

Abstract
Background
Acute myeloid leukemia (AML) remains a difficult disease to treat and requires new therapies to improve treatment outcome. Wee1 inhibitors have been used to prevent activation of the G2 cell cycle checkpoint, thus enhancing the antitumor activity of DNA damaging agents. In this study, we investigated MK-1775 in AML cell lines and diagnostic blast samples to identify sensitive subtypes as well as possible mechanisms of resistance.
Methods
In vitro MK-1775 cytotoxicities of AML cell lines and diagnostic blasts were measured using MTT assays. The effects of MK-1775 on cell cycle progression and related proteins were determined by propidium iodide (PI) staining and flow cytometry analysis and Western blotting. Drug-induced apoptosis was determined using annexin V/PI staining and flow cytometry analysis.
Results
We found that newly diagnosed and relapsed patient samples were equally sensitive to MK-1775. In addition, patient samples harboring t(15;17) translocation were significantly more sensitive to MK-1775 than non-t(15;17) samples. MK-1775 induced apoptosis in both AML cell lines and diagnostic blast samples, accompanied by decreased phosphorylation of CDK1 and CDK2 on Tyr-15 and increased DNA double-strand breaks (DSBs). Time-course experiments, using AML cell lines, revealed a time-dependent increase in DNA DSBs, activation of CHK1 and subsequent apoptosis following MK-1775 treatment, which could be attenuated by a CDK1/2 inhibitor, Roscovitine. Simultaneous inhibition of CHK1 and Wee1 resulted in synergistic anti-leukemic activity in both AML cell lines and primary patient samples ex vivo.
Conclusions
Our study provides compelling evidence that CHK1 plays a critical role in the anti-leukemic activity of MK-1775 and highlights a possible mechanism of resistance to MK-1775. In addition, our study strongly supports the use of MK-1775 to treat both newly diagnosed and relapsed AML, especially cases with t(15;17) translocation, and supports the development of combination therapies with CHK1 inhibitors.
Keywords: Wee1, MK-1775, CHK1, Acute myeloid leukemia
Introduction
Acute myeloid leukemia (AML) is a challenging disease to treat, with overall survival rates of 65% for children and 25% for adults [1],[2]. Resistance to cytarabine (ara-C) and anthracycline [e.g. daunorubicin (DNR)]-based chemotherapy remains a major cause of treatment failure. Therefore, new therapies are greatly needed to treat this disease.
One mechanism of chemotherapy resistance is the induction of G2/M cell cycle arrest, which allows cells to repair and survive DNA damage [3]. Wee1 kinase is a cell cycle checkpoint protein whose primary function is inhibitory phosphorylation of CDK1 and CDK2 on Tyr-15, preventing progression through G2/M and S phase, respectively [4],[5]. Inhibitory phosphorylation of CDK1 and CDK2 on Tyr-15 is removed by CDC25 phosphatases [6],[7]. CDC25 activity is inhibited when phosphorylated by CHK1, which is in turn controlled primarily by ATR kinase [8],[9]. Upon sensing DNA damage or replication stress ATR phosphorylates CHK1, activating it and eventually leading to S and G2 cell cycle arrest, allowing for DNA repair [8],[9].
The first selective and potent Wee1 inhibitor, MK-1775, has been primarily used to target the G2 checkpoint to exert toxicity in cells with impaired p53 function [10]. It has been demonstrated that when combined with DNA damaging agents it is able to abrogate the G2 checkpoint and enhance apoptosis [10]-[18]. In addition, Wee1 inhibition has been shown to induce DNA damage through the induction of replication stress secondary to overactive CDKs and inhibition of DNA repair [19].
In this study, we used AML cell lines and blast samples obtained either at diagnosis or at relapse to investigate the cytotoxic effects of MK-1775 on AML. Our results suggest that MK-1775 may be equally effective in newly diagnosed and relapsed AML. Additionally, we demonstrate that patient samples harboring t(15;17) translocation are significantly more sensitive to MK-1775 than others. Furthermore, our study suggests that MK-1775 treatment induces DNA damage which activates CHK1, CHK1 phosphorylates CDC25, inhibiting the dephosphorylation of CDK1/2, thus countering the effects of MK-1775. Activation of CHK1 can be overcome by the addition of a CHK1 inhibitor, resulting in synergistic anti-leukemic activity. This demonstrates a potential mechanism of resistance to MK-1775 treatment and highlights the importance of combination therapies.
Results
MK-1775 induces DNA damage and apoptosis in AML cells
To investigate the effects of MK-1775 in AML cells, first we determined the protein levels of Wee1, p-CDK1, CDK1, p-CDK2, CDK2 and MYT1 in six AML cell lines. The proteins were expressed at variable levels (Figure 1A). Next, we tested drug sensitivity by MTT assays. The IC50s ranged from about 200-400 nM after 72 h treatment (Figure 1B). To determine if MK-1775 induces cell death, we treated the cell lines with up to 500 nM MK-1775 for 48 h and assessed viability using the trypan blue exclusion assay. As shown in Figure 1C, there was a concentration-dependent increase in dead cells for all six cell lines tested. Annexin V/PI staining and flow cytometry analysis revealed that MK-1775 caused concentration-dependent increase in apoptotic cells (Figure 1D). This was accompanied by decreased p-CDK1 and p-CDK2 and increased total CDK1 and CDK2 (Figure 1E). There was also a concentration-dependent increase of phosphorylated H2AX (γH2AX), indicating increased DNA double-strand breaks (DSBs) [γH2AX is an established biomarker for DNA DSBs [20]]. The CTS and U937 cell lines were chosen as representative AML cell lines for further mechanistic studies.
Figure 1.
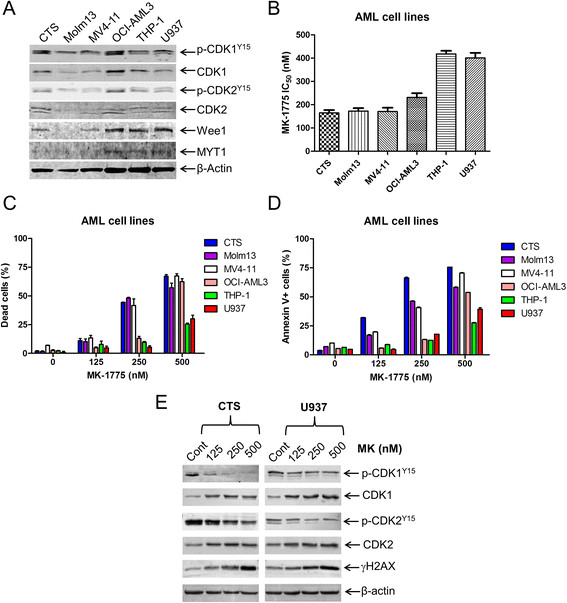
MK-1775 induces apoptosis in AML cells. Panel A: Protein extracts from 6 AML cell lines were subjected to Western blotting and probed with anti-Wee1, -Myt1, -p-CDK1 (Y15), -CDK1, -p-CDK2 (Y15), -CDK2, or -β-actin antibody. Panel B: AML cell lines were cultured for 72 h in complete medium with variable concentrations of MK-1775 (MK) and viable cell numbers were determined using MTT assays. IC50 values were calculated as drug concentration necessary to inhibit 50% growth compared to untreated control cells. The data for panels B and C are presented as mean values ± standard errors from at least 3 independent experiments. Panel C-E: AML cells were treated with MK-1775 for 48 h. The percentage of dead cells was determined by Trypan blue exclusion (Panel C). Apoptotic events were determined by annexin V/PI staining and flow cytometry analyses (Panel D). The data for panel D are presented as mean values ± standard errors of triplicates from one representative experiment which was repeated 3 independent times. Whole cell lysates were subjected to Western blotting, and probed with anti-p-CDK1, -CDK1, -p-CDK2, -CDK2, -γH2AX, or -β-actin antibody (Panel E). Representative Western blots are shown.
Next, we determined ex vivo MK-1775 sensitivity in freshly isolated AML blast samples (n = 29). MK-1775 IC50s ranged from 217 nM (AML#16) to 6.4 μM (AML#27, Table 1). Similar to the cell lines, we detected a concentration-dependent increase in apoptosis for three patient samples after MK-1775 treatment (Figure 2A). Interestingly, the median MK-1775 IC50 for the diagnostic (n = 23) and relapse samples (n = 6) were similar (1176 and 896.1 nM, respectively, p = 0.936, Figure 2B). In addition, we found that patient samples harboring t(15;17) translocation (n = 5) were significantly more sensitive to MK-1775 than non-t(15;17) samples (n = 24, p = 0.007, Figure 2C). Next, we generated a cytarabine resistant cell line (HL60/Ara-C) to determine if they would exhibit cross-resistance to MK-1775. Despite being approximately 600-times more resistant to cytarabine than the parental HL-60 cells (Figure 2D), HL-60/Ara-C cells were more sensitive to MK-1775 treatment (Figure 2E). There was a concentration-dependent increase in apoptotic cells for the HL60/Ara-C cell line, whereas the parental cell line remained relatively unaffected by MK-1775 concentrations up to 500 nM. A concentration-dependent decrease in p-CDK1 and p-CDK2 accompanied by increase of γH2AX was detected in cells from patient AML#10 as well as in HL60/Ara-C (Figure 2F). HL60 cells treated with 500 nM MK-1775 had a small increase of γH2AX and no change in p-CDK1 or p-CDK2, probably due to very low levels of expression prior to drug treatment.
Table 1.
Patient characteristics and MK-1775 sensitivity for primary AML patient samples
| Patient | Gender | Age (year) | Disease status | FAB subtype | WBC (×10 3 /μL) | Blast purity (%) | Cytogenetics | MK-1775 IC 50 (nM) |
|---|---|---|---|---|---|---|---|---|
| AML#0 |
Female |
13 |
At diagnosis |
M2 |
39.9 |
79 |
46, XX, t(8;21)(q22;q22) |
5996.5 |
| AML#1 |
Female |
50 |
At diagnosis |
M4 |
22.66 |
52 |
46, XX, del(5q), add(12p) |
2448.5 |
| AML#2 |
Male |
20 |
At diagnosis |
M2 |
23.55 |
42 |
45, X, -Y, t(8;21)(q22;q22) |
3204.0 |
| AML#3 |
Male |
4 |
At diagnosis |
M3 |
4.71 |
75 |
46, XY, t(15;17)(q22;q21) |
461.0 |
| AML#4 |
Male |
55 |
At relapse |
M2 |
5.64 |
14 |
46, XY, t(8;21)(q22;q22) |
844.0 |
| AML#5 |
Female |
57 |
At relapse |
M2 |
22.16 |
70 |
46, XX |
948.3 |
| AML#6 |
Male |
60 |
At diagnosis |
M3 |
1.83 |
86 |
46, XY, t(15;17)(q22;q21) |
233.7 |
| AML#7 |
Female |
53 |
At relapse |
M4 |
3.87 |
36 |
46, XX |
325.0 |
| AML#8 |
Male |
55 |
At diagnosis |
M2 |
3.87 |
32 |
46, XY |
887.9 |
| AML#9 |
Female |
35 |
At relapse |
M5 |
17.71 |
87 |
47, XX, +10, t(16;21)(p11;q22), add(11p) |
744.4 |
| AML#10 |
Male |
23 |
At diagnosis |
M2 |
25.89 |
53 |
46, XY, del(9q) |
439.7 |
| AML#11 |
Female |
43 |
At diagnosis |
M2 |
25.12 |
91 |
46, XX |
411.3 |
| AML#12 |
Male |
6 |
At diagnosis |
M3 |
93.99 |
85 |
46, XY, t(15;17)(q22;q21) |
277.5 |
| AML#13 |
Male |
52 |
At diagnosis |
M2 |
1.65 |
27 |
46, XY, t(11;15;17)(q25;q15;q21) |
3579.5 |
| AML#14 |
Female |
18 |
At diagnosis |
M3 |
1.62 |
64 |
46, XX |
9113 |
| AML#15 |
Female |
63 |
At diagnosis |
M2 |
0.6 |
23 |
46, XX |
344.1 |
| AML#16 |
Male |
38 |
At diagnosis |
M3 |
2.27 |
90 |
46, XY, t(15;17)(q22;q21) |
216.9 |
| AML#17 |
Female |
45 |
At diagnosis |
M4 |
1.52 |
36 |
46, XX |
3473.0 |
| AML#18 |
Female |
19 |
At relapse |
M2 |
21.55 |
52 |
46, XX, t(8;21)(q22;q22) |
1187.0 |
| AML#19 |
Male |
4 |
At diagnosis |
M4 |
77.32 |
41 |
46, XY |
5587.0 |
| AML#21 |
Male |
42 |
At diagnosis |
M4 |
10.60 |
51 |
46, XY/44, XY, -17, -19, (11q-?) |
1176.0 |
| AML#22 |
Female |
47 |
At diagnosis |
M5 |
244.53 |
80 |
46, XX |
1255.0 |
| AML#23 |
Male |
60 |
At diagnosis |
M5 |
2.10 |
81 |
46, XY, +2, +8, I(12)(q10) |
1480.0 |
| AML#25 |
Female |
12 |
At diagnosis |
M3 |
5.11 |
88 |
46, XX, t(15;17)(q22;q21) |
615.1 |
| AML#26 |
Male |
59 |
At diagnosis |
M2 |
129.98 |
81 |
46, XY |
5868.0 |
| AML#27 |
Female |
41 |
At relapse |
M4 |
40.20 |
24 |
47, XX,del(5q),+8,t(15;18)(q12,q23) |
6384.0 |
| AML#29 |
Male |
9 |
At diagnosis |
M4 |
68.14 |
31 |
46, XY, t(6;9)(p22;q34) |
880.4 |
| AML#31 |
Male |
17 |
At diagnosis |
M2 |
12.94 |
69 |
46, XY |
453.7 |
| AML#32 | Female | 76 | At diagnosis | M5 | 13.93 | 85 | 46, XX | 2888.0 |
Figure 2.
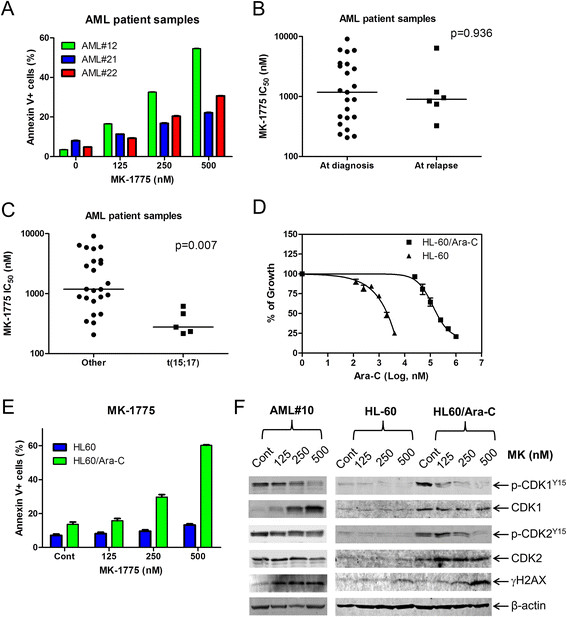
Diagnostic AML blasts from patients either at first diagnosis or at relapse are equally sensitive to MK-1775. Panel A: Freshly isolated AML patient samples were purified by standard Ficoll-Hypaque density centrifugation then treated with MK-1775 for 48 h and apoptotic events were determined by annexin V/PI staining and flow cytometry analyses. Panels B and C: Ex vivo MK-1775 (MK) sensitivity was determined using MTT assays. The horizontal lines indicate median MK-1775 IC50s in each group of AML patient samples. Panel D: Cytarabine sensitivity was determined using MTT assays for the HL-60 and HL-60/Ara-C cell lines. Panel E: The HL60 and HL60/Ara-C cells were treated with MK-1775 for 48 h and apoptotic events were determined by annexin V/PI staining and flow cytometry analyses. The data are presented as mean of triplicates ± standard errors from one representative experiment. Panel F: Freshly isolated cells from patient AML#10 were purified by standard Ficoll-Hypaque density centrifugation. AML#10, HL-60, and HL-60/Ara-C cells were treated with MK-1775 for 48 h. Whole cell lysates were subjected to Western blotting and probed with anti-p-CDK1, -CDK1, -p-CDK2, -CDK2, -γH2AX, or -β-actin antibody.
Next, we investigated the effects of MK-1775 on cell cycle progression in both CTS and U937 cells. Treatment with MK-1775 for 48 h revealed a concentration-dependent decrease of the G2/M population accompanied by concentration-dependent increase of the sub-G1 population (Additional file 1: Figure S1). Time course experiments revealed a time-dependent increase of the sub-G1 population and abrogation of the G2 checkpoint for both cell lines (Figure 3A&C and Additional file 2: Table S1). These changes were accompanied by a time-dependent increase of γH2AX as well as a decrease of p-CDK1 and p-CDK2 (Figure 3B&D). Increased total CDK1 levels were detected following MK-1775 treatment. Increased p-CHK1 was detected as early as 4 h following MK-1775 treatment.
Figure 3.
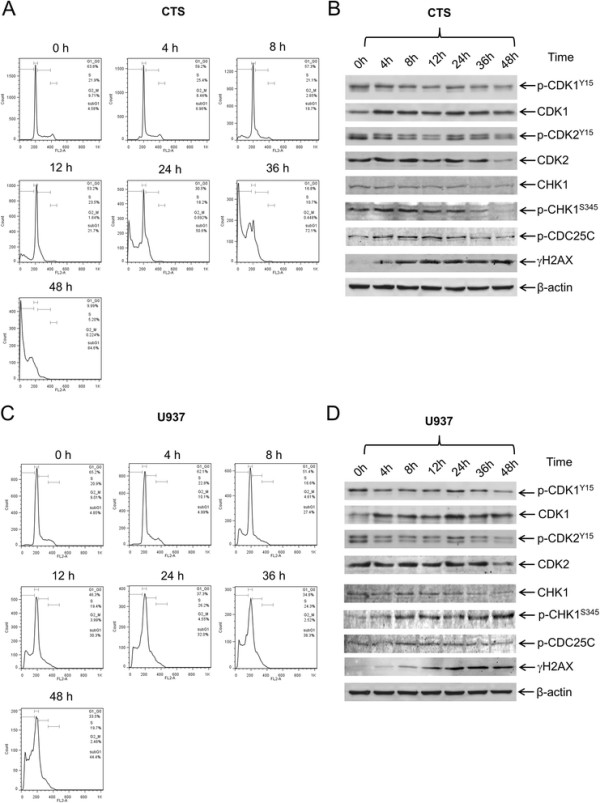
MK-1775 treatment abrogates the G2/M cell cycle checkpoint. CTS cells (Panels A and B) or U937 cells (Panels C and D) were treated with 500 nM MK-1775 for up to 48 h. Samples were taken at the indicated time points and fixed with ethanol for PI staining and cell cycle analysis (Panels A and C). Whole cell lysates were subjected to Western blotting and probed with anti-p-CDK1, -CDK1, -p-CDK2, -CDK2, -CHK1, -pCHK1, -p-cdc25c, -γH2AX, or -β-actin antibody (Panels B and D).
CDK activity is required for MK-1775 anti-leukemic activity
To determine if CDK activity is required for MK-1775 induced DNA damage and apoptosis, we treated AML cells with Roscovitine, a CDK inhibitor. There was a concentration-dependent decrease in viable cells after Roscovitine treatment for both CTS and U937 cells, as measured by MTT assays (Additional file 1: Figure S2A). Increased γH2AX and p-CHK1 was observed following 8 h MK-1775 treatment, which was substantially abolished by the addition of Roscovitine (Figure 4A and Additional file 1: Figure S3A). MK-1775 induced apoptosis at both 8 h and 24 h in both CTS and U937 cell lines. Combined MK-1775 and Roscovitine treatment abolished MK-1775 induced apoptosis (Figure 4B&C). MTT assays revealed clearly antagonistic anti-leukemic interactions as shown in the standard isobolograms (Figure 4D&E). Similar results were obtained with primary AML patient samples (Figure 4F-H). These results demonstrate that CDK activity is required for MK-1775 anti-leukemic activity in AML cells.
Figure 4.
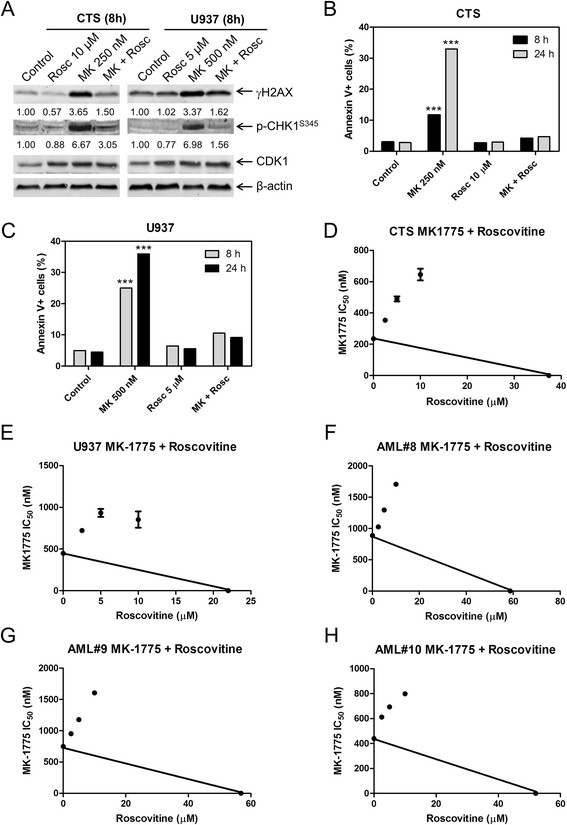
MK-1775-induced DNA damage and apoptosis are dependent on CDK activity. Panel A: U937 and CTS cell lines were treated for 8 h with the indicated concentrations of Roscovitine (Rosc) and MK-1775. Whole cell lysates were subjected to Western blotting and probed with anti-γH2AX, -p-CHK1, -CDK1, or -β-actin antibody. Densitometry measurements, as described in the Materials and methods section, are shown below the corresponding Western blot. Panels B and C: CTS and U937 cell lines were treated with MK-1775 and Roscovitine, alone or in combination, for 8 h and 24 h. Apoptosis was measured by annexin V/PI staining and flow cytometry analyses. The data are presented as mean of triplicates ± standard error from one representative experiment. ***indicates p < 0.0005. Panels D-H: Standard isobologram analyses of anti-leukemic interactions between MK-1775 and Roscovitine in the CTS cell line (Panel D), U937 cell line (Panel E), and patient samples (Panels F-H). The IC50 values of each drug are plotted on the axes; the solid line represents the additive effect, while the points represent the concentrations of each drug resulting in 50% inhibition of proliferation. Points falling below the line indicate synergism whereas those above the line indicate antagonism. The data for the CTS and U937 cell lines are presented as mean values ± standard errors from at least three independent experiments, while the data for the patient samples are presented as mean values of duplicates from one experiment.
CHK1 inhibitor LY2603618 synergizes with MK-1775 to induce apoptosis in AML cells
CTS and U937 cells were treated with MK-1775 plus the selective CHK1 inhibitor LY2603618 for 8 h. There was a concentration-dependent decrease in viable cells following LY2603618 treatment for both CTS and U937 cells (Additional file 1: Figure S2B). γH2AX levels were increased after MK-1775 treatment, which was further increased by the addition of LY2603618 (Figure 5A and Additional file 1: Figure S3B). CDK1 and CDK2 phosphorylation was lower after MK-1775 treatment compared to vehicle control and was further decreased in the combined MK-1775 and LY2603618 treatment. MK-1775-induced apoptosis was significantly enhanced by the addition of LY2603618 (Figure 5B&C). MTT assays revealed synergistic anti-leukemic interactions from the combined MK-1775 and LY2603618 treatment as displayed by the standard isobolograms (Figure 5D&E). Similar results were obtained with primary AML patient samples (Figure 5F-H).
Figure 5.
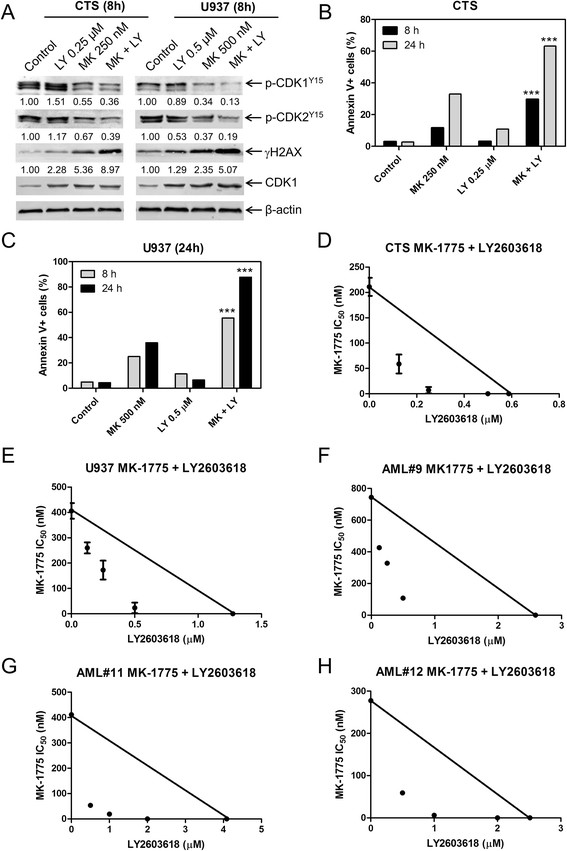
MK-1775 and LY2603618 synergize to induce apoptosis in AML cell lines and primary patient samples. Panel A: U937 and CTS cells were treated for 8 h. Whole cell lysates were subjected to Western blotting and probed with anti-γH2AX, -pCHK1, -p-cdc25c, -p-CDK1, -p-CDK2, -CDK1, or -β-actin antibody. Densitometry measurements, as described in the Materials and methods section, are shown below the corresponding Western blot. Panels B and C: CTS and U937 cells were treated with MK-1775 and LY2603618, alone or in combination, for 8 h and 24 h. Apoptotic events were determined by annexin V/PI staining and flow cytometry analyses. The data are presented as mean of triplicates ± standard error from one representative experiment. ***indicates p < 0.0005. Panels D-H: Standard isobologram analyses of antitumor interactions between MK-1775 and LY2603618 in the CTS cell line (Panel D), U937 cell line (Panel E), and patient samples (Panels F-H). The IC50 values of each drug are plotted on the axes; the solid line represents the additive effect, while the points represent the concentrations of each drug resulting in 50% inhibition of proliferation. Points falling below the line indicate synergism whereas those above the line indicate antagonism. The data for the CTS and U937 cell lines are presented as mean values ± standard errors from at least three independent experiments, while the data for the patient samples are presented as mean of duplicates from one experiment.
Discussion
Wee1 inhibitors have primarily been used to target the G2 cell cycle checkpoint, which is activated in response to DNA damaging agents, and has shown promising results when combined with standard chemotherapy drugs [10]-[18]. Using single agent treatment we demonstrate that AML patient samples with t(15;17) translocation are more sensitive to MK-1775 than non-t(15;17), suggesting a possible treatment for patients with t(15;17) who do not respond to all-trans retinoic acid and/or arsenic trioxide based therapies. The t(15;17) translocation results in rearrangement of the promyelocytic leukemia (PML) gene and the retinoic acid receptor α (RARα) gene, generating the PML/RARα fusion protein [21]. This fusion protein acts as a strong transcriptional repressor for numerous genes which are involved in proliferation, DNA repair, and cell death [22],[23], and thus may contribute to the elevated MK-1775 sensitivity. We also found that newly diagnosed and relapsed AML patient samples have similar ex vivo MK-1775 sensitivities, as measured by MTT assays. In addition, our studies in a cytarabine resistant cell line further support the preclinical development of MK-1775 for the treatment of relapsed AML.
In our study, we demonstrate that MK-1775 treatment results in increased phosphorylation of CHK1 and H2AX, in agreement with Chaudhuri et al.’s report [13]. Furthermore, increased phosphorylation of both CHK1 and H2AX is dependent on CDK activity. Inhibition of CHK1 in combination with MK-1775 resulted in synergistic anti-leukemic activities in both AML cell lines and primary patient samples. Although combined Wee1 and CHK1 inhibition has been reported in various cancer types [24]-[26], including AML [13], our study confirmed the enhanced anti-leukemic activity with a different CHK1 inhibitor, LY2603618, than has been previously reported (MK-8776, PF-00477736, AR458323) and found similar synergistic anti-leukemic activity. Thus, adding further evidence for the clinical efficacy of combined Wee1 and CHK1 inhibition for the treatment of AML.
Based on our own results and those previously reported, we propose a mechanism for the cooperative anti-leukemic activity of MK-1775 and LY2603618 in Figure 6. MK-1775 inhibits Wee1, inhibiting phosphorylation of CDK1/2, thus allowing CDK1/CDK2 to remain active. Overactive CDK1/2 eventually leads to DNA DSBs triggering activation of ATM/ATR and CHK1. Activated CHK1 phosphorylates CDC25s, leading to decreased removal of the inhibitory phosphorylation on CDK1/2, in turn limiting the amount of active CDK1/2, arresting the cell cycle to allow for adequate DNA damage repair. The addition of LY2603618 inhibits the inhibitory phosphorylation of CDC25s, accordingly maintaining their phosphatase activity, which maintains active CDK1/2 pools, perpetuating the DNA damage induced by MK-1775 treatment and eventually resulting in enhanced apoptosis.
Figure 6.
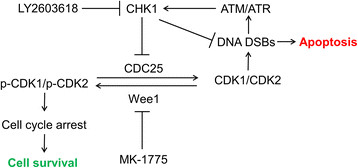
Proposed mechanisms for the anti-leukemic interaction of MK-1775 and LY2603618 in AML cells. MK-1775 inhibits Wee1, leading to decreased inhibitory phosphorylation of CDK1/2, allowing CDK1/CDK2 to remain active. This eventually leads to DNA DSBs which triggers activation of ATM/ATR and activates CHK1. Active CHK1 inhibits CDC25s leading to decreased removal of the inhibitory phosphorylation on CDK1/2, thus limiting the amount of active CDK1/2 and the resulting DNA damage from MK-1775 treatment. The addition of a CHK1 inhibitor (e.g. LY2603618) would inhibit the CHK1 DNA repair pathway, allowing for the DNA damage to accumulate and cause apoptosis.
In summary, our study demonstrates that MK-1775 has potential as an anti-leukemic drug in the treatment of AML. Particularly, our patient sample data supports the investigation of MK-1775 in relapsed AML, as they had similar sensitivities as newly diagnosed cases. In addition, AML cases with t(15;17) translocation were significantly more sensitive to MK-1775 treatment, suggesting a possible role for patients who do not respond to all-trans retinoic acid and/or arsenic trioxide-based therapies. We also demonstrate that MK-1775 in combination with cytarabine results in synergistic anti-leukemic activity in AML cell lines and patient samples (Additional file 1: Figure S4). Lastly, our results highlight CHK1 activation as a potential mechanism of resistance to MK-1775 treatment, thus supports the development of combination therapies.
Materials and methods
Drugs
MK-1775 (MK), Roscovitine (Rosc), and LY2603618 (LY) were purchased from Selleck Chemicals (Houston, TX, USA). Cytarabine (ara-C) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
The THP-1, MV4-11, U937, HL-60 cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). The OCI-AML3 cell line was purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). MOLM-13 cells were purchased from AddexBio (San Diego, CA, USA). The CTS cell line was a gift from Dr. A Fuse from the National Institute of Infectious Diseases, Tokyo, Japan. The cell lines were cultured in RPMI 1640 (except OCI-AML3, which was cultured in alpha-MEM) with 10-15% fetal bovine serum (Hyclone, Logan, UT, USA), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. All cells were cultured in a 37°C humidified atmosphere containing 5% CO2/95% air.
AML blast samples obtained either at diagnosis or at relapse were purified by standard Ficoll-Hypaque density centrifugation, then cultured in RPMI 1640 with 20% fetal bovine serum supplemented with ITS solution (Sigma-Aldrich) and 20% supernatant of the 5637 bladder cancer cell line [as a source of granulocyte-macrophage colony-stimulating factor [27],[28]].
HL-60 cytarabine resistant cells (designated HL-60/Ara-C) were generated by stepwise selection of HL-60 cells in the presence of cytarabine, until they could be maintained in the presence of 600 nM cytarabine.
Clinical samples
Diagnostic blast samples were obtained from the First Hospital of Jilin University. Written informed consent was provided according to the Declaration of Helsinki. This study was approved by the Human Ethics Committee of the First Hospital of Jilin University. Clinical samples were screened for FLT3-ITD, NPM1, C-kit, CEBPA, IDH1, IDH2 and DNMT3A gene mutations. The samples were also screened for the following fusion genes by real-time RT-PCR: PML-RARα, BCR-ABL, AML1-MDS1, MLL-AF10, MLL-AF4, MLL-ELL, SET-CAN, TLS-ERG, NPM-RARα, E2A-PBX1, AML1-EAP, MLL-AF17, MLL-AF6, MLL-ENL, SIL-TAL1, HOX11, PLZF-RARα, TEL-AML1, DEK-CAN, MLL-AF1p, MLL-AF9, NPM-ALK, TEL-ABL, EIP1L1-PDGFRA, AML1-ETO, CBFB-MYH11, E2A-HLF, MLL-AF1q, MLL-AFX, NPM-MLF1, dupMLL, and TEL-PDGFB.
In vitro cytotoxicity assays
In vitro cytotoxicities of the AML cells were measured by using MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazoliumbromide, Sigma-Aldrich) assays, as previously described [29],[30]. Briefly, the cells were treated with variable concentrations of MK-1775 for 72 hours. MTT was added to a final concentration of 1 mM and cells were incubated for 4 hours at 37°C. The cells were lysed overnight using 10% SDS in 10 mM HCL and plates were read at 590 nm using a microplate reader. IC50 values were calculated as drug concentrations necessary to inhibit 50% growth compared to vehicle control treated cells. The cell line IC50 values are presented as mean values ± standard errors from at least three independent experiments. The IC50 values for the patient samples are means of duplicates from one experiment, due to limited sample. Patient samples for the combined drug treatments were chosen based on sample availability.
Western blot analysis
Cells were lysed in the presence of protease and phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN, USA). Whole cell lysates were subjected to SDS-polyacrylamide gel electrophoresis, electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Inc., Rockford, IL, USA) and immunoblotted with anti-p-CDK1 (Y15), -CDK1, -p-CDK2 (Y15), -CDK2, -Wee1, -MYT1, -γH2AX, -CHK1, -p-CHK1 (S345), -p-CDC25C (S216) (Cell Signaling Technology, Danvers, MA, USA), or -β-actin (Sigma-Aldrich) antibody, as previously described [31],[32]. Immunoreactive proteins were visualized using the Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE, USA), as described by the manufacturer. Western blots were repeated at least three times and one representative blot is shown. Only one patient sample was used for MK-1775 treatment and subsequent Western blot analysis due to the limited amount of sample available. Densitometry measurements were made using Odyssey V3.0 (Li-Cor), normalized to β-actin, and then fold change relative to no drug control was calculated.
Apoptosis
AML cells were treated with MK-1775, Roscovitine, LY2603618, or the indicated combinations and subjected to flow cytometry analysis to determine drug-induced apoptosis using an annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis Kit (Beckman Coulter; Brea, CA, USA), as previously described [29],[33]. Results are expressed as percent of annexin V + cells. Experiments with AML cell lines were performed 3 independent times in triplicates and data presented are from one representative experiment, while patient sample experiments were performed once in triplicates. Data are presented as mean values ± standard errors from one representative experiment. Due to limited sample, only three patient samples were evaluated for MK-1775-induced apoptosis by flow cytometry.
Cell cycle progression
Cells were treated with the indicated drugs for up to 48 h. The cells were harvested and fixed with ice-cold 80% (v/v) ethanol for 24 h. The cells were pelleted, washed with PBS, and resuspended in PBS containing 50 μg/mL propidium iodide (PI), 0.1% Triton X-100 (v/v), and 1 μg/mL DNase-free RNase. DNA content was determined by flow cytometry analysis using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA) as previously described [34]. Cell cycle analysis was performed using Multicycle software (Phoenix Flow Systems, Inc., San Diego, CA, USA). Histograms were created using FlowJo v7.6.5 (Tree Star, Ashland, OR, USA).
Statistical analysis
Differences in cell apoptosis between treated (individually or combined) and untreated cells were compared using the pair-wise two-sample t-test. The p value for the differences between MK-1775 IC50s for the groups of patient samples was calculated using the Mann–Whitney U test. Statistical analyses were performed with GraphPad Prism 5.0.
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
WQ and CX performed the molecular biology studies. CL, JWT, YW, HL, and YG participated in the design and coordination of the study. CL, JTC, HE, JWT, YW, HL, and YG participated in the data analysis and interpretation. CL, JTC, HE, JWT, YW, HL, and YG helped to draft the manuscript. All authors read and approved the final manuscript.
Additional files
Supplementary Material
MK-1775 causes concentration-dependent abrogation of the G2/M cell cycle checkpoint. Figure S2. Concentration-dependent decrease in viable cells after roscovitine or LY2603618 treatment. Figure S3. Densitometry measurements for the western blot experiments corresponding to Figures 4A and 5A. Figure S4. MK-1775 and cytarabine synergize in AML cells.
Cell cycle distribution of CTS and U937 cells following MK-1775 treatment.
Contributor Information
Wenxiu Qi, Email: qiwenxiu0517@163.com.
Chengzhi Xie, Email: xiec@karmanos.org.
Chunhuai Li, Email: lichunhuai99@126.com.
J Timothy Caldwell, Email: jtcaldwe@med.wayne.edu.
Holly Edwards, Email: pitmanh@karmanos.org.
Jeffrey W Taub, Email: jtaub@med.wayne.edu.
Yue Wang, Email: wangyue7373@126.com.
Hai Lin, Email: maillinhai@sina.com.
Yubin Ge, Email: gey@karmanos.org.
Acknowledgments
This study was support by a Start-up Fund from Jilin University, Changchun, China, and grants from the National Natural Science Foundation of China (NSFC 31271477 and 81200363). Mr. JTC is a predoctoral trainee supported by T32 CA009531 from the National Cancer Institute. The funders had no role in study design, data collection, analysis and interpretation, writing of the manuscript, and in the decision to submit the article for publication. The authors would like to thank Dr. Mark Stout for generating the cytarabine resistant HL60 cells.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Rubnitz JE, Inaba H, Dahl G, Ribeiro RC, Bowman WP, Taub J, Pounds S, Razzouk BI, Lacayo NJ, Cao X, Meshinchi S, Degar B, Airewele G, Raimondi SC, Onciu M, Coustan-Smith E, Downing JR, Leung W, Pui CH, Campana D. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. The lancet oncology. 2010;11:543–552. doi: 10.1016/S1470-2045(10)70090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–1957. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, Itadani H, Takahashi-Suzuki I, Fukasawa K, Oki H, Nambu T, Jiang J, Sakai T, Arakawa H, Sakamoto T, Sagara T, Yoshizumi T, Mizuarai S, Kotani H. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A, Turner NC. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–539. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, Molkentine JM, Mason KA, Meyn RE. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17:5638–5648. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri L, Vincelette ND, Koh BD, Naylor RM, Flatten KS, Peterson KL, McNally A, Gojo I, Karp JE, Mesa RA, Sproat LO, Bogenberger JM, Kaufmann SH, Tibes R. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica. 2014;99:688–696. doi: 10.3324/haematol.2013.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, Hirsch HA, Feldman I, Benita Y, Toniatti C, Zawel L, Fawell SE, Gilliland DG, Shumway SD. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. 2013;12:1442–1452. doi: 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, Imagaki K, Ohtani J, Sakai T, Yoshizumi T, Mizuarai S, Iwasawa Y, Kotani H. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–522. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, Germing U, Haas R, Dohner H, Gattermann N. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 2006;106:112–119. doi: 10.1002/cncr.21552. [DOI] [PubMed] [Google Scholar]
- Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, Shumway SD, Mizuarai S, Hirai H, Maitra A, Hidalgo M. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–2806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, Reigan P, Porter CC. Inhibition of Wee1 Sensitizes Cancer Cells to Antimetabolite Chemotherapeutics In Vitro and In Vivo, Independent of p53 Functionality. Mol Cancer Ther. 2013;12:2675–2684. doi: 10.1158/1535-7163.MCT-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Sillje HH, de Vries EG, van Vugt MA. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2013;32:3001–3008. doi: 10.1038/onc.2012.296. [DOI] [PubMed] [Google Scholar]
- Redon CE, Nakamura AJ, Zhang YW, Ji JJ, Bonner WM, Kinders RJ, Parchment RE, Doroshow JH, Pommier Y. Histone gammaH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin Cancer Res. 2010;16:4532–4542. doi: 10.1158/1078-0432.CCR-10-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignani F, Ferrucci PF, Testa U, Talamo G, Fagioli M, Alcalay M, Mencarelli A, Grignani F, Peschle C, Nicoletti I, Pelicci PG. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell. 1993;74:423–431. doi: 10.1016/0092-8674(93)80044-F. [DOI] [PubMed] [Google Scholar]
- Lin RJ, Sternsdorf T, Tini M, Evans RM. Transcriptional regulation in acute promyelocytic leukemia. Oncogene. 2001;20:7204–7215. doi: 10.1038/sj.onc.1204853. [DOI] [PubMed] [Google Scholar]
- Hoemme C, Peerzada A, Behre G, Wang Y, McClelland M, Nieselt K, Zschunke M, Disselhoff C, Agrawal S, Isken F, Tidow N, Berdel WE, Serve H, Muller-Tidow C. Chromatin modifications induced by PML-RARalpha repress critical targets in leukemogenesis as analyzed by ChIP-Chip. Blood. 2008;111:2887–2895. doi: 10.1182/blood-2007-03-079921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, Pawel B, Shumway SD, Maris JM, Cole KA. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73:776–784. doi: 10.1158/0008-5472.CAN-12-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrassa L, Chila R, Lupi M, Ricci F, Celenza C, Mazzoletti M, Broggini M, Damia G. Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012;11:2507–2517. doi: 10.4161/cc.20899. [DOI] [PubMed] [Google Scholar]
- Davies KD, Cable PL, Garrus JE, Sullivan FX, von Carlowitz I, Huerou YL, Wallace E, Woessner RD, Gross S. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol Ther. 2011;12:788–796. doi: 10.4161/cbt.12.9.17673. [DOI] [PubMed] [Google Scholar]
- Taub JW, Matherly LH, Stout ML, Buck SA, Gurney JG, Ravindranath Y. Enhanced metabolism of 1-beta-D-arabinofuranosylcytosine in Down syndrome cells: a contributing factor to the superior event free survival of Down syndrome children with acute myeloid leukemia. Blood. 1996;87:3395–3403. [PubMed] [Google Scholar]
- Niu X, Wang G, Wang Y, Caldwell JT, Edwards H, Xie C, Taub JW, Li C, Lin H, Ge Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia. 2014;28:1557–1560. doi: 10.1038/leu.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Edwards H, Xu X, Zhou H, Buck SA, Stout ML, Yu Q, Rubnitz JE, Matherly LH, Taub JW, Ge Y. Mechanisms of synergistic antileukemic interactions between valproic acid and cytarabine in pediatric acute myeloid leukemia. Clin Cancer Res. 2010;16:5499–5510. doi: 10.1158/1078-0432.CCR-10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Xie C, Edwards H, Zhou H, Buck SA, Ge Y. Inhibition of histone deacetylases 1 and 6 enhances cytarabine-induced apoptosis in pediatric acute myeloid leukemia cells. PLoS One. 2011;6:e17138. doi: 10.1371/journal.pone.0017138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dombkowski AA, LaFiura KM, Tatman D, Yedidi RS, Stout ML, Buck SA, Massey G, Becton DL, Weinstein HJ, Ravindranath Y, Matherly LH, Taub JW. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006;107:1570–1581. doi: 10.1182/blood-2005-06-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Stout ML, Tatman DA, Jensen TL, Buck S, Thomas RL, Ravindranath Y, Matherly LH, Taub JW. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst. 2005;97:226–231. doi: 10.1093/jnci/dji026. [DOI] [PubMed] [Google Scholar]
- Edwards H, Xie C, LaFiura KM, Dombkowski AA, Buck SA, Boerner JL, Taub JW, Matherly LH, Ge Y. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009;114:2744–2752. doi: 10.1182/blood-2008-09-179812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, He J, Zhao J, Yun W, Xie C, Taub JW, Azmi A, Mohammad RM, Dong Y, Kong W, Guo Y, Ge Y. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PloS One. 2012;7:e52095. doi: 10.1371/journal.pone.0052095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MK-1775 causes concentration-dependent abrogation of the G2/M cell cycle checkpoint. Figure S2. Concentration-dependent decrease in viable cells after roscovitine or LY2603618 treatment. Figure S3. Densitometry measurements for the western blot experiments corresponding to Figures 4A and 5A. Figure S4. MK-1775 and cytarabine synergize in AML cells.
Cell cycle distribution of CTS and U937 cells following MK-1775 treatment.