (m, 2H), 2.34–2.30 (m, 2H), 1.93–1.90 (m, 2H), 1.75–1.71 (m, 2H).
tert-Butyl 2-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methylamino)-2-oxoethylcarbamate (16b)
Prepared according to general procedure 4, method A, using Boc-gly-OH to afford 3.99 g (65%) of the title material as a white solid. 1H NMR (CDCl3): δ 7.30–7.23 (m, 5H), 7.16 (t, J = 7.6 Hz, 2H), 6.79 (t, J = 7.3 Hz, 1H), 6.73 (d, J = 8.0 Hz, 2H), 6.66 (br s, 1H), 5.45 (br s, 1H), 3.72 (d, J = 5.5 Hz, 2H), 3.53 (d, J = 5.4 Hz, 2H), 3.49 (s, 2H), 3.22 (br s, 1H), 2.58–2.55 (m, 2H), 2.31 (t, J = 10.1 Hz, 2H), 1.89–1.86 (m, 2H), 1.73–1.68 (m, 2H), 1.40 (s, 9H). MS (ESI) m/z 453 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(phenylamino)piperidin-4-yl)-12,12-dimethyl-3,10-dioxo-11-oxa-2,7,9-triazatridecan-8-ylidenecarbamate (16c)
Prepared according to general procedure 4, method A, using 4-(aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (5b) (0.25 g, 0.85 mmol) and 4-(2,3-bis(tert-butoxycarbonyl)guanidino)butanoic acid (0.38 g, 1.11 mmol) to afford 0.41 g (77%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.48 (s, 1H), 8.37 (br s, 1H), 7.39–7.13 (m, 7H), 6.79–6.72 (m, 3H), 6.35 (br s, 1H), 3.55–3.49 (m, 4H), 3.43 (q, J = 4.8 Hz, 2H), 2.57–2.54 (m, 2H), 2.33 (t, J = 7.2 Hz, 2H), 2.21 (t, J = 5.4 Hz, 2H), 1.92–1.34 (m, 24 H). MS (ESI) m/z 623 (M+ + 1). 4-(2,3-Bis(tert-butoxycarbonyl)guanidino)butanoic acid was prepared from 4-aminobutanoic acid according to general procedure 4, method B, also using HgCl2 (1.7 equiv), and matched reported values.30
tert-Butyl 1-(1-Benzyl-4-(phenylamino)piperidin-4-yl)-13,13-dimethyl-3,11-dioxo-12-oxa-2,8,10-triazatetradecan-9-ylidenecarbamate (16d)
Prepared according to general procedure 4, method A, using 4-(aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (5b) (0.25 g, 0.85 mmol) and 5-(2,3-bis(tert-butoxycarbonyl)guanidino)pentanoic acid (0.41 g, 1.11 mmol) to afford 0.43 g (80%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.46 (s, 1H), 8.29 (s, 1H), 7.35–7.14 (m, 7H), 6.79 (t, J = 5.7 Hz, 1H), 6.72–6.70 (m, 2H), 5.96–5.93 (m, 1H), 3.54–3.48 (m, 4H), 3.38 (q, J = 5.1 Hz, 2H), 2.58–2.55 (m, 2H), 2.30 (t, J = 6.9 Hz, 2H), 2.18 (t, J = 5.4 Hz, 2H), 1.86–1.36 (m, 26 H). MS (ESI) m/z 637 (M+ + 1). 5-(2,3-Bis(tert-butoxycarbonyl)guanidino)pentanoic acid was prepared from 5-aminopentanoic acid according to general procedure 4, method B, also using HgCl2 (1.7 equiv), and matched reported values.31
tert-Butyl N-[(1Z)-{[3-({[1-Benzyl-4-(phenylamino)piperidin-4-yl]methyl}carbamoyl)phenyl]amino}({[(tert-butoxy)carbonyl]imino})methyl]carbamate (16e)
Prepared according to general procedure 4, method A, using 4-(aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (5b) and 3-(2,3-bis(tert-butoxycarbonyl)guanidino)benzoic acid (1.67 g, 4.41 mmol) to yield 1.7 g (75% yield) of the title material. 1H NMR (400 MHz, CDCl3): δ 11.65 (s, 1H), 10.45 (s, 1H), 7.86 (s, 2H), 7.54–7.14 (m, 9H), 6.86–6.79 (m, 4H), 3.75 (d, J = 4.9 Hz, 2H), 3.55 (s, 2H), 3.46 (s, 1H), 2.65–2.62 (m, 2H), 2.42–2.38 (m, 2H), 1.97–1.82 (m, 4H), 1.52 (d, J = 27.2 Hz, 18H). MS(ESI) 657.7 [M + H]+. 3-(2,3-Bis(tert-butoxycarbonyl)guanidino)benzoic acid was prepared from 3-aminobenzoic acid according to general procedure 4, method B, also using HgCl2 (1.7 equiv), and matched reported values.32
tert-Butyl N-[(1Z)-{[4-({[1-Benzyl-4-(phenylamino)piperidin-4-yl]methyl}carbamoyl)phenyl]amino}({[(tert-butoxy)carbonyl]imino})methyl]carbamate (16f)
Prepared according to general procedure 4, method A, using 4-(aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (5b) and 4-(2,3-bis(tert-butoxycarbonyl)guanidino)benzoic acid (1 g, 3.45 mmol) to afford 1.5 g (66%) of the title material. 1H NMR (400 MHz): δ 11.61 (s, 1 H), 10.50 (s, 1 H), 7.73–7.67 (m, 4 H), 7.40–7–28 (m, 5 H), 7.21 (t, J = 7.7, 2 H), 6.88–6.82 (m, 3 H), 6.71 (s, 1 H), 3.75 (d, J = 5.1, 2 H), 3.66 (s, 2 H), 2.73 (s, 2 H), 2.56 (s, 2 H), 2.11–1.83 (m, 4 H), 1.53 (d, J = 13.6, 19 H). MS(ESI) 657.4 [M + H]+. 4-(2,3-Bis(tert-butoxycarbonyl)guanidino)benzoic acid was prepared from was prepared from 4-aminobenzoic acid according to general procedure 4, method B, also using HgCl2 (1.7 equiv), and matched reported values.33
tert-Butyl N-[(1Z)-({4-[({[1-Benzyl-4-(phenylamino)piperidin-4-yl]methyl}carbamoyl)methyl]phenyl}amino)({[(tert-butoxy)carbonyl]imino})methyl]carbamate (16g)
Prepared according to general procedure 4, method A, using 4-(aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (5b) (1.08 g, 3.66 mmol) and 4-((2,3-bis(tert-butoxycarbonyl)guanidino)methyl)benzoic acid (1.7 g, 4.49 mmol) to afford the title material in 84% yield. 1H NMR (400 MHz, CDCl3): δ 11.66 (s, 1H), 10.31 (s, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.28–7.11 (m, 9H), 6.77 (t, J = 7.0 Hz, 1H), 6.59 (d, J = 7.5 Hz, 2H), 5.88 (s, 1H), 3.48 (s, 7H), 2.54–2.51 (m, 2H), 2.30–2.28 (m, 2H), 1.79–1.76 (m, 2H), 1.68–1.63 (m, 2H), 1.53 (d, J = 9.9 Hz, 16H). MS(ESI) 671.7 [M + H]+. 4-((2,3-Bis(tert-butoxycarbonyl)guanidino)methyl)benzoic acid was prepared from 4-aminophenylacetic acid (2.42 g, 16 mmol) according to general procedure 4, method B, also using HgCl2 (1.7 equiv), to afford 1.02 g of target compound (16% yield). 1H NMR (400 MHz, CDCl3): δ 10.31 (s, 2H), 7.53 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 8.2 Hz, 2H), 3.57 (s, 2H), 1.51 (s, 18H). MS(ESI) 394.5 [M + H]+.
N-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methyl)-3-guanidinopropanamide Tritriflate (17a)
Prepared according to general procedure 5, method B, using 19a (0.3 g, 0.49 mmol) to afford 0.29 g (77%) of the title material. 1H NMR (500 MHz, DMSO-d6): δ 9.97 (s, 1H), 8.13–7.97 (m, 1H), 7.50–6.61 (m, 15H), 5.26 (br s, 1H), 4.30 (s, 2H), 3.32–3.11 (m, 8H), 2.45–2.37 (m, 2H), 2.11–1.84 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 170.70, 158.81 (q, J = 34.4 Hz), 156.99, 145.85, 131.32, 129.77, 129.55, 128.83, 128.81, 117.29, 116.25 (q, J = 292.2 Hz), 116.14, 58.91, 53.16, 47.14, 44.49, 37.19, 34.40, 29.17. MS (ESI) m/z 409 (M+ + 1).
2-Amino-N-((1-benzyl-4-(phenylamino)piperidin-4-yl)methyl)acetamide (17b)
Prepared according to general procedure 5, method B, using 16b (5.5 g, 12.15 mmol) using the following modified workup and purification: Methylene chloride (150 mL) and a 10% solution of NaOH (80 mL) were added. The layers were separated, and the organic layer was washed with water and brine. The solvent was dried and evaporated. The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol saturated with ammonia (10:0 → 8:2) as the eluent to give 4.02 g (92%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.48–7.46 (m, 1H), 7.27–7.21 (m, 5H), 7.14 (t, J = 7.7 Hz, 2H), 6.77 (t, J = 7.2 Hz, 1H), 6.72 (d, J = 7.7 Hz, 2H), 3.53 (d, J = 5.7 Hz, 2H), 3.47 (s, 2H), 3.28 (s, 2H), 2.57–2.55 (m, 2H), 2.30 (t, J = 9.7 Hz, 2H), 1.88–1.85 (m, 2H), 1.73–1.68 (m, 2H), 1.62 (br s, 2H). 13C NMR (125 MHz, CDCl3): δ 173.15, 145.53, 138.28, 129.37, 129.19, 128.33, 127.18, 119.31, 117.62, 63.12, 54.51, 49.12, 45.60, 44.92, 33.79. MS (ESI) m/z 353 (M+ + 1).
N-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methyl)-4-guanidinobutanamide (17c)
Prepared according to general procedure 5, method B, (no purification) using 16c (0.19 g, 0.30 mmol). 1H NMR (DMSO-d6): δ 10.12–10.10 (m, 1H), 7.99–7.89 (m, 2H), 7.50–7.07 (m, 10H), 6.81–6.63 (m, 3H), 4.37–4.30 (m, 2H), 3.32–3.05 (m, 8H), 2.23–1.66 (m, 8H). 13C NMR (DMSO-d6): δ 172.18, 158.93, 158.58, 156.99, 145.74, 131.20, 129.63, 129.42, 128.74, 128.69, 117.50, 117.47, 117.27, 116.15, 114.58, 114.56, 58.88, 53.09, 47.09, 44.48, 31.83, 29.18, 24.60. MS (ESI) m/z 423 (M+ + 1).
N-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methyl)-5-guanidinopentanamide (17d)
Prepared according to general procedure 5, method B, (no purification) using 16d (0.20 g, 0.31 mmol). 1H NMR (CD3OD): δ 7.51–7.45 (m, 5H), 7.21–7.16 (m, 3H), 6.88–6.76 (m, 2H), 4.32 (s, 2H), 3.49–3.17 (m, 7H), 2.29–2.26 (m, 5H), 1.99–1.94 (m, 2H), 1.64–1.57 (m, 4H). 13C NMR (CD3OD): δ 174.79, 157.28, 145.09, 130.94, 129.81, 128.93, 128.91, 128.81, 118.57, 116.79, 60.14, 53.23, 45.04, 40.68, 34.74, 29.87, 27.99, 22.31. HRMS (ESI) calcd for C25H37N6O [M + H]+ 437.3029, found 437.3044.
N-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methyl)-3-guanidinobenzamide (17e)
Prepared according to general procedure 5, method B, (no purification) using 16e (0.1 g, 0.152 mmol). 1H NMR (500 MHz, DMSO-d6): δ 10.33 (s, 1H), 9.70 (s, 1H), 8.59 (s, 1H), 7.78–7.73 (m, 5H), 7.51–7.39 (m, 6H), 7.19–7.05 (m, 2H), 6.89 (d, J = 7.9 Hz, 2H), 6.66 (t, J = 7.1 Hz, 1H), 4.32 (d, J = 3.6 Hz, 2H), 3.55 (d, J = 5.7 Hz, 1H), 3.38–3.08 (m, 4H), 2.20 (d, J = 14.5 Hz, 2H), 2.04–1.90 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 166.76, 156.64, 146.39, 136.33, 136.07, 131.74, 130.17, 130.10, 129.96, 129.31, 129.22, 127.70, 125.74, 123.77, 117.78, 116.64, 59.42, 54.07, 47.65, 45.61, 29.77. HRMS (ESI) calcd for C27H33N6O 457.2716, found 457.2708 [M + H]+.
N-{[1-Benzyl-4-(phenylamino)piperidin-4-yl]methyl}-4-carbamimidamidobenzamide (17f)
Prepared according to general procedure 5, method B, (no purification) using 16f (0.1 g, 0.152 mmol). 1H NMR (500 MHz, DMSO-d6): δ 10.36–10.31 (m, 1H), 9.65 (s, 1H), 8.54 (s, 1H), 7.93–7.92 (m, 2H), 7.79 (s, 4H), 7.58–7.41 (m, 4H), 7.32–7.31 (m, 2H), 7.14–7.09 (m, 2H), 6.89–6.82 (m, 2H), 6.66 (t, J = 6.3 Hz, 1H), 4.32 (s, 2H), 3.55–3.54 (m, 2H), 3.25–3.23 (m, 2H), 3.18–3.13 (m, 2 H), 2.20–2.17 (m, 2H), 1.96 (t, J = 13.4 Hz, 2H). 13C NMR (126 MHz, DMSO-d6): δ 166.64, 156.27, 146.39, 139.04, 131.89, 131.75, 130.16, 129.98, 129.31, 129.24, 123.31, 118.24, 117.71, 116.57, 115.86, 59.40, 54.01, 47.67, 45.58, 29.69. HRMS (ESI) calcd for C27H33N6O 457.2716, found 457.2697 [M + H]+.
N-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methyl)-2-(4-guanidinophenyl)acetamide (17g)
Prepared according to general procedure 5, method B, (no purification) using 16g (0.19 g, 0.283 mmol). 1H NMR (500 MHz, DMSO-d6): δ 10.02 (s, 1H), 9.89 (s, 1H), 8.17 (s, 1H), 7.65–7.42 (m, 9H), 7.30 (d, J = 7.9 Hz, 2H), 7.15 (d, J = 7.8 Hz, 2H), 7.09 (t, J = 7.5 Hz, 2H), 6.81 (d, J = 7.8 Hz, 2H), 6.64 (t, J = 7.2 Hz, 1H), 4.32 (d, J = 2.3 Hz, 2H), 3.47 (s, 2H), 3.33 (d, J = 5.8 Hz, 2H), 3.25–3.22 (m, 2H), 3.15–3.09 (m, 2H), 2.15–2.12 (m, 2H), 1.87 (t, J = 13.3 Hz, 2H). 13C NMR (126 MHz, DMSO-d6): δ 156.56, 146.34, 134.89, 134.22, 131.66, 130.74, 129.89, 129.20, 124.54, 118.96, 117.62, 116.52, 116.45, 116.00, 59.31, 55.01, 53.47, 49.01, 47.53, 41.88, 29.70. HRMS (ESI) calcd for C28H35N6O 471.2872 [M + H]+, found 471.2856.
3-Amino-N-((1-benzyl-4-(phenylamino)piperidin-4-yl)methyl)propanamide (18a)
Prepared according to general procedure 2, method B, using 16a (0.5 g, 1.38 mmol). The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol saturated with ammonia (9:1 → 8:2) as the eluent to give 0.38 g (76%) of the title material as a white solid. 1H NMR (CDCl3): δ 7.30–7.23 (m, 5H), 7.16 (t, J = 8.0 Hz, 2H), 7.07–7.05 (m, 1H), 6.78 (t, J = 7.3 Hz, 1H), 6.73 (d, J = 7.8 Hz, 2H), 3.55 (d, J = 5.5 Hz, 2H), 3.49 (s, 2H), 2.94 (t, J = 5.7 Hz, 2H), 2.58–2.55 (m, 2H), 2.34–2.27 (m, 4H), 1.90–1.87 (m, 2H), 1.79–1.69 (m, 2H), 1.65 (br s, 2H). MS (ESI) m/z 367 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(phenylamino)piperidin-4-yl)-11,11-dimethyl-3,9-dioxo-10-oxa-2,6,8-triazadodecan-7-ylidenecarbamate (19a)
Prepared according to general procedure 4, method B, using 18a (0.32 g, 0.88 mmol) to afford 0.47 g (87%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.47 (s, 1H), 8.77–8.75 (m, 1H), 7.31–7.23 (m, 5H), 7.18 (t, J = 7.6 Hz, 2H), 6.81 (t, J = 7.2 Hz, 1H), 6.74 (d, J = 8.0 Hz, 2H), 6.15–6.13 (m, 1H), 3.74–3.70 (m, 2H), 3.57 (d, J = 5.8 Hz, 2H), 3.51 (s, 2H), 2.60–2.57 (m, 2H), 2.44 (t, J = 6.0 Hz, 2H), 2.35–2.31 (m, 2H), 1.89–1.87 (m, 2H), 1.76–1.72 (m, 2H), 1.50 (s, 9H), 1.46 (s, 9H). MS (ESI) m/z 609 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(phenylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (20)
Prepared according to general procedure 4, method B, using 17b (0.2 g, 0.57 mmol) to afford 0.285 g (84%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.36 (s, 1H), 8.87 (s, 1H), 7.30–7.25 (m, 5H), 7.16 (s, 2H), 6.79 (s, 1H), 6.72 (s, 2H), 6.54 (s, 1H), 4.00 (s, 2H), 3.56 (s, 2H), 3.51 (s, 2H), 3.50 (br s, 1H), 2.57 (s, 2H), 2.37 (s, 2H), 1.90–1.88 (m, 2H), 1.76–1.74 (m, 2H), 1.49 (s, 9H), 1.47 (s, 9H). MS (ESI) m/z 595 (M+ + 1).
N-((1-Benzyl-4-(phenylamino)piperidin-4-yl)methyl)-2-guanidinoacetamide tritriflate (21)
Prepared according to general procedure 5, method B, using 20 (0.18 g, 0.3 mmol) to afford 0.2 g (90%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 7.49–7.42 (m, 5H), 7.12–7.09 (m, 2H), 6.90–6.69 (m, 3H), 4.27 (s, 2H), 3.91 (s, 2H), 3.48–3.29 (m, 6H), 2.26–1.99 (m, 4H). 13C NMR (125 MHz, CD3OD): δ 170.14, 163.18, 159.37, 146.64, 132.33, 131.10, 130.34, 130.23, 130.16, 119.86, 118.25 (q, J = 282.7 Hz), 118.15, 61.47, 54.60, 49.01, 46.59, 44.74, 31.21. MS (ESI) m/z 395 (M+ + 1).
1-Benzyl-3-(phenylamino)piperidine-3-carbonitrile (23)
To a mixture of 1-benzyl-3-piperidone (2 g, 1.06 mmol) and aniline (0.96 mL, 1.06 mmol) in 1,2-dichloroethane (40 mL) was added titanium isopropoxide (4 mL, 1.37 mmol). The reaction mixture was stirred at room temperature for 21 h and then diethylaluminum cyanide (1 M in toluene, 21 mL, 2.11 mmol) was added. After 3 h of stirring, ethyl acetate (100 mL) and a solution of saturated sodium bicarbonate were added. The mixture was stirred for 10 min and filtered through a pad of Celite. The Celite was washed with water and ethyl acetate, and the layers of the filtrate were separated. The organic layer was washed with brine, dried, and evaporated. The residue was purified by chromatography on a silica gel column using ethyl acetate and hexanes (4:6) as the eluent to give 2.36 g (74%) of the title material as a white solid. 1H NMR (CDCl3): δ 7.41–7.32 (m, 5H), 7.26 (t, J = 8.0 Hz, 2H), 6.96 (d, J = 7.7 Hz, 2H), 6.90 (t, J = 7.4 Hz, 1H), 4.35 (s, 1H), 3.62 (s, 2H), 2.88–2.80 (m, 2H), 2.60 (s, 1H), 2.40 (s, 1H), 2.23 (s, 1H), 1.91 (s, 1H), 1.67 (s, 2H). MS (ESI) m/z 292 (M+ + 1).
3-(Aminomethyl)-1-benzyl-N-phenylpiperidin-3-amine (24)
Prepared according to general procedure 2, method B, to afford 1.34 g (78%) of the title material as a yellow oil. 1H NMR (500 MHz, CDCl3): δ 7.38–7.21 (m, 7H), 6.80 (br s, 3H), 4.25 (br s, 1H), 3.62–3.53 (m, 2H), 3.23 (br s, 2H), 2.91–2.68 (m, 3H), 2.02 (s, 3H), 1.58–1.15 (m, 4H). MS (ESI) m/z 296 (M+ + 1).
tert-Butyl 1-(1-Benzyl-3-(phenylamino)piperidin-3-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (25)
Prepared according to general procedure 4, method C, to afford 0.23 g (37%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.36 (s, 1H), 8.94 (s, 1H), 7.38–7.28 (m, 5H), 7.19 (t, J = 7.6 Hz, 2H), 6.82 (t, J = 7.2 Hz, 1H), 6.73 (d, J = 7.7 Hz, 2H), 6.55–6.53 (m, 1H), 4.50 (br s, 1H), 4.05–4.04 (m, 2H), 3.59–3.42 (m, 4H), 2.79–2.78 (m, 1H), 2.70–2.68 (m, 1H), 2.06–1.98 (m, 4H), 1.63–1.40 (m, 21H). MS (ESI) m/z 595 (M+ + 1).
N-((1-Benzyl-3-(phenylamino)piperidin-3-yl)methyl)-2-guanidinoacetamide Tritriflate (26)
Prepared according to general procedure 5, method B, to afford 0.16 g (65%) of the title material as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 9.26 (br s, 1H), 8.26 (s, 1H), 7.90 (s, 1H), 7.57–7.17 (m, 8H), 7.00 (s, 2H), 6.68–6.65 (m, 3H), 5.24 (br s, 1H), 4.32–4.17 (m, 2H), 3.88 (s, 2H), 3.50–2.95 (m, 6H), 2.00–1.59 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 168.15, 158.65 (q, J = 31.3 Hz), 157.81, 145.12, 131.49, 129.53, 129.14, 128.73, 128.72, 118.46, 117.23, 117.15 (q, J = 297.2 Hz), 59.81, 55.93, 55.02, 52.47, 43.68, 43.38, 28.62, 18.28. MS (ESI) m/z 395 (M+ + 1).
1-Benzyl-4-(naphthalen-1-ylamino)piperidine-4-carbonitrile (27a)
Prepared according to general procedure 1, using N-benzylpiperidone, 1-naphthylamine, and KCN to afford 3.2 g (59%) of the target material as a thick oil. 1H NMR (CDCl3): δ 7.91–7.84 (m, 2H), 7.52–7.27 (m, 10H), 4.24 (s, 1H), 3.58 (s, 2H), 2.85–2.82 (m, 1H), 2.77–2.74 (m, 1H), 2.57–2.43 (m, 4H), 2.14–2.09 (m, 2H). MS (ESI) m/z 342 (M+ + 1).
1-Benzyl-4-(3,5-dimethoxyphenylamino)piperidine-4-carbonitrile (27b)
Prepared according to general procedure 1, using N-benzylpiperidone, 3,5-dimethoxyaniline, and TMSCN to afford 6.44 g (69%) of the title material as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 7.34–7.29 (m, 5H), 6.11–6.07 (m, 3H), 3.78 (s, 6H), 3.58 (s, 2H), 2.83–2.81 (m, 2H), 2.49 (t, J = 10.6 Hz, 2H), 2.38 (d, J = 12.7 Hz, 2H), 1.96–1.94 (m, 2H). MS (ESI) m/z 352 (M+ + 1).
1-Benzyl-4-(3-methoxyphenylamino)piperidine-4-carbonitrile (27c)
Prepared according to general procedure 1, using N-benzylpiperidone, m-anisidine, and TMSCN to afford 4.26 g (63%) of the title material as a white solid. 1H NMR (DMSO-d6): δ 7.31–7.25 (m, 5H), 7.07 (t, J = 8.0 Hz, 1H), 6.44 (d, J = 8.0 Hz, 1H), 6.40 (s, 1H), 6.32 (d, J = 8.0 Hz, 1H), 6.06 (s, 1H), 3.68 (s, 3H), 3.51 (s, 2H), 2.76–2.74 (m, 2H), 2.29–2.27 (m, 4H), 1.85–1.80 (m, 2H). MS (ESI) m/z 322 (M+ + 1).
1-Benzyl-4-(3,4-dichlorophenylamino)piperidine-4-carbonitrile (27d)
Prepared according to general procedure 1, using N-benzylpiperidone, 3,4-dichloroaniline, and TMSCN to afford 7.31 g (77%) of the title material as a light brown solid. 1H NMR (DMSO-d6): δ 7.39–7.24 (m, 6H), 7.01 (d, J = 2.3 Hz, 1H), 6.83 (dd, J = 8.8, 2.4 Hz, 1H), 6.56 (s, 1H), 3.50 (s, 2H), 2.76–2.73 (m, 2H), 2.32–2.29 (m, 4H), 1.85–1.82 (m, 2H). MS (ESI) m/z 360 (M+ + 1).
4-(Aminomethyl)-1-benzyl-N-(naphthalen-1-yl)piperidin-4-amine (28a)
Prepared from 27a (3 g, 8.7 mmol) according to general procedure 2, method B, to afford 2.7 g (89%) of the title material as a light brown solid. 1H NMR (CDCl3): δ 7.95–7.93 (m, 1H), 7.86–7.83 (m, 1H), 7.52–7.50 (m, 2H), 7.35–7.30 (m, 7H), 6.87–6.85 (m, 1H), 4.24 (br s, 1H), 3.52 (s, 2H), 3.06 (s, 2H), 2.70–2.67 (m, 2H), 2.39 (t, J = 10.7 Hz, 2H), 2.25–2.18 (m, 2H), 1.79–1.73 (m, 2H), 1.45 (br s, 2H). MS (ESI) m/z 346 (M+ + 1).
4-(Aminomethyl)-1-benzyl-N-(3,5-dimethoxyphenyl)piperidin-4-amine (28b)
Prepared from 27b (2.89 g, 8.24 mmol) according to general procedure 2, method B, to afford 2.8 g (97%) of the title material as a greenish oil. 1H NMR (CDCl3, 50 °C): δ 7.31–7.22 (m, 5H), 5.97–5.93 (m, 3H), 3.73 (s, 6H), 3.51 (s, 2H), 2.92 (br s, 2H), 2.59–2.53 (m, 3H), 2.35 (br s, 2H), 2.15–1.69 (m, 6H). MS (ESI) m/z 356 (M+ + 1).
4-(Aminomethyl)-1-benzyl-N-(3-methoxyphenyl)piperidin-4-amine (28c)
Prepared from 27c (2.3 g, 7.16 mmol) according to general procedure 2, method B, to afford 2.2 g (95%) of the title material as a brown oil. 1H NMR (500 MHz, DMSO-d6): δ 7.29–7.22 (m, 5H), 6.92 (t, J = 7.8 Hz, 1H), 6.34–6.31 (m, 2H), 6.14 (d, J = 6.3 Hz, 1H), 4.82 (s, 1H), 3.65 (s, 3H), 3.18 (s, 2H), 2.70–2.75 (s, 2H), 2.50–2.28 (m, 6H), 1.91–1.89 (m, 2H), 1.55–1.53 (m, 2H). MS (ESI) m/z 326 (M+ + 1).
4-(Aminomethyl)-1-benzyl-N-(3,4-dichlorophenyl)piperidin-4-amine (28d)
Prepared from 27d (3 g, 8.3 mmol) according to general procedure 2, method B, to afford 2.8 g (95%) of the title material as a thick colorless oil. 1H NMR (CDCl3): δ 7.31–7.24 (m, 5H), 7.16 (d, J = 8.7 Hz, 1H), 6.81 (d, J = 2.6 Hz, 1H), 6.55 (dd, J = 8.7, 2.6 Hz, 1H), 3.55–3.43 (m, 3H), 2.86 (s, 2H), 2.63–2.60 (m, 2H), 2.26 (t, J = 10.4 Hz, 2H), 1.94 (d, J = 13.6 Hz, 2H), 1.67–1.60 (m, 2H), 1.44 (s, 2H). MS (ESI) m/z 364 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(naphthalen-1-ylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (29a)
Prepared from 28a (0.35 g, 1.02 mmol) according to general procedure 4, method C, to afford 0.33 g (50%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.24 (s, 1H), 8.79 (s, 1H), 7.90–7.88 (m, 1H), 7.82–7.81 (m, 1H), 7.51–7.47 (m, 2H), 7.31–7.25 (m, 7H), 6.86–6.84 (m, 1H), 6.46–6.45 (m, 1H), 3.96 (d, J = 5.1 Hz, 2H), 3.72 (d, J = 5.6 Hz, 2H), 3.51 (s, 2H), 2.64–2.62 (m, 2H), 2.44–2.40 (m, 2H), 2.17–2.14 (m, 2H), 1.89–1.85 (m, 2H), 1.46 (m, 18H). MS (ESI) m/z 645 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(3,5-dimethoxyphenylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (29b)
Prepared from 28b (0.39 g, 1.10 mmol) according to general procedure 4, method C, to afford 0.34 g (48%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.36 (s, 1H), 8.85 (s, 1H), 7.31–7.25 (m, 5H), 6.42 (s, 1H), 5.94–5.89 (m, 3H), 3.99 (d, J = 5.3 Hz, 2H), 3.76 (s, 6H), 3.59 (d, J = 5.6 Hz, 2H), 3.52 (s, 2H), 2.59–2.57 (m, 2H), 2.37 (t, J = 9.3 Hz, 2H), 1.92 (d, J = 13.5 Hz, 2H), 1.78–1.74 (m, 2H), 1.51 (s, 9H), 1.49 (s, 9H). MS (ESI) m/z 655 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(3-methoxyphenylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (29c)
Prepared from 28c (0.39 g, 1.10 mmol) according to general procedure 4, method C, to afford 0.46 g (67%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.36 (s, 1H), 8.85 (s, 1H), 7.31–7.25 (m, 5H), 7.07 (t, J = 8.0 Hz, 1H), 6.48 (s, 1H), 6.36–6.32 (m, 2H), 6.29 (s, 1H), 3.99 (d, J = 5.0 Hz, 2H), 3.77 (s, 3H), 3.58 (d, J = 5.3 Hz, 2H), 3.52 (br s, 1H), 3.51 (s, 2H), 2.58–2.56 (m, 2H), 2.37 (t, J = 9.5 Hz, 2H), 1.91 (d, J = 13.0 Hz, 2H), 1.77–1.73 (m, 2H), 1.51 (s, 9H), 1.48 (s, 9H). MS (ESI) m/z 625 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(3,4-dichlorophenylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (29d)
Prepared from 28d (0.4 g, 1.10 mmol) according to general procedure 4, method C, to afford 0.24 g (33%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.31 (s, 1H), 8.82 (s, 1H), 7.30–7.24 (m, 5H), 7.17 (d, J = 8.7 Hz, 1H), 6.80 (d, J = 2.6 Hz, 1H), 6.66–6.63 (m, 1H), 6.56 (dd, J = 8.7, 2.6 Hz, 1H), 3.97 (d, J = 5.5 Hz, 2H), 3.62 (s, 1H), 3.52 (d, J = 5.7 Hz, 2H), 3.50 (s, 2H), 2.57–2.55 (m, 2H), 2.35–2.30 (m, 2H), 1.90–1.86 (m, 2H), 1.77–1.71 (m, 2H), 1.49 (s, 9H), 1.46 (s, 9H). MS (ESI) m/z 663 (M+ + 1).
N-((1-Benzyl-4-(naphthalen-1-ylamino)piperidin-4-yl)methyl)-2-guanidinoacetamide Tritriflate (30a)
Prepared from 29a (0.2 g, 0.31 mmol) according to general procedure 5, method B, to afford 0.18 g (74%) of the title material as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 10.35 (s, 1H), 8.29–8.16 (m, 2H), 7.91–7.22 (m, 14H), 6.90 (d, J = 7.23 Hz, 1H), 5.33 (s, 1H), 4.40–4.27 (m, 2H), 3.96–3.74 (m, 2H), 3.55–3.53 (m, 2H), 3.25–3.07 (m, 4H), 2.52–2.49 (m, 2H), 2.03–1.97 (m, 2H). 13C NMR (125 MHz, DMSO-d6): δ 167.98, 158.95 (q, J = 31.9 Hz), 157.74, 140.42, 134.39, 131.37, 129.64, 129.52, 128.72, 128.05, 126.10, 125.72, 125.34, 124.48, 118.26, 117.74, 115.89, 115.63 (q, J = 526.6 Hz), 58.80, 53.60, 47.18, 44.28, 43.36, 28.94. MS (ESI) m/z 445 (M+ + 1).
N-((1-Benzyl-4-(3,5-dimethoxyphenylamino)piperidin-4-yl)methyl)-2-guanidinoacetamide Tritriflate (30b)
Prepared from 29b (0.17 g, 0.26 mmol) according to general procedure 5, method B, to afford 0.17 g (83%) of the title material as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 10.10 (s, 1H), 8.24 (s, 1H), 7.75 (s, 1H), 7.50–7.43 (m, 9H), 5.97 (s, 2H), 5.81 (s, 1H), 5.46 (s, 1H), 4.30 (s, 2H), 3.83–3.09 (m, 14H), 2.12–1.88 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 167.89, 160.89, 158.88 (q, J = 31.0 Hz), 157.60, 147.54, 131.29, 129.80, 129.52, 128.78, 116.11 (q, J = 296.3 Hz), 94.27, 89.50, 58.86, 54.77, 53.15, 47.09, 44.48, 43.34, 29.18. MS (ESI) m/z 455 (M+ + 1).
N-((1-Benzyl-4-(3-methoxyphenylamino)piperidin-4-yl)methyl)-2-guanidinoacetamide Tritriflate (30c)
Prepared from 29c (0.2 g, 0.32 mmol) according to general procedure 5, method B, to afford 0.13 g (53%) of the title material as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 10.24 (s, 1H), 8.21 (s, 1H), 7.80 (s, 1H), 7.51–7.18 (m, 9H), 6.98 (t, J = 7.9 Hz, 1H), 6.40–6.37 (m, 2H), 6.21 (d, J = 7.8 Hz, 1H), 5.39 (br s, 1H), 4.30 (s, 2H), 3.85–3.83 (m, 2H), 3.67 (s, 3H), 3.36–3.08 (m, 6H), 2.14–1.88 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 167.93, 160.05, 158.95 (q, J = 31.9 Hz), 157.71, 147.20, 131.32, 131.10, 129.83, 129.54, 128.80, 117.09 (q, J = 297.0 Hz), 108.51, 102.65, 101.81, 58.87, 54.68, 53.18, 47.11, 44.60, 43.36, 29.20. MS (ESI) m/z 425 (M+ + 1).
N-((1-Benzyl-4-(3,4-dichlorophenylamino)piperidin-4-yl)methyl)-2-guanidinoacetamide Tritriflate (30d)
Prepared from 29d (0.22 g, 0.33 mmol) according to general procedure 5, method B, to afford 0.19 g (72%) of the title material as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 10.13 (s, 1H), 8.25 (s, 1H), 7.70 (s, 1H), 7.49–7.13 (m, 10H), 6.98 (s, 1H), 6.77–6.75 (m, 1H), 5.90 (s, 1H), 4.29 (s, 2H), 3.83 (s, 2H), 3.59–3.04 (m, 6H), 2.14–1.88 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 167.85, 158.57 (q, J = 31.2 Hz), 157.52, 146.16, 131.24, 130.97, 130.27, 129.74, 129.50, 128.76, 117.65, 117.13 (q, J = 297.3 Hz), 116.34, 115.10, 58.82, 53.25, 46.96, 44.00, 43.33, 28.96. MS (ESI) m/z 463 (M+ + 1).
tert-Butyl 2-((1-Benzyl-4-phenylpiperidin-4-yl)methylamino)-2-oxoethylcarbamate (32)
Prepared according to general procedure 4, method D (step I), using 1-(1-benzyl-4-phenylpiperidin-4-yl)methanamine (31, 0.75 g, 2.66 mmol) to afford 0.9 g (78%) of the title material as a white solid. 1H NMR (CDCl3): δ 7.39–7.35 (m, 2H), 7.31–7.23 (m, 8H), 5.73 (br s, 1H), 5.11 (br s, 1H), 3.65 (d, J = 5.8 Hz, 2H), 3.45 (s, 4H), 2.61–2.59 (m, 2H), 2.34 (br s, 2H), 2.34–2.10 (m, 2H), 1.93–1.89 (m, 2H), 1.42 (s, 9H). MS (ESI) m/z 438 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-phenylpiperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (33)
Prepared according to general procedure 4, method D (step II), from intermediate 32 (0.3 g, 0.685 mmol) to afford 0.35 g (89%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.36 (s, 1H), 8.74–8.72 (m, 1H), 7.36–7.22 (m, 10H), 5.74–5.73 (m, 1H), 3.89 (d, J = 5.3 Hz, 2H), 3.47–3.46 (m, 4H), 2.61–2.59 (m, 2H), 2.36 (br s, 2H), 2.14–2.12 (m, 2H), 1.94–1.91 (m, 2H), 1.50 (s, 9H), 1.47 (s, 9H). MS (ESI) m/z 580 (M+ + 1).
N-((1-Benzyl-4-phenylpiperidin-4-yl)methyl)-2-guanidinoacetamide Ditriflate (34)
Prepared according to general procedure 5, method B, from intermediate 33 (0.12 g, 0.207 mmol) to afford the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 7.41–7.25 (m, 10H), 4.03 (s, 2H), 3.85 (s, 2H), 3.37–3.23 (m, 4H), 2.79 (br s, 2H), 2.39–2.37 (m, 2H), 2.15–2.11 (m, 2H). 13C NMR (125 MHz, CD3OD): δ 169.96, 163.14 (q, J = 34.2 Hz), 159.32, 142.36, 132.09, 131.93, 130.48, 130.19, 129.98, 128.11, 127.87, 118.12 (q, J = 290.9 Hz), 61.84, 50.34, 44.70, 42.28, 31.13. MS (ESI) m/z 380 (M+ + 1).
1,4-Dibenzylpiperidine-4-carbonitrile (36)
Prepared according to general procedure 6 using benzyl bromide to afford 1.29 g (89%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.39–7.28 (m, 10H), 3.56 (s, 2H), 2.91–2.89 (m, 4H), 2.34 (t, J = 12.1 Hz, 2H), 1.88 (d, J = 13.2 Hz, 2H), 1.73–1.67 (m, 2H). MS (ESI) m/z 291 (M+ + 1).
1-Benzyl-4-phenethylpiperidine-4-carbonitrile (37)
Prepared according to general procedure 6 using (2-bromoethyl)benzene to afford 1.25 g (86%) of the title material as a colorless oil. 1H NMR (CDCl3): δ 7.35–7.20 (m, 10H), 3.55 (s, 2H), 2.89–2.81 (m, 4H), 2.37 (t, J = 12 Hz, 2H), 1.96 (d, J = 13.1 Hz, 2H), 1.88–1.83 (m, 2H), 1.64–1.57 (m, 2H). MS (ESI) m/z 305 (M+ + 1).
4-(Aminomethyl)-1-benzyl-N-phenylpiperidin-4-amine (38)
Prepared from 36 (0.53 g, 1.82 mmol) using general procedure 2, method B, to afford 0.33 g (61%) of the title material as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.35–7.17 (m, 10H), 3.56 (s, 2H), 2.69–2.45 (m, 8H), 1.56–1.47 (m, 4H), 1.18 (br s, 2H). MS (ESI) m/z 295 (M+ + 1).
(1-Benzyl-4-phenethylpiperidin-4-yl)methanamine (39)
Prepared from 37 (0.436 g, 1.43 mmol) using general procedure 2, method B, to afford 0.4 g (90%) of the title material as a colorless oil. 1H NMR (CDCl3): δ 7.36–7.23 (m, 10H), 3.55 (s, 2H), 2.69 (s, 2H), 2.57–2.43 (m, 6H), 1.70–1.66 (m, 2H), 1.56–1.54 (m, 4H), 1.18 (br s, 2H). MS (ESI) m/z 309 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(phenylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (40)
Prepared from 38 (0.32 g, 1.08 mmol) according to general procedure 4, method C, to afford 0.31 g (48%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.45 (s, 1H), 8.89 (s, 1H), 7.33–7.20 (m, 7H), 7.10 (d, J = 7.1 Hz, 2H), 6.89–6.88 (m, 1H), 4.04 (d, J = 5.5 Hz, 2H), 3.54 (s, 2H), 3.20(d, J = 5.4 Hz, 2H), 2.63–2.58 (m, 4H), 2.43–2.41 (m, 2H), 1.52–1.43 (m, 22H). MS (ESI) m/z 594 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-phenethylpiperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (41)
Prepared from 39 (0.374 g, 1.21 mmol) according to general procedure 4, method D, to afford 0.258 g (76%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.41 (s, 1H), 8.93 (s, 1H), 7.34–7.14 (m, 10H), 6.97 (s, 1H), 4.06–4.05 (m, 2H), 3.55 (s, 2H), 3.33–3.32 (m, 2H), 2.56–2.50 (m, 6H), 1.57–1.55 (m, 6H), 1.51 (s, 9H), 1.41 (s, 9H). MS (ESI) m/z 608 (M+ + 1).
N-((1,4-Dibenzylpiperidin-4-yl)methyl)-2-guanidinoacetamide Ditriflate (42)
Prepared from 40 (0.13 g, 0.22 mmol) according to general procedure 5, method B, to afford 0.096 g (71%) of the title material as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 9.89–9.48 (m, 1H), 8.29–8.15 (m, 1H), 7.79–7.71 (m, 1H), 7.52–7.13 (m, 14H), 4.38–4.27 (m, 2H), 3.98–3.88 (m, 2H), 3.28–3.11 (m, 4H), 2.93–2.82 (m, 2H), 2.54–2.50 (m, 2H), 1.64–1.44 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 168.23, 168.02, 158.60 (q, J = 31.2 Hz), 137.00, 136.52, 131.17, 130.91, 130.59, 130.04, 129.83, 129.50, 128.90, 128.80, 128.15, 127.95, 126.42, 126.32, 116.16 (q, J = 297.5 Hz), 58.88, 58.63, 47.44, 47.08, 45.55, 43.52, 41.12, 36.38, 35.44, 35.31, 28.32, 28.16. MS (ESI) m/z 394 (M+ + 1).
N-((1-Benzyl-4-phenethylpiperidin-4-yl)methyl)-2-guanidinoacetamide Ditriflate (43)
Prepared from 41 (0.1 g, 0.197 mmol) according to general procedure 5, method B, to afford 0.11 g (85%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 7.54–7.52 (m, 5H), 7.29–7.18 (m, 5H), 4.37–4.36 (m, 2H), 4.01 (m, 2H), 3.52–3.18 (m, 6H), 2.67–2.63 (m, 2H), 1.81–1.60 (m, 6H). 13C NMR (125 MHz, 45 °C, CD3OD): δ 170.25, 163.06 (q, J = 34.5 Hz), 159.49, 143.32, 132.19, 131.02, 130.37, 130.18, 129.37, 129.33, 126.79, 118.25 (q, J = 289.2 Hz), 61.29, 49.62, 44.96, 44.86, 41.94, 35.98, 30.66, 30.15. MS (ESI) m/z 408 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(phenylamino)piperidin-4-yl)-10,10-dimethyl-1,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (45)
To a stirred mixture of 1-benzyl-4-phenylamino-4-carboxy-piperidine (44, 0.3 g, 0.9 mmol), 1-hydroxy-benzotriazole hydrate (0.2 g, 1.46 mmol), 2-(2-aminoethyl)-1,3-di-Boc-guanidine (0.34 g, 1.1 mmol), and triethylamine (0.6 mL, 4.5 mmol) in 1-methyl-2-pyrrolidone (15 mL) was added N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (0.22 g, 1.1 mmol). The reaction mixture was stirred at room temperature for 24 h. The mixture was then poured into 80 mL of water, extracted with ethyl acetate (3 × 30 mL), washed with saturated aqueous NaCl, and dried. The solvent was removed in vacuo, and the residue was chromatographed on a silica gel column using a gradient of methylene chloride and methanol (10:0 → 9.5:0.5) as the eluent to give 0.37 g (70%) of tert-butyl 1-(1-benzyl-4-(phenylamino)piperidin-4-yl)-10,10-dimethyl-1,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.19 (s, 1H), 8.42 (s, 1H), 7.77 (s, 1H), 7.32–7.25 (m, 5H), 7.14 (t, J = 7.4 Hz, 2H), 6.75 (t, J = 7.2 Hz, 1H), 6.54 (d, J = 7.8 Hz, 2H), 3.99 (s, 1H), 3.55–3.43 (m, 6H), 2.74 (d, J = 11.6 Hz, 2H), 2.33 (t, J = 11.2 Hz, 2H), 2.16 (t, J = 11.6 Hz, 2H), 1.90 (d, J = 13.2 Hz, 2H), 1.53 (s, 9H), 1.49 (s, 9H). MS (ESI) m/z 595 (M+ + 1).
1-Benzyl-N-(2-guanidinoethyl)-4-(phenylamino)piperidine-4-carboxamide Tritriflate (46)
Prepared from 45 (0.12 g, 0.20 mmol) according to general procedure 5, method B, to afford 0.08 g (54%) of the title material as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 10.40 (s, 1H), 8.28–7.91 (m, 2H), 7.50–7.07 (m, 12H), 6.63–6.55 (m, 3H), 6.06 (s, 1H), 4.29 (s, 2H), 3.36–3.10 (m, 8H), 2.25–1.95 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 174.46, 158.77 (q, J = 31.4 Hz), 157.32, 144.44, 131.30, 129.53, 128.79, 128.71, 117.52, 117.12 (q, J = 296.6 Hz), 115.02, 114.52, 59.02, 55.95, 46.73, 40.19, 38.00, 28.07. MS (ESI) m/z 395 (M+ + 1).
tert-Butyl 10,10-Dimethyl-3,8-dioxo-1-(4-(phenylamino)piperidin-4-yl)-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (47)
Intermediate 20 (1 g, 1.68 mmol) was dissolved in methanol (65 mL) and hydrogenated over 10% Pd/C (0.4 g) under pressure (4 bar) for 7 days. The catalyst was removed by filtration, and the solvent was evaporated in vacuo. The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol saturated with ammonia (10:0 → 9:1) as the eluent to give 0.57 g (68%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.34 (br s, 1H), 8.83 (br s, 1H), 7.15 (t, J = 7.6 Hz, 2H), 6.79 (t, J = 7.4 Hz, 1H), 6.72 (d, J = 7.8 Hz, 2H), 6.53–6.55 (m, 1H), 3.98 (d, J = 5.2 Hz, 2H), 3.55 (d, J = 5.6 Hz, 2H), 3.42 (br s, 1H), 2.89–2.87 (m, 4H), 1.86–1.82 (m, 2H), 1.68–1.63 (m, 3H), 1.49 (s, 9H), 1.47 (s, 9H). MS (ESI) m/z 505 (M+ + 1).
tert-Butyl 1-(1-(Cyclohexylmethyl)-4-(phenylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (48b)
Prepared according to general procedure 7 using intermediate 47 (0.13 g, 0.257 mmol) and cyclohexylmethyl bromide (0.055 g, 0.308 mmol) in anhydrous NMP (5 mL) to afford 0.04 g (27%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.31 (br s, 1H), 8.84 (br s, 1H), 7.10 (t, J = 7.6 Hz, 2H), 6.76–6.69 (m, 4H), 3.97 (d, J = 4.9 Hz, 2H), 3.50 (d, J = 5.3 Hz, 2H), 2.67 (s, 2H), 2.48 (s, 2H), 2.24 (s, 2H), 1.91–1.59 (m, 10H), 1.45 (s, 9H), 1.43 (s, 9H), 1.21–1.08 (m, 4H), 0.89–0.84 (m, 2H). MS (ESI) m/z 601 (M+ + 1).
tert-Butyl 10,10-Dimethyl-3,8-dioxo-1-(1-phenethyl-4-(phenylamino)piperidin-4-yl)-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (48c)
Prepared according to general procedure 7 using intermediate 47 (0.15 g, 0.297 mmol) and 2-(bromoethyl)benzene (0.055 g, 0.297 mmol) to afford 0.13 g (72%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.35 (s, 1H), 8.85 (s, 1H), 7.29–7.25 (m, 2H), 7.19–7.14 (m, 5H), 6.79 (t, J = 7.2 Hz, 1H), 6.73 (d, J = 7.8 Hz, 2H), 6.62–6.60 (m, 1H), 3.99 (d, J = 5.1 Hz, 2H), 3.56 (d, J = 5.3 Hz, 2H), 3.56 (br s, 1H), 2.84–2.80 (m, 2H), 2.67–2.64 (m, 4H), 2.53–2.52 (m, 2H), 1.97–1.94 (m, 2H), 1.85–1.80 (m, 2H), 1.49 (s, 9H), 1.48 (s, 9H). MS (ESI) m/z 609 (M+ + 1).
tert-Butyl 10,10-Dimethyl-1-(1-(naphthalen-1-ylmethyl)-4-(phenylamino)piperidin-4-yl)-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (48d)
Prepared according to general procedure 7 using intermediate 47 (0.13 g, 0.257 mmol) and 1-(chloromethyl)naphthalene (0.041 g, 0.231 mmol) to afford 0.096 g (65%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.37 (s, 1H), 8.86–8.84 (m, 1H), 8.28 (d, J = 8.0 Hz, 1H), 7.83 (t, J = 7.2 Hz, 1H), 7.76 (d, J = 7.2 Hz, 1H), 7.51–7.47 (m, 2H), 7.40–7.37 (m, 2H), 7.16 (t, J = 7.7 Hz, 2H), 6.79 (t, J = 7.3 Hz, 1H), 6.73 (d, J = 7.9 Hz, 2H), 6.56–6.53 (m, 1H), 4.00 (d, J = 5.2 Hz, 2H), 3.90 (s, 2H), 3.55 (d, J = 5.4 Hz, 2H), 3.43 (br s, 1H), 2.64–2.61 (m, 2H), 2.43 (t, J = 9.5 Hz, 2H), 1.89–1.85 (m, 2H), 1.73–1.68 (m, 2H), 1.49 (s, 9H), 1.47 (s, 9H). MS (ESI) m/z 645 (M+ + 1).
tert-Butyl 10,10-Dimethyl-1-(1-(naphthalen-2-ylmethyl)-4-(phenylamino)piperidin-4-yl)-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (48e)
Prepared according to general procedure 4, method C, using 5d (0.3 g, 0.87 mmol) to afford 0.31 g (48%) of the title material as a yellow solid. 1H NMR (500 MHz, CDCl3): δ 11.30 (s, 1H), 8.80 (s, 1H), 7.77–7.67 (m, 4H), 7.43–7.39 (m, 3H), 7.11 (t, J = 7.4 Hz, 2H), 6.74 (t, J = 7.2 Hz, 1H), 6.68 (d, J = 7.7 Hz, 2H), 6.54 (s, 1H), 3.95 (br s, 3H), 3.62–3.51 (m, 4H), 2.56–2.55 (m, 2H), 2.38–2.37 (m, 2H), 1.87–1.84 (m, 2H), 1.74–1.72 (m, 2H), 1.44 (s, 9H), 1.40 (s, 9H). MS (ESI) m/z 645 (M+ + 1).
tert-Butyl 1-(1-(4-Bromobenzyl)-4-(phenylamino)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (48f)
Prepared according to general procedure 7 using intermediate 47 (0.13 g, 0.257 mmol) and 4-bromobenzyl bromide (0.058 g, 0.231 mmol) to afford 0.12 g (69%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.33 (s, 1H), 8.82 (s, 1H), 7.40 (d, J = 8.2 Hz, 2H), 7.18–7.12 (m, 4H), 6.78 (t, J = 7.1 Hz, 1H), 6.70 (d, J = 8.0 Hz, 2H), 6.51–6.50 (m, 1H), 3.98 (d, J = 5.2 Hz, 2H), 3.54 (d, J = 5.3 Hz, 2H), 3.44 (s, 2H), 3.42 (br s, 1H), 2.53–2.51 (m, 2H), 2.37–2.33 (m, 2H), 1.88–1.85 (m, 2H), 1.75–1.70 (m, 2H), 1.48 (s, 9H), 1.45 (s, 9H). MS (ESI) m/z 673 (M+ + 1) for 79Br, 675 (M+ + 1) for 81Br.
Methyl 4-((4-(6-(tert-Butoxycarbonylamino)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundec-6-enyl)-4-(phenylamino)piperidin-1-yl)methyl)benzoate (48g)
Prepared according to general procedure 7 using intermediate 47 (0.13 g, 0.257 mmol) and methyl 4-(bromomethyl)benzoate (0.053 g, 0.231 mmol) to afford 0.14 g (96%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.33 (s, 1H), 8.82 (s, 1H), 7.96 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.1 Hz, 2H), 7.14 (t, J = 7.7 Hz, 2H), 6.78 (t, J = 7.2 Hz, 1H), 6.70 (d, J = 7.8 Hz, 2H), 6.52–6.50 (m, 1H), 3.98 (d, J = 5.3 Hz, 2H), 3.89 (s, 3H), 3.55–3.46 (m, 5H), 2.54–2.52 (m, 2H), 2.39–2.36 (m, 2H), 1.89–1.86 (m, 2H), 1.76–1.72 (m, 2H), 1.48 (s, 9H), 1.45 (s, 9H). MS (ESI) m/z 653 (M+ + 1).
2-Guanidino-N-((4-(phenylamino)piperidin-4-yl)methyl)acetamide Tritriflate (49a)
Prepared according to general procedure 5, method B, from 47 (0.1 g, 0.198 mmol) to afford 0.11 g (85%) of the title material as a brown solid. 1H NMR (DMSO-d6): δ 10.85 (br s, 2H), 8.74–8.63 (m, 2H), 8.18 (s, 1H), 7.74 (s, 1H), 7.41 (br s, 3H), 7.10 (t, J = 6 Hz, 2H), 6.85 (d, J = 6.2 Hz, 2H), 6.66–6.64 (m, 1H), 3.86–3.85 (m, 2H), 3.42–3.41 (m, 2H), 3.12–3.03 (m, 4H), 2.07–2.06 (m, 2H), 1.80–1.75 (m, 2H). 13C NMR (DMSO-d6): δ 168.01, 158.70 (q, J = 34.2 Hz), 157.60, 145.43, 128.87, 117.59, 116.33, 116.23 (q, J = 290.6 Hz), 53.58, 44.46, 43.42, 30.65, 28.66. MS (ESI) m/z 305 (M+ + 1).
N-((1-(Cyclohexylmethyl)-4-(phenylamino)piperidin-4-yl)methyl)-2-guanidinoacetamide Tritriflate (49b)
Prepared according to general procedure 5, method B, from 48b (0.04 g, 0.066 mmol) to afford 0.04 g (81%) of the title material as a yellow solid. 1H NMR (400 MHz, CD3OD): δ 7.14 (t, J = 4 Hz, 2H), 6.90–6.88 (m, 2H), 6.73 (t, J = 4 Hz, 1H), 3.97 (s, 2H), 3.53 (s, 2H), 3.45–3.42 (m, 2H), 3.26–3.20 (m, 2H), 2.95 (d, J = 8 Hz, 2H), 2.30–2.26 (m, 2H), 2.16–2.09 (m, 3H), 1.84–1.70 (m, 7H), 1.38–1.28 (m, 3H). MS (ESI) m/z 401 (M+ + 1).
2-Guanidino-N-((1-phenethyl-4-(phenylamino)piperidin-4-yl)methyl)acetamide Tritriflate (49c)
Prepared according to general procedure 5, method B, from 48c (0.12 g, 0.197 mmol) to afford 0.076 g (51%) of the title material as a yellow solid. 1H NMR (400 MHz, CD3OD): δ 7.35–7.24 (m, 5H), 7.16 (t, J = 7.6 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 6.75 (t, J = 7.2 Hz, 1H), 3.96 (s, 2H), 3.56–3.51 (m, 4H), 3.35–3.31 (m, 4H), 3.10–3.05 (m, 2H), 2.35–2.31 (m, 2H), 2.12–2.01 (m, 2H). 13C NMR (125 MHz, CD3OD): δ 170.15, 163.05 (q, J = 37.0 Hz), 159.37, 146.68, 137.65, 130.19, 129.93, 129.79, 128.21, 119.85, 118.26 (q, J = 290.9 Hz), 118.12, 59.02, 54.58, 46.61, 44.72, 43.00, 31.39, 30.62. MS (ESI) m/z 409 (M+ + 1).
2-Guanidino-N-((1-(naphthalen-1-ylmethyl)-4-(phenylamino)piperidin-4-yl)methyl)acetamide Tritriflate (49d)
Prepared according to general procedure 5, method B, from 48d (0.09 g, 0.139 mmol) to afford 0.07 g (65%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 8.15 (d, J = 8.3 Hz, 1H), 7.92 (d, J = 8.2 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 6.8 Hz, 1H), 7.57–7.46 (m, 3H), 7.09–7.06 (m, 2H), 6.80–6.65 (m, 3H), 4.71 (s, 2H), 3.84 (s, 2H), 3.40–3.25 (m, 6H), 2.17–1.92 (m, 4H). 13C NMR (125 MHz, CD3OD): δ 170.05, 163.07 (q, J = 34.4 Hz), 159.33, 146.63, 135.33, 133.46, 132.45, 132.13, 130.16, 130.07, 128.50, 127.53, 126.32, 124.23, 120.36, 119.83, 118.15 (q, J = 291.0 Hz), 118.09, 57.85, 54.50, 49.56, 46.47, 44.66, 31.17. MS (ESI) m/z 445 (M+ + 1).
2-Guanidino-N-((1-(naphthalen-2-ylmethyl)-4-(phenylamino)piperidin-4-yl)methyl)acetamide Tritriflate (49e)
Prepared according to general procedure 5, method B, from 48e (0.1 g, 0.15 mmol) to afford 0.08 g (66%) of the title material as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 10.44 (s, 1H), 8.19 (s, 1H), 8.07 (s, 1H), 7.97–7.78 (m, 4H), 7.66–7.17 (m, 8H), 7.08–7.05 (t, J = 7.4 Hz, 2H), 6.80 (d, J = 8.0 Hz, 2H), 6.61 (t, J = 7.0 Hz, 1H), 5.32 (br s, 1H), 4.46 (s, 2H), 3.84–3.83 (m, 2H), 3.58–3.16 (m, 6H), 2.14–2.11 (m, 2H), 1.98–1.92 (m, 2H). 13C NMR (125 MHz, DMSO-d6): δ 167.86, 158.65 (q, J = 31.6 Hz), 157.69, 145.90, 133.07, 132.53, 131.11, 128.81, 128.37, 128.08, 127.99, 127.71, 127.42, 127.07, 126.72, 117.32, 117.13 (q, J = 295.4 Hz), 116.19, 59.02, 53.19, 47.28, 44.74, 43.38, 29.21. MS (ESI) m/z 445 (M+ + 1).
N-((1-(4-Bromobenzyl)-4-(phenylamino)piperidin-4-yl)methyl)-2-guanidinoacetamide Tritriflate (49f)
Prepared according to general procedure 5, method B, from 48f (0.1 g, 0.148 mmol) to afford 0.066 g (54%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 7.50 (d, J = 8.1 Hz, 2H), 7.32 (d, J = 8.1 Hz, 2H), 7.10 (t, J = 7.4 Hz, 2H), 6.81 (d, J = 8.1 Hz, 2H), 6.68 (t, J = 7.1 Hz, 1H), 3.92 (s, 2H), 3.84 (s, 2H), 3.51 (s, 2H), 2.89–2.81 (m, 4H), 2.11–2.09 (m, 2H), 1.86–1.84 (m, 2H). 13C NMR (125 MHz, CD3OD): δ 170.00, 163.18 (q, J = 35.8 Hz), 159.33, 146.87, 134.00, 133.36, 132.83, 130.03, 123.64, 119.60, 118.26, 118.09 (q, J = 290.7 Hz), 61.81, 54.99, 49.46, 46.62, 44.78, 32.36. MS (ESI) m/z 473 (M+ + 1) for 79Br, 475 (M+ + 1) for 81Br.
Methyl 4-((4-((2-Guanidinoacetamido)methyl)-4-(phenylamino)piperidin-1-yl)methyl)benzoate Tritriflate (49g)
Prepared according to general procedure 5, method B, from 48g (0.13 g, 0.199 mmol) to afford 0.09 g (56%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 8.10 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.1 Hz, 2H), 7.16 (t, J = 7.8 Hz, 2H), 6.87 (d, J = 7.8 Hz, 2H), 6.76 (s, 1H), 4.42 (s, 2H), 3.94 (s, 5H), 3.54 (s, 2H), 3.36–3.33 (m, 4H), 2.30–2.06 (m, 4H). 13C NMR (125 MHz, CD3OD): δ 170.14, 167.70, 163.09 (q, J = 35.6 Hz), 159.38, 146.64, 135.40, 132.89, 132.58, 131.16, 130.18, 119.90, 118.29 (q, J = 288.8 Hz), 118.15, 60.77, 54.61, 52.89, 49.34, 46.61, 44.72, 31.22. MS (ESI) m/z 453 (M+ + 1).
1-Benzyl-4-(naphthalen-1-ylmethyl)piperidine-4-carbonitrile (50a)
Prepared according to general procedure 6 using 1-benzylpiperidine-4-carbonitrile (35, 0.9 g, 4.49 mmol) and 1-(chloromethyl)naphthalene (1 g, 5.84 mmol) to afford 1.4 g (91%) of the title material as a white solid. 1H NMR (CDCl3): δ 8.11 (d, J = 8.2 Hz, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.58–7.47 (m, 4H), 7.36–7.27 (m, 5H), 3.53 (s, 2H), 3.40 (s, 2H), 2.87 (d, J = 12.1 Hz, 2H), 2.32 (t, J = 10.0 Hz, 2H), 1.88–1.78 (m, 4H). MS (ESI) m/z 341 (M+ + 1).
1-Benzyl-4-(3,4-dichlorobenzyl)piperidine-4-carbonitrile (50b)
Prepared according to general procedure 6 using 1-benzylpiperidine-4-carbonitrile (35, 0.9 g, 4.49 mmol) and 3,4-dichlorobenzyl bromide (1.4 g, 5.84 mmol) to afford 0.98 g (61%) of the title material as a white solid. 1H NMR (DMSO-d6): δ 7.59 (d, J = 8.2 Hz, 1H), 7.55 (s, 1H), 7.32–7.23 (m, 6H), 3.46 (s, 2H), 2.91 (s, 2H), 2.78 (d, J = 11.9 Hz, 2H), 2.07 (t, J = 11.0 Hz, 2H), 1.72–1.61 (m, 4H). MS (ESI) m/z 359 [M + H]+.
1-Benzyl-4-(naphthalen-2-ylmethyl)piperidine-4-carbonitrile (50c)
Prepared according to general procedure 6 using 1-benzylpiperidine-4-carbonitrile (35, 0.7 g, 3.49 mmol) and 2-(bromomethyl)naphthalene (1 g, 4.54 mmol) to afford 0.85 g (71%) of the title material as a white solid. 1H NMR (CDCl3): δ 7.86–7.83 (m, 3H), 7.75 (s, 1H), 7.53–7.43 (m, 3H), 7.34–7.27 (m, 5H), 3.54 (s, 2H), 3.04 (s, 2H), 2.89 (d, J = 12.3 Hz, 2H), 2.33 (t, J = 12.2 Hz, 2H), 1.90 (d, J = 12.4 Hz, 2H), 1.74 (td, J = 12.5, 2.9 Hz, 2H). MS (ESI) m/z 341 [M + H]+.
1-Benzyl-4-(biphenyl-4-ylmethyl)piperidine-4-carbonitrile (50d)
Prepared according to general procedure 6 using 1-benzylpiperidine-4-carbonitrile (35, 0.7 g, 3.49 mmol) and 4-(bromomethyl)biphenyl (1.12 g, 4.54 mmol) to afford 0.76 g (59%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.59–7.55 (m, 4H), 7.44 (t, J = 7.5 Hz, 2H), 7.35–7.26 (m, 8H), 3.53 (s, 2H), 2.89–2.86 (m, 4H), 2.31 (d, J = 10.6 Hz, 2H), 1.88 (d, J = 12.4 Hz, 2H), 1.71–1.66 (m, 2H). MS (ESI) m/z 367 (M+ + 1).
(1-Benzyl-4-(naphthalen-1-ylmethyl)piperidin-4-yl)methanamine (51a)
Prepared according to general procedure 2, method B, from 50a (1.3 g, 3.81 mmol) at 2.38 atm for 15 h. The catalyst was removed by filtration, and the solvent was evaporated in vacuo. The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol (10:0 → 8:2) as the eluent to give 0.49 g (38%) of the title material as a white solid. 1H NMR (CDCl3): δ 8.29 (d, J = 8.2 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.55–7.43 (m, 3H), 7.38–7.27 (m, 6H), 3.52 (s, 2H), 3.17 (s, 2H), 2.72 (br s, 2H), 2.65–2.62 (m, 2H), 2.30 (t, J = 9.8 Hz, 2H), 1.70–1.64 (m, 4H), 1.57–1.54 (m, 2H). MS (ESI) m/z 345 (M+ + 1).
(1-Benzyl-4-(3,4-dichlorobenzyl)piperidin-4-yl)methanamine (51b)
Prepared according to general procedure 2, method B, from 50b (0.9 g, 2.5 mmol) to afford 0.85 g (93%) of the title material as a white solid. 1H NMR (DMSO-d6): δ 7.49–7.43 (m, 2H), 7.29–7.15 (m, 6H), 3.43 (s, 2H), 3.17 (s, 2H), 2.60–2.26 (m, 8H), 1.31 (s, 4H). MS (ESI) m/z 363 (M+ + 1), 365 (M+ + 1).
(1-Benzyl-4-(naphthalen-2-ylmethyl)piperidin-4-yl)methanamine (51c)
Prepared according to general procedure 2, method B, from 50c (0.8 g, 2.35 mmol) to afford 0.62 g (77%) of the title material as a colorless oil. 1H NMR (CDCl3): δ 7.84–7.76 (m, 3H), 7.62 (s, 1H), 7.50–7.44 (m, 2H), 7.37–7.27 (m, 6H), 3.57 (s, 2H), 2.85 (s, 2H), 2.63–2.58 (m, 4H), 2.48–2.46 (m, 2H), 1.63–1.50 (m, 4H), 1.21 (br s, 2H). MS (ESI) m/z 345 [M + H]+.
(1-Benzyl-4-(biphenyl-4-ylmethyl)piperidin-4-yl)methanamine (51d)
Prepared according to general procedure 2, method B, from 50d (0.69 g, 1.88 mmol) to afford 0.67 g (96%) of the title material as a colorless oil. 1H NMR (CDCl3): δ 7.60 (d, J = 7.5 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.45 (t, J = 7.4 Hz, 2H), 7.35–7.32 (m, 4H), 7.29–7.23 (m, 3H), 3.56 (s, 2H), 2.72 (s, 2H), 2.60–2.57 (m, 4H), 2.47–2.46 (m, 2H), 1.59–1.52 (m, 4H), 1.32 (br s, 2H). MS (ESI) m/z 371 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(naphthalen-1-ylmethyl)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (52a)
Prepared according to general procedure 4, method C, from 51a (0.35 g, 1.02 mmol). The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride and methanol (10:0 → 97:3) to give 0.27 g (41%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.45 (s, 1H), 8.87 (s, 1H), 8.07 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 7.4 Hz, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.49–7.42 (m, 2H), 7.39 (t, J = 7.2 Hz, 1H), 7.27–7.22 (m, 6H), 6.91–6.89 (m, 1H), 3.98 (d, J = 5.4 Hz, 2H), 3.48 (s, 2H), 3.35 (d, J = 5.6 Hz, 2H), 3.09 (s, 2H), 2.62–2.59 (m, 2H), 2.31 (t, J = 10.3 Hz, 2H), 1.70–1.65 (m, 2H), 1.51–1.47 (m, 11H), 1.40 (s, 9H). MS (ESI) m/z 644 (M+ + 1).
(tert-Butyl 1-(1-Benzyl-4-(3,4-dichlorobenzyl)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (52b)
Prepared according to general procedure 4, method C, from 51b (0.37 g, 1.02 mmol) to afford 0.33 g (49%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.42 (s, 1H), 8.92 (s, 1H), 7.33–7.19 (m, 8H), 6.94 (d, J = 8.1 Hz, 1H), 4.05 (d, J = 5.8 Hz, 2H), 3.50 (s, 2H), 3.15 (d, J = 5.4 Hz, 2H), 2.57–2.52 (m, 4H), 2.39–2.37 (m, 2H), 1.50–1.40 (m, 22H). MS (ESI) m/z 662 (M+ + 1), 664 (M+ + 1).
tert-Butyl 1-(1-Benzyl-4-(naphthalen-2-ylmethyl)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (52c)
Prepared according to general procedure 4, method C, from 51c (0.35 g, 1.02 mmol) to afford 0.38 g (72%) of the title material as a white solid. 1H NMR (CDCl3): δ 11.47 (s, 1H), 8.97–8.95 (m, 1H), 7.80–7.72 (m, 3H), 7.53 (s, 1H), 7.46–7.42 (m, 2H), 7.31–7.19 (m, 7H), 4.08 (d, J = 5.6 Hz, 2H), 3.52 (s, 2H), 3.21 (d, J = 5.6 Hz, 2H), 2.94 (s, 1H), 2.88 (s, 1H), 2.78 (s, 2H), 2.60–2.58 (m, 2H), 2.43–2.40 (m, 2H), 1.63–1.56 (m, 2H), 1.51 (s, 9H), 1.40 (s, 9H). MS (ESI) m/z 644 (M+ + 1).
(tert-Butyl 1-(1-Benzyl-4-(biphenyl-4-ylmethyl)piperidin-4-yl)-10,10-dimethyl-3,8-dioxo-9-oxa-2,5,7-triazaundecan-6-ylidenecarbamate (52d)
Prepared according to general procedure 4, method D, from 51d (0.413 g, 1.11 mmol) to afford 0.223 g (80%) of the title material as a white solid. 1H NMR (500 MHz, CDCl3): δ 11.47 (s, 1H), 8.95 (s, 1H), 7.59 (d, J = 7.3 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.44 (t, J = 7.5 Hz, 2H), 7.36–7.31 (m, 5H), 7.28–7.25 (m, 1H), 7.18 (d, J = 8.0 Hz, 2H), 7.12 (br s, 1H), 4.09 (d, J = 6.0 Hz, 2H), 3.57 (s, 2H), 3.22 (d, J = 5.7 Hz, 2H), 2.68 (s, 2H), 2.61 (s, 2H), 2.46 (s, 2H), 1.64–1.60 (m, 4H), 1.52 (s, 9H), 1.44 (s, 9H). MS (ESI) m/z 670 (M+ + 1).
N-((1-Benzyl-4-(naphthalen-1-ylmethyl)piperidin-4-yl)methyl)-2-guanidinoacetamide Tritriflate (53a)
Prepared according to general procedure 5, method B, from 52a (0.12 g, 0.189 mmol). The residue was purified by chromatography on a silica gel column using column using a gradient of methylene chloride and methanol (95:5 → 8:2) as the eluent to give 0.07 g (54%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 8.03 (d, J = 8.3 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.74 (t, J = 8.1 Hz, 1H), 7.48–7.27 (m, 9H), 4.27–4.13 (m, 2H), 4.08–3.84 (m, 2H), 3.59 (s, 2H), 3.35–3.02 (m, 6H), 1.88–1.61 (m, 4H). 13C NMR (125 MHz, CD3OD): δ 170.65, 163.05 (q, J = 34.2 Hz), 159.53, 135.52, 134.74, 134.49, 133.79, 132.13, 131.09, 131.00, 130.21, 129.86, 128.69, 127.06, 126.57, 126.15, 125.34, 118.24 (q, J = 292.9 Hz), 61.40, 49.47, 44.84, 44.13, 40.42, 38.22, 29.86. MS (ESI) m/z 444 (M+ + 1).
N-((1-Benzyl-4-(3,4-dichlorobenzyl)piperidin-4-yl)methyl)-2-guanidinoacetamide Ditriflate (53b)
Prepared according to general procedure 5, method B, from 52b (0.1 g, 0.151 mmol) to afford 0.07 g (72%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 7.57–7.41 (m, 7H), 7.22–7.13 (m, 1H), 4.39–4.29 (m, 2H), 4.07–4.00 (m, 2H), 3.46–3.14 (m, 6H), 2.91–2.67 (m, 2H), 1.88–1.85 (m, 2H), 1.69–1.68 (m, 2H). 13C NMR (125 MHz, 45 °C, CD3OD): δ 170.49, 163.06 (q, J = 34.4 Hz), 159.56, 138.37, 133.71, 133.10, 132.18, 131.82, 131.75, 131.24, 131.08, 130.33, 130.23, 118.56 (q, J = 280.0 Hz), 61.43, 49.45, 44.90, 44.14, 42.65, 36.89, 29.81. MS (ESI) m/z 462 (M+ + 1) 464 (M+ + 1).
N-((1-Benzyl-4-(naphthalen-2-ylmethyl)piperidin-4-yl)methyl)-2-guanidinoacetamide Ditriflate (53c)
Prepared according to general procedure 5, method B, from 52c (0.13 g, 0.202 mmol) to afford 0.10 g (75%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 7.85–7.69 (m, 4H), 7.52–7.32 (m, 8H), 4.42–4.26 (m, 2H), 4.11–3.99 (m, 2H), 3.52–3.27 (m, 6H), 3.09–2.86 (m, 2H), 1.85–1.76 (m, 4H). MS (ESI) m/z 444 (M+ + 1).
N-((1-Benzyl-4-(biphenyl-4-ylmethyl)piperidin-4-yl)methyl)-2-guanidinoacetamide Ditriflate (53d)
Prepared according to general procedure 5, method B, from 52d (0.1 g, 0.149 mmol) to afford 0.08 g (76%) of the title material as a white solid. 1H NMR (500 MHz, CD3OD): δ 7.62–7.57 (m, 4H), 7.51–7.43 (m, 6H), 7.36–7.34 (m, 2H), 7.28–7.27 (m, 2H), 4.40–4.29 (m, 2H), 4.09–4.00 (m, 2H), 3.50–3.20 (m, 6H), 2.96–2.73 (m, 2H), 1.78–1.70 (m, 4H). 13C NMR (125 MHz, 45 °C, CD3OD): δ 170.41, 163.02 (q, J = 32.4 Hz), 159.53, 141.89, 140.86, 136.46, 132.34, 132.16, 131.05, 130.32, 130.20, 129.78, 128.24, 127.75, 127.74, 118.56 (q, J = 290.0 Hz), 61.38, 49.45, 44.92, 44.91, 42.81, 36.91, 29.93. MS (ESI) m/z 470 (M+ + 1).
Biology
NPFF1 and NPFF2 Receptor Binding Studies
Compounds 7a–d, 9a–c, and 15a,b were tested against [125I]YVP and [125I]EYF for NPFF1 and NPFF2 affinity, respectively, as described in Mollereau et al.21 The affinities of 17a–g, 21, 30a–d, 34, 42–43, 46, 49a–g, and 53a–c on NPFF1 and NPFF2 receptors were evaluated by competition experiments using the selective NPFF1 and NPFF2 radioligands [3H]-NPVF and [3H]-EYF, respectively, in membranes of CHO cells stably expressing each receptor. For membrane preparation, CHO cells expressing human NPFF receptors were harvested in phosphate buffer saline (PBS), frozen at least for 1 h at −70 °C, and then homogenized in 50 mM Tris-HCl, pH 7.4, in a Potter Elvehjem tissue grinder. The nuclear pellet was discarded by centrifugation at 1000g for 15 min at 4 °C, and the membrane fraction was collected upon centrifugation of the supernatant at 100000g for 30 min at 4 °C. Membranes were aliquoted and stored at −80 °C in Tris 50 mM, pH 7.4, and the protein concentration was determined by the Lowry method. [3H]-NPVF and [3H]-EYF were custom-made by RC TRITEC AG (Teufen, Switzerland) by hydrogenation of the unsaturated peptide precursors with 99% tritium gas. Tritiated products were purified (>98%) by HPLC and dissolved in ethanol to obtain 1 mCi/mL (37 MBq/mL). Binding of [3H]-NPVF and [3H]-EYF was measured by rapid filtration. Membranes (5–15 μg of protein) were incubated in polypropylene tubes in a final volume of 500 μL containing 50 mM Tris-HCl, pH 7.4, 0.1% bovine serum albumin, 60 mM NaCl (for NPFF2 receptors only), the radioligand at 0.5–1 nM, and compounds to be tested at the desired concentration. The nonspecific binding was determined in the presence of 1 μM YVPNLPQRFa (for hNPFF1 receptor) and 1 μM EYWSLAAPQRFa (for hNPFF2 receptor). After 1 h incubation at 25 °C, samples were rapidly filtered on Whatman GF/B filters preincubated in 0.3% polyethylenimine. The filters were rinsed three times with 4 mL of ice cold buffer containing 0.1% bovine serum albumin, and the bound radioactivity was counted in a liquid scintillation spectrophotometric counter (50% efficiency, Packard).
cAMP Assay at NPFF1 and NPFF2 Receptors
Recombinant cells (200000) were seeded in glass tubes and incubated overnight as usual. The culture medium was then removed and replaced by fresh medium (200 μL) containing 0.1 μM adenine and 0.6 μCi of [3H]adenine (26 Ci/mmole, Amersham, France). After 1 h incubation at 37 °C in the incubator under 5% CO2 atmosphere, cells were rinsed two times with 400 μL of HEPES-buffered Krebs–Ringer saline (KRH, 124 mM NaCl, 5 mM KCl, 1.25 mM MgSO4, 1.5 mM CaCl2, 1.25 mM KH2PO4, 25 mM HEPES, 8 mM glucose, 0.5 mg/mL bovine serum albumin; pH 7.4). Prewarmed KRH (150 μL) was added to each tube, and the reaction was initiated by the addition of 50 μL of KRH containing 20 μM forskolin (Sigma, France), 0.4 mM IBMX (3-isobutyl-1-methylxanthine; Sigma, France), 0.4 mM Ro-20 1724 (4-(3-butoxy-4methoxyphenyl)methyl-2-imidazolidone; Fisher, France), and the ligand to be tested. To evaluate the antagonist activity, the compound was tested in the presence of the NPFF agonists NPVF and 1DMe for hNPFF-1 and hNPFF-2 receptors, respectively. After 10 min at 37 °C, the reaction was stopped by addition of 20 μL of HCl (2.2 N) with rapid mixing. The [3H]cAMP content of each tube was isolated by chromatographic procedures on acid alumina columns as described by Alvarez and Daniels34 and counted in a liquid scintillation analyzer (Packard).
[35S]GTPγS Binding Assay at NPFF1 and NPFF2 Receptors
The assay buffer consisted of 20 mM HEPES, pH 7.4, 20 mM NaCl, 3 mM MgCl2, and 0.1% BSA. Aliquots (50 μL, equivalent to 1.5 μg and to 0.7 μg protein, for NPFF1 and NPFF2, respectively) were incubated in polypropylene tubes at 30 °C for 60 min in 500 μL of buffer with 5 μg of saponin, 0.1 μM (NPFF1) or 1 μM (NPFF2) GDP, the ligand to be tested, and 0.066 nM [35S]GTPγS. Reaction was stopped by rapid vacuum filtration through GF/B Whatman glass fiber filters, preincubated in the buffer at room temperature for 1 h, and washed three times with 4 mL of ice-cold buffer. Membrane-bound radioactivity retained on the filters was determined by liquid scintillation spectrophotometry (94% efficiency, Packard counter) after overnight extraction of the filters in 4 mL of Ready Protein scintillation fluid (Beckman).
Opioid Receptor (μ, δ, κ) Binding Studies
Reagents
Buffer reagents were purchased from Sigma-Aldrich (St. Louis, MO). All radioligands and MicroScint were purchased from PerkinElmer (Waltham, MA). Nonlabeled controls were purchased from Tocris Bioscience (Minneapolis, MN). Membrane preparation and all assay dilutions were made using a Tris-HCl buffer (50 mM Tris-HCl), pH 7.4.
Cell Culture
HEK293 cells stably transfected with human opioid receptor subtypes μ, δ, or κ were used to perform the opioid receptor binding assays. These cells were maintained at 37 °C and 5% CO2 in a DMEM nutrient mixture supplemented with 2 mM l-glutamine, 10% fetal bovine serum, 1% penicillin–streptomycin, and G418 antibiotic solutions. When the cells reached 90% confluency, the media was removed, and the cells were washed with cold PBS, pH 7.4. The cells were lysed and scraped in cold Tris-HCl, pH 7.4, and then centrifuged at 5200g for 10 min at 4 °C. The supernatant was discarded ,and the pellet was resuspended in the same buffer and homogenized via Sonic Dismembrator model 100 (Fisher Scientific, Pittsburgh, PA) for 30 s and then centrifuged at 1000g for 10 min at 4 °C. The supernatant was saved, and the pellet underwent the suspension and homogenization process two more times with the same conditions. The supernatants were combined and centrifuged at 23300g for 40 min at 4 °C. The pellet was resuspended in cold Tris-HCl buffer, aliquoted into 2 mL vials, and stored at −80 °C. The total protein concentration was determined using a Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL) using manufacturer’s instructions.
Radioligand Binding Assays
For each assay, nonspecific binding was determined using 10 μM of a positive control [U-69,593 (κ), DPDPE (δ), or DAMGO (μ)], and total binding was ascertained with 0.1% DMSO in Tris-HCl buffer. Each test well contained 50 μL of its respective radioligand (U-69,593,[phenyl-3,4-3H] (κ), DAMGO,[tyrosyl-3,5-3H(N)] (μ) or enkephalin(DPDPE),[tyrosyl-3,5-3H(N)] (δ)), 50 μL of compound, control, or vehicle, and 100 μL of cell membrane. The assays were incubated for 60 min at room temperature. The reaction was terminated via rapid filtration with cold Tris-HCl buffer through a UniFilter GF/B 96-well plate presoaked with 0.3% BSA. When the filters were dry, 50 μL of MicroScint-20 was applied to each filter, and the plates were read on a TopCount NXT HTS microplate scintillation counter (PerkinElmer, Waltham, MA) where the counts per minute (CPM) were recorded.
The Kd for ligands for each receptor was established through a membrane evaluation and saturation binding experiment. For the membrane evaluation experiment, 1–15 μg of membrane was incubated with 0.6 nM of its respective radioligand. Total, specific, and nonspecific binding were used to calculate the % binding of the nonlabeled control to receptor. The membrane concentration exhibiting good % binding (>90%), and total binding with high signal (thousands of CPM) was used as the optimal membrane concentration for the assay. For the saturation assay, the optimal membrane concentration and 0.25–10 nM of its respective radioligand were incubated with 10 μM of a nonlabeled positive control or 0.1% DMSO in buffer. Data was analyzed by a nonlinear curve fit model using GraphPad Prizm 5.04 software (GraphPad, La Jolla, CA), and the Kd value was calculated.
The competitive binding assay was performed used the optimal concentration of membrane with a radioligand concentration ≤Kd and 12 concentrations of each compound ranging from 0.00001 to 1000 μM. Each compound was tested in triplicate. The assays were performed as stated above. The Ki values were calculated by a nonlinear curve fit model using GraphPad Prizm 5.0 software.
In Vivo Characterization
Animals
Adult (8–11 weeks old) male C57BL/6J were obtained from the Jackson Laboratory, Bar Harbor, Maine, USA. All mice were housed four to a cage in a temperature- and humidity-controlled room at the Torrey Pines Institute for Molecular Studies (Port Saint Lucie, Florida, USA) vivarium on a 12:12-h light/dark cycle (lights off at 19:00 h) with free access to food and water except during experimental sessions. All procedures were preapproved and carried out in accordance with the Institutional Animal Care and Use Committees at the Torrey Pines Institute for Molecular Studies as specified by the 2008 National Institutes of Health Guide for the Care and Use of Laboratory Animals. Consistent with these guidelines, ongoing statistical testing of data collected was used to minimize the number of animals used, within the constraints of necessary statistical power.
Injection Techniques
Intracerebroventricular (icv) injections were made directly into the lateral ventricle according to the modified method of Haley and McCormick.35 The volume of all icv injections was 5 μL, using a 10-μL Hamilton microliter syringe. The mouse was lightly anesthetized with isoflurane, an incision was made in the scalp, and the injection was made 2 mm lateral and 2 mm caudal to bregma at a depth of 3 mm.
Hyperalgesic Testing: The 48 °C Warm-Water Tail-Withdrawal Assay
The nociceptive stimulus was 48 °C water, with the latency to withdraw the tail taken as the end point.36,37 The water temperature of 48 °C was selected for this work to ensure a moderate tail-withdrawal response, with a measurable decrease in withdrawal time possible but also a significant temperature for hyperalgesic testing. After determining control latencies, mice received a single icv dose of vehicle (50% DMSO) or novel ligand pretreatment as discussed above. The dose of RF9 used in this study (10 nmol, icv) was selected because it was previously reported effective in antagonizing the hyperalgesic response of NPFF.38 The dose of compound 46 (30 nmol, icv) was selected on the basis of similar affinity for NPFF1-R as RF9 (75 nM vs the reported 58 nM).15 Animals showing an initial baseline latency fewer than 4 s or more than 15 s were removed from the study; two mice were so excluded. Mice administered the test compound were tested for analgesia every 10 min for 80 min postinjection. If the mouse failed to display a tail-flick in 30 s, the tail was removed from the water to minimize tissue damage. Experimentally induced increases in tail-withdrawal latencies provide a measure of antinociceptive effect, whereas decreases in tail-withdrawal latency indicate hyperalgesic effects.39−41
Data Analysis
All tail-withdrawal latency data for hyperalgesic testing are reported as percent baseline response to control for each animal’s baseline latency, ±SEM. Percent baseline response is calculated by the following equation: % baseline response = 100(test latency – baseline latency). Responses to treatment over time were analyzed by one- or two-way ANOVA followed by Tukey’s post hoc test as appropriate, with factors of treatment and time as required.
Acknowledgments
The work described in this paper was partially supported by Grants DA29738 (C. Mesangeau), GM104932 (S. J. Cutler), DA034777 (C. R. McCurdy) and the State of Florida, Executive Office of the Governor’s Dept. of Economic Opportunity (J. P. McLaughlin). The authors are grateful to Sara Pettaway for conducting the opioid binding experiments as part of the COBRE in vitro research core. This investigation was conducted in a facility constructed with support from research facilities improvement program Grant Number RR104503 from the National Center for Research Resources, National Institutes of Health. Cell lines used for the opioid binding were a generous gift from Dr. B. Roth (Department of Pharmacology, School of Medicine, University of North Carolina, Chapel Hill).
Glossary
Abbreviations
- NPFF
neuropeptide FF
- 1DMe
(D.YL(N-Me)FQPQRFa)
- RF9
n-adamantanecarbonyl-Arg-Phe-NH2
- AC-099
1-(3-bromo-4,5-dimethoxybenzylideneamino)guanidine hydrochloride
- EDCI
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride
- HOBt
1-hydroxybenzotriazole hydrate
- [35S]GTPγS
guanosine-5′-O-(3-[35S]thio)triphosphate
- cAMP
cyclic adenosine monophosphate
- YVP
YVPNLPQRFa
- EYF
EYWSLAAPQRFa (EYW-NPSF)
- NPVF
VPNLPQRFa
- CHO
Chinese hamster ovary
- ANOVA
analysis of variance
- HEK
human embryonic kidney
- DAMGO
(d-Ala2,MePhe4,Gly-ol5)enkephalin
- DPDPE
[D-Pen2,D-Pen5]enkephalin
- U69,593
(5α,7α,8β)-(−)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4,5]dec-8-yl]benzeneacetamide
- TMSCN
trimethylsilyl cyanide
- DMF
dimethylformamide
- THF
tetrahydrofuran
- NMP
N-methyl-2-pyrrolidone
- HEPES
4-(2-hydroxyethyl)-1-piperazine-1-ethanesulfonic acid
- DMEM
Dulbecco’s Modified Eagle’s medium
- Boc
tert-butyloxycarbonyl
- TFA
trifluoroacetic acid
- DCM
dichloromethane
- LDA
lithium diisopropylamide
- icv
intracerebroventricular
- it.
intrathecal
- OIH
opioid-induced hyperalgesia
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Mollereau C.; Roumy M.; Zajac J. M. Opioid-modulating peptides: Mechanisms of action. Curr. Top. Med. Chem. 2005, 5, 341–355. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Guan X. M.; Martin W. J.; McDonald T. P.; Clements M. K.; Jiang Q.; Zeng Z.; Jacobson M.; Williams D. L. Jr.; Yu H.; Bomford D.; Figueroa D.; Mallee J.; Wang R.; Evans J.; Gould R.; Austin C. P. Identification and characterization of novel mammalian neuropeptide FF-like peptides that attenuate morphine-induced antinociception. J. Biol. Chem. 2001, 276, 36961–36969. [DOI] [PubMed] [Google Scholar]
- Gouarderes C.; Quelven I.; Mollereau C.; Mazarguil H.; Rice S. Q.; Zajac J. M. Quantitative autoradiographic distribution of NPFF1 neuropeptide FF receptor in the rat brain and comparison with NPFF2 receptor by using [125I]YVP and [(125I]EYF as selective radioligands. Neuroscience 2002, 115, 349–361. [DOI] [PubMed] [Google Scholar]
- Bonini J. A.; Jones K. A.; Adham N.; Forray C.; Artymyshyn R.; Durkin M. M.; Smith K. E.; Tamm J. A.; Boteju L. W.; Lakhlani P. P.; Raddatz R.; Yao W. J.; Ogozalek K. L.; Boyle N.; Kouranova E. V.; Quan Y.; Vaysse P. J.; Wetzel J. M.; Branchek T. A.; Gerald C.; Borowsky B. Identification and characterization of two G protein-coupled receptors for neuropeptide FF. J. Biol. Chem. 2000, 275, 39324–39331. [DOI] [PubMed] [Google Scholar]
- Talmont F.; Mouledous L.; Piedra-Garcia L.; Schmitt M.; Bihel F.; Bourguignon J. J.; Zajac J. M.; Mollereau C. Pharmacological characterization of the mouse NPFF2 receptor. Peptides 2010, 31, 215–220. [DOI] [PubMed] [Google Scholar]
- Yang H. Y.; Fratta W.; Majane E. A.; Costa E. Isolation, sequencing, synthesis, and pharmacological characterization of two brain neuropeptides that modulate the action of morphine. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 7757–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gicquel S.; Mazarguil H.; Allard M.; Simonnet G.; Zajac J. M. Analogues of F8Famide resistant to degradation, with high affinity and in vivo effects. Eur. J. Pharmacol. 1992, 222, 61–67. [DOI] [PubMed] [Google Scholar]
- Oberling P.; Stinus L.; Le Moal M.; Simonnet G. Biphasic effect on nociception and antiopiate activity of the neuropeptide FF (FLFQPQRFamide) in the rat. Peptides 1993, 14, 919–924. [DOI] [PubMed] [Google Scholar]
- Desprat C.; Zajac J. M. Differential modulation of mu- and delta-opioid antinociception by neuropeptide FF receptors in young mice. Neuropeptides 1997, 31, 1–7. [DOI] [PubMed] [Google Scholar]
- Gouarderes C.; Sutak M.; Zajac J. M.; Jhamandas K. Antinociceptive effects of intrathecally administered F8Famide and FMRFamide in the rat. Eur. J. Pharmacol. 1993, 237, 73–81. [DOI] [PubMed] [Google Scholar]
- Kontinen V. K.; Kalso E. A. Differential modulation of alpha 2-adrenergic and mu-opioid spinal antinociception by neuropeptide FF. Peptides 1995, 16, 973–977. [DOI] [PubMed] [Google Scholar]
- Xu M.; Kontinen V. K.; Panula P.; Kalso E. Effects of (1DMe)NPYF, a synthetic neuropeptide FF analogue, in different pain models. Peptides 1999, 20, 1071–1077. [DOI] [PubMed] [Google Scholar]
- Altier N.; Dray A.; Menard D.; Henry J. L. Neuropeptide FF attenuates allodynia in models of chronic inflammation and neuropathy following intrathecal or intracerebroventricular administration. Eur. J. Pharmacol. 2000, 407, 245–255. [DOI] [PubMed] [Google Scholar]
- Lameh J.; Bertozzi F.; Kelly N.; Jacobi P. M.; Nguyen D.; Bajpai A.; Gaubert G.; Olsson R.; Gardell L. R. Neuropeptide FF receptors have opposing modulatory effects on nociception. J. Pharmacol. Exp. Ther. 2010, 334, 244–254. [DOI] [PubMed] [Google Scholar]
- Simonin F.; Schmitt M.; Laulin J. P.; Laboureyras E.; Jhamandas J. H.; MacTavish D.; Matifas A.; Mollereau C.; Laurent P.; Parmentier M.; Kieffer B. L.; Bourguignon J. J.; Simonnet G. RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhabazi K.; Trigo J. M.; Mollereau C.; Mouledous L.; Zajac J. M.; Bihel F.; Schmitt M.; Bourguignon J. J.; Meziane H.; Petit-demouliere B.; Bockel F.; Maldonado R.; Simonin F. Involvement of neuropeptide FF receptors in neuroadaptive responses to acute and chronic opiate treatments. Br. J. Pharmacol. 2012, 165, 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gealageas R.; Schneider S.; Humbert J. P.; Bertin I.; Schmitt M.; Laboureyras E.; Dugave C.; Mollereau C.; Simonnet G.; Bourguignon J. J.; Simonin F.; Bihel F. Development of sub-nanomolar dipeptidic ligands of neuropeptide FF receptors. Bioorg. Med. Chem. Lett. 2012, 22, 7471–7474. [DOI] [PubMed] [Google Scholar]
- Mouledous L.; Froment C.; Dauvillier S.; Burlet-Schiltz O.; Zajac J. M.; Mollereau C. GRK2 protein-mediated transphosphorylation contributes to loss of function of mu-opioid receptors induced by neuropeptide FF (NPFF2) receptors. J. Biol. Chem. 2012, 287, 12736–12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaubert G.; Bertozzi F.; Kelly N. M.; Pawlas J.; Scully A. L.; Nash N. R.; Gardell L. R.; Lameh J.; Olsson R. Discovery of selective nonpeptidergic neuropeptide FF2 receptor agonists. J. Med. Chem. 2009, 52, 6511–6514. [DOI] [PubMed] [Google Scholar]
- Scully A. L.; Davis R. E.; Vanover K. E.; Gardell L. R.; Lameh J.; Kelly N. M.; Bertozzi F.. Treating neuropathic pain with neuropeptide FF receptor 2 agonists. International Patent WO 2005031000 A2, 2005.
- Mollereau C.; Mazarguil H.; Marcus D.; Quelven I.; Kotani M.; Lannoy V.; Dumont Y.; Quirion R.; Detheux M.; Parmentier M.; Zajac J. M. Pharmacological characterization of human NPFF(1) and NPFF(2) receptors expressed in CHO cells by using NPY Y(1) receptor antagonists. Eur. J. Pharmacol. 2002, 451, 245–256. [DOI] [PubMed] [Google Scholar]
- Findeisen M.; Wurker C.; Rathmann D.; Meier R.; Meiler J.; Olsson R.; Beck-Sickinger A. G. Selective mode of action of guanidine-containing non-peptides at human NPFF receptors. J. Med. Chem. 2012, 55, 6124–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankus J. V.; McCurdy C. R. Nonpeptide ligands of neuropeptide FF: Current status and structural insights. Future Med. Chem. 2012, 4, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gicquel S.; Mazarguil H.; Desprat C.; Allard M.; Devillers J. P.; Simonnet G.; Zajac J. M. Structure–activity study of neuropeptide FF: Contribution of N-terminal regions to affinity and activity. J. Med. Chem. 1994, 37, 3477–3481. [DOI] [PubMed] [Google Scholar]
- Mazarguil H.; Gouarderes C.; Tafani J. A.; Marcus D.; Kotani M.; Mollereau C.; Roumy M.; Zajac J. M. Structure-activity relationships of neuropeptide FF: Role of C-terminal regions. Peptides 2001, 22, 1471–1478. [DOI] [PubMed] [Google Scholar]
- Schwyzer R. ACTH: A short introductory review. Ann. N.Y. Acad. Sci. 1977, 297, 3–26. [DOI] [PubMed] [Google Scholar]
- Nieto M. J.; Philip A. E.; Poupaert J. H.; McCurdy C. R. Solution-phase parallel synthesis of spirohydantoins. J. Comb. Chem. 2005, 7, 258–263. [DOI] [PubMed] [Google Scholar]
- McCurdy C. R.; Prisinzano T. E.. Opioid Receptor Ligands. In Burger’s Medicinal Chemistry, Drug Discovery and Development, 7 ed.; Abraham D. J., Rotella D. P., Eds.; Wiley: Hoboken, NJ, 2010; Vol. 8, pp 569–736. [Google Scholar]
- Sharma S. K.; Jones R. M.; Metzger T. G.; Ferguson D. M.; Portoghese P. S. Transformation of a kappa-opioid receptor antagonist to a kappa-agonist by transfer of a guanidinium group from the 5′- to 6′-position of naltrindole. J. Med. Chem. 2001, 44, 2073–2079. [DOI] [PubMed] [Google Scholar]
- Thomas J. B.; Atkinson R. N.; Namdev N.; Rothman R. B.; Gigstad K. M.; Fix S. E.; Mascarella S. W.; Burgess J. P.; Vinson N. A.; Xu H.; Dersch C. M.; Cantrell B. E.; Zimmerman D. M.; Carroll F. I. Discovery of an opioid kappa receptor selective pure antagonist from a library of N-substituted 4β-methyl-5-(3-hydroxyphenyl)morphans. J. Med. Chem. 2002, 45, 3524–3530. [DOI] [PubMed] [Google Scholar]
- Costanzo M. J.; Yabut S. C.; Almond H. R. Jr.; Andrade-Gordon P.; Corcoran T. W.; De Garavilla L.; Kauffman J. A.; Abraham W. M.; Recacha R.; Chattopadhyay D.; Maryanoff B. E. Potent, small-molecule inhibitors of human mast cell tryptase. Antiasthmatic action of a dipeptide-based transition-state analogue containing a benzothiazole ketone. J. Med. Chem. 2003, 46, 3865–3876. [DOI] [PubMed] [Google Scholar]
- Riches A. G.; Cablewski T.; Glattauer V.; Thissen H.; Meagher L. Scalable synthesis of an integrin-binding peptide mimetic for biomedical applications. Tetrahedron 2012, 68, 9448–9455. [Google Scholar]
- Kayser K. J.; Glenn M. P.; Sebti S. M.; Cheng J. Q.; Hamilton A. D. Modifications of the GSK3beta substrate sequence to produce substrate-mimetic inhibitors of Akt as potential anti-cancer therapeutics. Bioorg. Med. Chem. Lett. 2007, 17, 2068–2073. [DOI] [PubMed] [Google Scholar]
- Alvarez R.; Daniels D. V. A single column method for the assay of adenylate cyclase. Anal. Biochem. 1990, 187, 98–103. [DOI] [PubMed] [Google Scholar]
- Haley T. J.; McCormick W. G. Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br. J. Pharmacol. Chemother. 1957, 12, 12–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J. P.; Hill K. P.; Jiang Q.; Sebastian A.; Archer S.; Bidlack J. M. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: in vivo and in vitro characterization of mu-selective agonist and antagonist activity. J. Pharmacol. Exp. Ther. 1999, 289, 304–311. [PubMed] [Google Scholar]
- Vaught J. L.; Takemori A. E. Differential effects of leucine and methionine enkephalin on morphine-induced analgesia, acute tolerance and dependence. J. Pharmacol. Exp. Ther. 1979, 208, 86–90. [PubMed] [Google Scholar]
- Fang Q.; Wang Y. Q.; He F.; Guo J.; Guo J.; Chen Q.; Wang R. Inhibition of neuropeptide FF (NPFF)-induced hypothermia and anti-morphine analgesia by RF9, a new selective NPFF receptors antagonist. Regul. Pept. 2008, 147, 45–51. [DOI] [PubMed] [Google Scholar]
- Crain S. M.; Shen K. F. Acute thermal hyperalgesia elicited by low-dose morphine in normal mice is blocked by ultra-low-dose naltrexone, unmasking potent opioid analgesia. Brain Res. 2001, 888, 75–82. [DOI] [PubMed] [Google Scholar]
- Crain S. M.; Shen K. F. Naloxone rapidly evokes endogenous kappa opioid receptor-mediated hyperalgesia in naive mice pretreated briefly with GM1 ganglioside or in chronic morphine-dependent mice. Brain Res. 2007, 1167, 31–41. [DOI] [PubMed] [Google Scholar]
- Dubois D.; Gendron L. Delta opioid receptor-mediated analgesia is not altered in preprotachykinin A knockout mice. Eur. J. Neurosci. 2010, 32, 1921–1929. [DOI] [PubMed] [Google Scholar]