

Background: Mesotrypsin targets protease inhibitors as substrates; substrate recognition depends on sequence and conformational motifs.
Results: Mesotrypsin cleaves three human Kunitz domains as specific substrates, but other similarly shaped substrates are cleaved less efficiently.
Conclusion: Substrate conformation aids recognition by mesotrypsin but is not sufficient for efficient cleavage.
Significance: Mesotrypsin cleavage of human Kunitz domains may contribute to cancer progression.
Keywords: Crystal Structure, Enzyme Kinetics, Protease Inhibitor, Protein Degradation, Protein Structure, Protein-Protein Interaction, Proteolysis, Serine Protease, Substrate Specificity, Trypsin
Abstract
Mesotrypsin is an isoform of trypsin that is uniquely resistant to polypeptide trypsin inhibitors and can cleave some inhibitors rapidly. Previous studies have shown that the amyloid precursor protein Kunitz protease inhibitor domain (APPI) is a specific substrate of mesotrypsin and that stabilization of the APPI cleavage site in a canonical conformation contributes to recognition by mesotrypsin. We hypothesized that other proteins possessing potential cleavage sites stabilized in a similar conformation might also be mesotrypsin substrates. Here we evaluated a series of candidate substrates, including human Kunitz protease inhibitor domains from amyloid precursor-like protein 2 (APLP2), bikunin, hepatocyte growth factor activator inhibitor type 2 (HAI2), tissue factor pathway inhibitor-1 (TFPI1), and tissue factor pathway inhibitor-2 (TFPI2), as well as E-selectin, an unrelated protein possessing a potential cleavage site displaying canonical conformation. We find that Kunitz domains within APLP2, bikunin, and HAI2 are cleaved by mesotrypsin with kinetic profiles of specific substrates. TFPI1 and TFPI2 Kunitz domains are cleaved less efficiently by mesotrypsin, and E-selectin is not cleaved at the anticipated site. Cocrystal structures of mesotrypsin with HAI2 and bikunin Kunitz domains reveal the mode of mesotrypsin interaction with its canonical substrates. Our data suggest that major determinants of mesotrypsin substrate specificity include sequence preferences at the P1 and P′2 positions along with conformational stabilization of the cleavage site in the canonical conformation. Mesotrypsin up-regulation has been implicated previously in cancer progression, and proteolytic clearance of Kunitz protease inhibitors offers potential mechanisms by which mesotrypsin may mediate pathological effects in cancer.
Introduction
Mesotrypsin, encoded by the PRSS3 gene, is a minor human trypsin that represents less than 10% of trypsinogen secreted by the pancreas; like other pancreatic trypsinogens, it undergoes proteolytic activation by enteropeptidase in the duodenum (1, 2). Alternately spliced isoforms of mesotrypsinogen that differ in exon 1 are expressed in the brain (3, 4) and in keratinocytes (5). These alternative isoforms, trypsinogens 4 and 5, do not possess classic signal sequences, and the mechanisms by which they are trafficked and activated are not yet fully elucidated, but functional studies provide evidence for significant roles for active mesotrypsin in keratinocyte differentiation and skin desquamation (6, 7). PRSS3 transcripts, primarily encoding trypsinogen 4, are also expressed in many epithelial cells and cancers, and functional studies likewise provide evidence for extracellular roles for active mesotrypsin in tumor progression of breast (8), pancreatic (9), and prostate cancers (10, 11). An unusual feature of mesotrypsin is its extreme resistance to endogenous proteinaceous trypsin inhibitors (1, 12, 13) and its ability to digest some of these inhibitors as substrates (6, 14, 15). We and others have hypothesized that physiological functions of mesotrypsin may derive from its ability to degrade protein protease inhibitors, for example by digesting plant-derived serine protease inhibitors, such as soy bean trypsin inhibitor in the diet (14), or by degrading the lymphoepithelial Kazal-type inhibitor LEKTI to promote desquamation (6). Of note, the specific mesotrypsin substrates responsible for its role in promoting invasion and metastasis of prostate and pancreatic cancers (9, 10) have not been identified.
One of the most highly represented families of mammalian serine protease inhibitors is the I2 or Kunitz-BPTI family (MEROPS database designation (16)). The inhibitory domains of these inhibitors, as exemplified by the archetypal example of bovine pancreatic trypsin inhibitor (BPTI),3 are composed of about 58 amino acid residues that assume a very compact pear-shaped fold stabilized by three intramolecular disulfide bonds (17). Like other serine protease inhibitors belonging to the canonical inhibitor class, they feature a protease binding loop that is preconfigured in a conserved canonical backbone conformation, highly complementary to the serine protease active site (18, 19). These inhibitors follow the Laskowski mechanism of inhibition, wherein they bind very tightly to a target protease in a substrate-like fashion, positioning a specific “reactive site” bond for cleavage in the active site, but are cleaved many orders of magnitude more slowly than a normal substrate (19–21).
In biochemical and structural studies to explore the mechanism by which mesotrypsin targets protease inhibitors as substrates, we have focused on two inhibitors from the Kunitz-BPTI family: BPTI and the Kunitz protease inhibitor domain of the human amyloid precursor protein (APPI). Both of these potent trypsin inhibitors are bound orders of magnitude more weakly and cleaved orders of magnitude more rapidly by mesotrypsin than by typical trypsins (15, 22). Furthermore, the canonical conformation of the binding loop was found to be critical for recognition as a substrate by mesotrypsin, because an unstructured peptide containing the same reactive site sequence as APPI was not bound or cleaved by mesotrypsin (15). Surprisingly, we found considerable differences between BPTI and APPI in their relative susceptibility to cleavage by mesotrypsin. Whereas APPI was efficiently cleaved by mesotrypsin in seconds, with kinetics resembling those of a good substrate (15), BPTI behaved as a weak temporary inhibitor of mesotrypsin and was only gradually cleaved over the course of hours (22). Mutagenesis studies targeting the canonical binding loops of APPI and BPTI revealed that these differences were not solely determined by the cleavage site sequences, but rather a large proportion of the superior proteolytic stability of BPTI was endemic to the protein scaffold (23), suggesting that the resistance of different Kunitz domains to proteolysis is quite variable.
The human proteome reveals many other Kunitz domain-containing proteins in addition to APP, some of which have been found to inhibit trypsin and/or other serine proteases of tryptic-like specificity and some of which are coexpressed with mesotrypsin in epithelial tissues and cancer cells. However, it is not known whether these inhibitors are substrates or inhibitors of mesotrypsin. If cleaved rapidly like APPI, these Kunitz domain-containing proteins may represent physiological substrates of mesotrypsin, and their clearance or inactivation by mesotrypsin could represent mechanisms of importance in understanding the role of mesotrypsin in physiology and disease. If more resistant to cleavage like BPTI, these proteins may represent physiological inhibitors of mesotrypsin and might also offer candidate scaffolds for the development of human recombinant protein therapeutics targeting mesotrypsin. Here, we have produced and evaluated a series of recombinant human Kunitz domains as candidate substrates or inhibitors of mesotrypsin. Our results, identifying new mesotrypsin substrates, offer insights into the biochemical and structural basis of substrate recognition by mesotrypsin and highlight potential biological pathways through which mesotrypsin may exert its cancer-promoting activities.
EXPERIMENTAL PROCEDURES
Generation of Expression Constructs for Human Kunitz Domains
To generate expression constructs for human Kunitz domains, human amino acid sequences were identified for entries in the UniProtKB/Swiss-Prot database, and corresponding synthetic genes, codon-optimized for expression in Pichia pastoris, were synthesized by Mr. Gene GmbH (Regensburg, Germany). Kunitz domain boundaries were standardized to include four native amino acid residues preceding the first Cys residue and three native amino acid residues following the sixth/last Cys residue. Inhibitor residue numbering used within this work is similarly standardized to the BPTI numbering scheme for consistency; a single-residue insertion of Ala between residues 5 and 6 of bikunin-KD2 was numbered with the insertion code 5A. Synthetic genes contained the 5′-flanking sequence CTCGAGAAAAGAGAG, containing an XhoI restriction site (underlined) and encoding the yeast Kex2 cleavage site Leu-Glu-Lys-Arg↓Glu, and the 3′-flanking sequence TGATAAGCGGCCGC, encoding tandem stop codons and containing a NotI restriction site (underlined). Synthetic genes were subcloned into the pPICZαA vector (Invitrogen) using XhoI and NotI restriction sites, and sequences were confirmed by DNA sequencing (Mayo Clinic Sequencing Core, Rochester, MN). Expression constructs were generated for human amyloid-like protein 2 Kunitz domain (APLP2-KD; UniProt: Q06481), bikunin Kunitz domain 2 (bikunin-KD2, inter-α-trypsin inhibitor light chain Kunitz domain 2; Uniprot: P02760), hepatocyte growth factor activator inhibitor type 1 Kunitz domain 1 (HAI1-KD1; Uniprot: O43278), hepatocyte growth factor activator inhibitor type 2 Kunitz domains 1 and 2 (HAI2-KD1 and HAI2-KD2; Uniprot: O43291), tissue factor pathway inhibitor Kunitz domains 1 and 2 (TFPI1-KD1 and TFPI1-KD2; Uniprot: P10646), and tissue factor pathway inhibitor 2 Kunitz domain 1 (TFPI2-KD1; Uniprot: P48307).
Protein Expression and Purification
Recombinant human mesotrypsinogen was produced in Escherichia coli, isolated from inclusion bodies, refolded, purified by ecotin affinity chromatography, activated by bovine enteropeptidase cleavage, and further purified by benzamidine affinity chromatography as described previously (22). Active mesotrypsin concentrations were determined by active site titration with 4-nitrophenyl 4-guanidinobenzoate (24). For crystallization studies, a catalytically inactive active site mutant enzyme, mesotrypsin-S195A, was produced and purified following similar procedures.
Recombinant Kunitz domains were expressed and purified from the methylotropic yeast P. pastoris under control of the alcohol oxidase (AOX1) promotor, following protocols described previously for BPTI (23, 25) and APPI (15, 26) with minor modifications. Briefly, expression cultures grown in BMMY medium (buffered medium containing methanol and yeast nitrogen base) at 30 °C were harvested after 72 h. The supernatant was subjected to ammonium sulfate precipitation (95% saturation) at room temperature. Pellets were resuspended and dialyzed using 3.5 kDa cut-off dialysis tubing (Thermo Scientific) first against double-distilled H2O and then against 20 mm buffer appropriate for the subsequent ion exchange chromatography. Dialyzed samples were purified by ion exchange chromatography using resins and gradients as follows: APLP2-KD, SP-Sepharose FF (GE Healthcare), and 20 mm bis-tris, pH 5.8, eluted with a gradient of 0–1 m NaCl; bikunin-KD2, SP-Sepharose FF, and 20 mm bis-tris, pH 6.5, eluted with a gradient of 0–1 m NaCl; HAI2-KD1, SP-Sepharose FF, and 20 mm bis-tris, pH 5.8, eluted with a gradient 0–1 m NaCl; TFPI1-KD1, Q-Sepharose FF, and 20 mm Tris, pH 8.5, eluted with a gradient of 0–1 m NaCl; TFPI1-KD2, Q-Sepharose FF (GE Healthcare), and 50 mm potassium phosphate, pH 6.7, eluted with a gradient of 0–1 m NaCl; TFPI2-KD1, Q-Sepharose FF, and 50 mm potassium phosphate, pH 7.0, eluted with a gradient of 0–1 m NaCl. All proteins were further purified on a bovine trypsin affinity column and eluted with a gradient of 0–100% 50 mm HCl. Concentrations of Kunitz domains were determined by titration with bovine trypsin (Sigma) (22).
Mesotrypsin Inhibition Studies
For competitive inhibition studies, initial rates for cleavage of the chromogenic substrate benzoxycarbonyl-Gly-Pro-Arg-p-nitroanilide (Z-GPR-pNA; Sigma) by 0.25 nm mesotrypsin were determined at varying substrate and inhibitor concentrations. For APLP2-KD, bikunin-KD2, HAI2-KD1, and TFPI1-KD2, assays were conducted as described previously in a Varian Cary-100 spectrophotometer at 37 °C using 40× working stocks of mesotrypsin, Z-GPR-pNA, and Kunitz domains (22). Assay buffer (740 μl of 100 mm Tris, pH 8.0, 1 mm CaCl2), substrate (20 μl of 40× working stock made in DMSO), and inhibitor (20 μl of 40× working stock prepared in 100 mm Tris, pH 8.0) were mixed and equilibrated in cuvettes prior to initiation of the reaction by the addition of mesotrypsin (20 μl of 40× working stock prepared in 20 mm sodium acetate, pH 4.5, 1 mg/ml bovine serum albumin, and 10 mm CaCl2). Reactions were followed spectroscopically for 3 min, and initial rates were determined from the absorbance increase caused by the release of p-nitroaniline (ϵ410 = 8480 m−1 cm−1) (27). For TFPI1-KD1 and TFPI2-KD1, assays were conducted similarly but in a volume of 80 μl in an Agilent 8453 spectrophotometer (Agilent Technologies, Santa Clara, CA) using 4× working stocks of mesotrypsin, Z-GPR-pNA, and Kunitz domains at 37 °C. In this case, 20 μl of assay buffer, 20 μl of 4× substrate, 20 μl of 4× inhibitor, and 20 μl of 4× mesotrypsin were used to make up the reaction. Duplicate reactions were run for each combination of substrate and inhibitor concentrations. Data were globally fitted by multiple regression to Equation 1, the classic competitive inhibition equation, using Prism (GraphPad Software, San Diego CA). Reported inhibition constants represent average values obtained from 2–3 independent experiments.
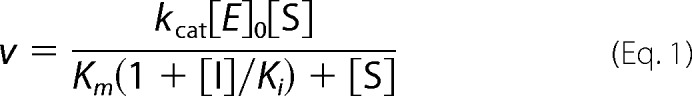 |
Kunitz Domain Hydrolysis Studies
The depletion of intact Kunitz domain in time course incubations with active mesotrypsin was monitored by HPLC. Reaction of each purified recombinant Kunitz domain with mesotrypsin was carried out in 0.1 m Tris, pH 8.0, and 1 mm CaCl2 at 37 °C. Initial studies evaluated for each substrate whether better HPLC resolution of intact Kunitz domain from cleaved products was achieved using a reducing/denaturing sample workup versus a nondenaturing/nonreducing sample workup. For final kinetics studies using bikunin-KD2, HAI2-KD1, TFPI1-KD1, TFPI1-KD2, and TFPI2-KD1 as substrates, aliquots for HPLC analysis were withdrawn at periodic intervals, adjusted to 6 m urea and 2 mm DTT, incubated for 10 min at 37 °C, quenched by acidification with HCl to pH 1, and then frozen at −20 °C until analyzed, as reported previously for hydrolysis studies with BPTI (22). For APLP2-KD, reactions were carried out similarly, but aliquots for analysis were not subjected to reduction/denaturation with DTT/urea, as described previously for hydrolysis studies with APPI (15). Enzyme, intact Kunitz domains, and hydrolysis products were resolved on a 50 × 2.0–33 Jupiter 4μ 90-Å C12 column (Phenomenex) with a gradient of 0–100% acetonitrile in 0.1% trifluoroacetic acid at a flow rate of 0.6 ml/min over 50 min. Peak integration to quantify the disappearance of intact Kunitz domains was carried out as described previously for BPTI (22). Initial rates were obtained by linear regression using a minimum of six data points within the initial linear phase of the reaction; the hydrolysis rates reported present the average of 2–3 independent experiments.
E-selectin Hydrolysis Studies
Soluble recombinant human E-selectin (535 amino acid residues), produced in CHO cells and lacking the transmembrane and cytoplasmic domains, was purchased from R&D Systems (catalogue no. ADP1). Time course incubations of E-selectin (5.5 μm final concentration) with mesotrypsin (0.275 μm final concentration) were carried out in 0.1 m Tris, pH 8.0, and 1 mm CaCl2 at 37 °C. Samples were withdrawn at various time points and frozen until analysis by SDS-PAGE. Gels were electroblotted to Immobilon P-FL membrane (EMD Millipore), membranes were stained with Coomassie, and bands were excised for N-terminal chemical sequencing using a Procise cLC protein sequencing system (Applied Biosystems). Alternatively, gels were stained with Coomassie, and bands of interest were excised for in-gel digestion with chymotrypsin and proteomic analysis by nanoflow liquid chromatography tandem mass spectrometry (nanoLC-MS/MS) using a ThermoFinnigan LTQ Orbitrap hybrid mass spectrometer, essentially as we have described previously (15). The MS/MS raw files were searched with Mascot (Matrix Science) against the human NCBI database without specifying enzyme cleavage specificity. The matches to E-selectin were manually reviewed.
Protein Crystallization, X-ray Data Collection, Structure Determination, and Refinement
Complexes of catalytically inactive mesotrypsin-S195A with bikunin-KD2 were crystallized by vapor diffusion. Mesotrypsin-S195A and bikunin-KD2 were mixed in a 1:1 stoichiometric molar ratio to achieve a total protein concentration of 3.3 mg/ml. Crystals were grown at room temperature in hanging drops over a reservoir of 0.1 m HEPES, pH 7.0, and 1 m sodium citrate. Drops (4 μl) were prepared by mixing equal volumes of protein and reservoir solutions. Crystals appeared within 3 days; grew over the course of several weeks; and then were harvested, cryoprotected by a brief soak in reservoir solution containing 20% glycerol, and flash-cooled in liquid N2. Complexes of catalytically inactive mesotrypsin-S195A with HAI2-KD1 were crystallized similarly in hanging drops over a reservoir of 0.1 m trisodium citrate, pH 5.5, 10% (w/v) PEG 4000, and 0.2 m sodium acetate. Crystals formed over the course of several months were likewise cryoprotected by a brief soak in reservoir solution containing 20% glycerol and were then flash-cooled in liquid N2.
Crystals were screened at beam line X29 (bikunin-KD2) and X25 (HAI2-KD1) of the National Synchrotron Light Source, Brookhaven National Laboratory. Data were collected at 100 K from one crystal of mesotrypsin-S195A·bikunin-KD2 that diffracted to 1.9 Å resolution and one crystal of mesotrypsin-S195A·HAI2-KD1 that diffracted to 1.6 Å resolution. Crystals of mesotrypsin-S195A·bikunin-KD2 belong to the space group P3, with unit cell dimensions a = 164.0, b = 164.0, c = 81.0, and contain four copies of the complex in the asymmetric unit. Crystals of mesotrypsin-S195A·HAI2-KD1 belong to the space group P212121, with unit cell dimensions a = 37.2, b = 70.2, c = 123.0, and contain one copy of the complex in the asymmetric unit. The x-ray data were merged and scaled using DENZO/SCALEPACK (28). Data were cut at high resolution limits beyond which the Rmerge statistic indicated poor merging of symmetry-related reflections.
The x-ray structures of mesotrypsin-S195A·bikunin-KD2 and mesotrypsin-S195A·HAI2-KD1 were solved by molecular replacement using MOLREP in CCP4 (29). The structure of mesotrypsin previously solved in complex with BPTI (PDB entry 2R9P, chain A) was used as a search model. A test set of 5% of the total reflections was excluded from refinement. Iterative model building using COOT (30) and refinement using Refmac5 (31) were used to build the structures of bikunin-KD2 and HAI2-KD1, refine the models, and add water molecules into difference peaks (Fo − Fc) greater than 2σ. For mesotrypsin-S195A·bikunin-KD2, non-crystallographic symmetry restraints were employed to refine the structure. The final stage of refinement included the addition of solvent molecules into peaks greater than 1σ and within acceptable hydrogen-bonding distance from neighboring protein atoms. The quality of the final models was analyzed using wwPDB validation tools (32). The coordinates and structure factors have been submitted to the RCSB Protein Data Bank under the accession codes 4U30 for mesotrypsin-S195A·bikunin-KD2 and 4U32 for mesotrypsin-S195A·HAI2-KD1. Structure figures were generated using PyMOL (Schrodinger, LLC).
Mathematical Analysis and Modeling
Solvent accessibility of the available Kunitz domain structures was calculated by DSSP (33). B-Factor values were taken from PDB files. Multiple linear regression models were constructed using MATLAB Statistical Toolbox (The MathWorks Inc., Natick, MA). A presence of particular amino acids in the P1 and P′2 positions was represented in the regression model by independent indicator variables. Impact of amino acids in other positions was modeled by a constant. Two regression models were constructed for kinetic constants kcat and Km, considering them as dependent variables.
RESULTS
Human Kunitz Protease Inhibitor Domains
Using the MEROPS database (available on the World Wide Web) (16), we identified 14 inhibitory human Kunitz domains with distinct MEROPS identifiers and found that 11 of 14 possessed Lys or Arg at the P1 position, making them potential candidates for mesotrypsin substrates or inhibitors: APPI, APLP2-KD, bikunin-KD2, HAI1-KD1, HAI1-KD2, HAI2-KD1, HAI2-KD2, TFPI1-KD1, TFPI1-KD2, TFPI2-KD1, and Kunitz domain 2 of the WAP, Kazal, immunoglobulin, and Kunitz and NTR domain-containing protein-1 (WFIKKN1) (Fig. 1 and Table 1). We previously have studied the interaction of mesotrypsin with APPI in detail, finding it to be a good substrate (15). Of the remaining 10 Kunitz domains, we were primarily interested in pursuing those representing potentially relevant substrates or inhibitors of mesotrypsin in the context of prostate cancer, because we have recently identified mesotrypsin as a driver of metastatic progression and a potential therapeutic target in prostate cancer (10, 11). The majority of the Kunitz domain-containing proteins were seen to be expressed at moderate or high levels in prostate cancer cell lines; however, WFIKKN1 was not expressed in prostate tissue, tumors, or tumor cell lines and was not considered further in our study. Next we looked at the reported trypsin affinities of the isolated Kunitz domains, reasoning that any proteins incapable of inhibiting classic trypsins would be unlikely to bind to mesotrypsin either as an inhibitor or a substrate, because mesotrypsin typically binds to canonical trypsin inhibitors 3–6 orders of magnitude more weakly than other mammalian trypsins (2). Most of the human Kunitz domains are reported to have antitrypsin inhibitory activity in the picomolar to low nanomolar range (Table 1). However, studies with recombinant HAI1 have found that mutation of the P1 residue of Kunitz domain 1 reduces the trypsin inhibitory potential of the compound inhibitor by 2 orders of magnitude to ∼200 nm, revealing that HAI1-KD2 has poor trypsin-inhibitory activity (34); accordingly, we did not pursue studies with HAI1-KD2.
FIGURE 1.
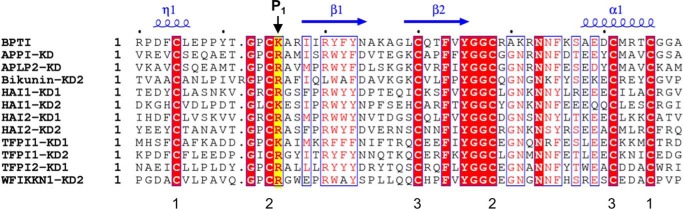
Sequence alignment of I2 Kunitz protease inhibitor domains. Sequences are shown for the 11 inhibitory human Kunitz domains that possess Arg or Lys at the P1 position, defining specificity for inhibition of trypsin and similar proteases. The sequence of bovine pancreatic trypsin inhibitor, archetypal example of the I2 family of inhibitors, is shown for comparison. Elements of secondary structure are indicated in blue above the alignment. The position of the P1 specificity residue is highlighted in yellow and indicated with a black arrow. Numbers below the six conserved Cys residues indicate the connectivity of the three conserved disulfide bonds. This figure was generated using ESPript (82).
TABLE 1.
Characteristics of human Kunitz protease inhibitor domains
| Kunitz domain | MEROPS ID | Gene | UNIPROT ID | aa position in precursora | P1 residue | Reported Ki for trypsin inhibition |
|---|---|---|---|---|---|---|
| m | ||||||
| APPI | I02.015 | APP | P05067 | 287–344 | Arg | 1.7 × 10−10b (15); 3.1 × 10−10c (54) |
| APLP2-KD | I02.016 | APLP2 | Q06481 | 306–363 | Arg | 3.2 × 10−10c (55) |
| Bikunin-KD2 | I02.006 | AMBP | P02760 | 283–340 | Arg | 2.5 × 10−8b; 1.3 × 10−10d (35) |
| HAI1-KD1 | I02.007 | SPINT1 | O43278 | 246–303 | Arg | 2.4 × 10−9c (34) |
| HAI1-KD2 | I02.008 | SPINT1 | O43278 | 387–444 | Lys | >2 × 10−7c (34) |
| HAI2-KD1 | I02.009 | SPINT2 | O43291 | 34–91 | Arg | 3 × 10−11c (66) |
| HAI2-KD2 | I02.0010 | SPINT2 | O43291 | 129–186 | Arg | 5 × 10−11c (66) |
| TFPI1-KD1 | I02.0011 | TFPI | P10646 | 50–107 | Lys | 1.2 × 10−8d (83) |
| TFPI1-KD2 | I02.0012 | TFPI | P10646 | 121–178 | Arg | 1 × 10−10d (83) |
| TFPI2-KD1 | I02.0013 | TFPI2 | P48307 | 32–89 | Arg | 1.3 × 10−8d (84) |
| WFIKKN1-KD2 | I02.0033 | WFIKKN1 | Q96NZ8 | 355–412 | Arg | 9.6 × 10−9c (85) |
a aa, amino acid; Kunitz domain boundaries here were defined to include 4 residues preceding the initial Cys residue and 3 residues following that sixth/final Cys residue for consistency.
b Measured versus human cationic trypsin.
c Measured versus bovine trypsin.
d Measured versus porcine trypsin.
The remaining eight Kunitz domains (APLP2-KD, bikunin-KD2, HAI1-KD1, HAI2-KD1, HAI2-KD2, TFPI1-KD1, TFPI1-KD2, and TFPI2-KD1) were synthesized as synthetic genes, and expression constructs for recombinant protein production using P. pastoris were generated and verified. Efforts to identify highly or moderately expressing P. pastoris clones and to produce and purify sufficient quantities of recombinant protein for biochemical studies were successful for six Kunitz domains: APLP2-KD, bikunin-KD2, HAI2-KD1, TFPI1-KD1, TFPI1-KD2, and TFPI2-KD1. For HAI1-KD1, screening large numbers of P. pastoris transformants failed to identify a clone with adequate expression, whereas for HAI2-KD2, a suitable clone was identified, and initial expression was successful, but loss of inhibitor activity was observed in repeated attempts at protein purification. Ultimately, expression and purification attempts were abandoned for these two domains.
Human Kunitz Domains Inhibit Mesotrypsin with Nanomolar to Micromolar Affinities
To assess the binding affinity of each recombinant Kunitz domain for mesotrypsin, we conducted enzyme inhibition experiments, monitoring cleavage of a chromogenic peptide substrate by mesotrypsin in the presence of varying concentrations of the Kunitz domain proteins. In each case, our data were well fitted by the classic competitive model of inhibition and gave inhibition constants (Ki) in the mid-nanomolar to mid-micromolar range (Fig. 2). Consistent with previous reports of the inhibitor resistance of mesotrypsin, the observed inhibition constants were all 2–4 orders of magnitude weaker than reported affinities of the same inhibitors for classic mammalian trypsins (Table 1), with the unique exception of bikunin-KD2. With a Ki of 138 nm, bikunin-KD2 inhibited mesotrypsin only 4-fold more weakly than was previously reported for inhibition of human cationic trypsin by intact bikunin consisting of multiple domains (35).
FIGURE 2.

Competitive inhibition of mesotrypsin by recombinant Kunitz domains. For each Kunitz domain, initial reaction rates are plotted for a representative experiment in which peptide substrate Z-GPR-pNA concentration and Kunitz domain concentration were varied; fitted lines obtained from multiple regression using the competitive inhibition equation are superposed on the data. All studies were performed with a final mesotrypsin concentration of 0.25 nm and with substrate concentration ranging from 10 to 200 μm as indicated on the x axis. A, mesotrypsin is competitively inhibited by APLP2-KD with a Ki of 633 nm. APLP2 concentration was 0 nm (○), 400 nm (■), 800 nm (▵), 1600 nm (▾), or 2000 nm (♢). B, mesotrypsin is competitively inhibited by bikunin-KD2 with a Ki of 138 nm. Bikunin-KD2 concentration was 0 nm (○), 50 nm (■), 100 nm (▵), 200 nm (▾), or 400 nm (♢). C, HAI2-KD1 competitively inhibits mesotrypsin with a Ki of 379 nm. HAI2-KD1 concentration was 0 nm (○), 400 nm (■), 800 nm (▵), 1200 nm (▾), or 2000 nm (♢). D, mesotrypsin is competitively inhibited by TFPI1-KD1 with a Ki of ∼75 μm. Concentrations of TFPI1-KD1 were 0 μm (○), 20 μm (■), 30 μm (▵), or 45 μm (▾). Higher concentrations of TFPI1-KD1 could not be tested due to the limited quantities of recombinant protein available, and thus the calculated Ki represents a rough approximation. E, TFPI1-KD2 competitively inhibits mesotrypsin with a Ki of 42.6 nm. TFPI1-KD2 concentrations were 0 nm (○), 50 nm (■), 100 nm (▵), 200 nm (▾), or 400 nm (♢). F, TFPI2-KD1 competitively inhibits mesotrypsin with a Ki of 2.75 μm. TFPI2-KD1 concentrations were 0 μm (○), 1 μm (■), or 3 μm (▵). Error bars, S.E.
Mesotrypsin Cleaves Human Kunitz Domains Rapidly
Based on prior observations that mesotrypsin cleaves several canonical serine protease inhibitors with accelerated kinetics (6, 14, 15, 22), we anticipated that some or all of the human Kunitz domains may be susceptible to rapid proteolysis by mesotrypsin. To quantify rates of mesotrypsin proteolysis, we used HPLC assays to monitor the disappearance of intact recombinant Kunitz domains in time course incubations with mesotrypsin. The Ki values measured for mesotrypsin inhibition by Kunitz domains also describe the Km values for cleavage of the Kunitz domains as alternative mesotrypsin substrates (36). Therefore, by carrying out proteolysis time courses at Kunitz domain concentrations well in excess of these measured Ki (Km) values, we anticipate that the rates of proteolysis observed will reflect enzyme behavior under saturating conditions, giving rates that approximate kcat. An exception to this approach was required for TFPI1-KD1; because Ki for this inhibitor was estimated at 75 μm, and limitations in the availability of recombinant TFPI1-KD1 precluded proteolysis studies under saturating conditions, for this domain, it was necessary to estimate kcat from experiments conducted under subsaturating conditions. Chromatograms, data plots, and calculated hydrolysis rates from representative proteolysis experiments for each Kunitz domain are shown in Fig. 3. Catalytic rate constants ranged from ∼0.1 s−1 for APLP2-KD and TFPI1-KD1, corresponding to a catalytic turnover time of ∼10 s, to 0.0058 s−1 for TFPI1-KD2, corresponding to a catalytic turnover time of ∼3 min. Notably, all of the human Kunitz domains evaluated were hydrolyzed considerably more rapidly than BPTI, for which we have previously demonstrated a kcat of 1.85 × 10−4, corresponding to a catalytic turnover time of 1.5 h (23).
FIGURE 3.

Proteolysis of recombinant Kunitz domains by mesotrypsin. For each Kunitz domain, the first and last sample traces from the linear phase of a representative hydrolysis time course are shown. The depletion of intact Kunitz domain was quantified by peak integration, as shown plotted below the chromatograms for a representative time course, allowing calculation of hydrolysis rates. Catalytic turnover rates (kcat) were calculated from the linear plot, taking into account the molar ratio of substrate to enzyme. For each study, [S] shows the Kunitz domain concentration, and [E] is the mesotrypsin concentration in hydrolysis time course reactions. A, mesotrypsin cleaved APLP2-KD with kcat = 1.1 × 10−1 s−1. B, mesotrypsin cleaved bikunin-KD2 with kcat = 4.1 × 10−2 s−1. C, mesotrypsin cleaved HAI2-KD1 with kcat = 4.4 × 10−2 s−1. D, mesotrypsin cleaved TFPI1-KD1 with an observed rate of 3.1 × 10−2 s−1. Because the substrate concentration in this study was below the estimated Km for TFPI1-KD1 of 75 μm, an approximate kcat of 1.2 × 10−1 s−1 was calculated using the Michaelis-Menten equation. E, mesotrypsin cleaved TFPI1-KD2 with kcat = 5.8 × 10−3 s−1. F, mesotrypsin cleaved TFPI2-KD1 with kcat = 2.6 × 10−2 s−1.
APLP2, Bikunin-KD2, and HAI2-KD1 Are Specific Substrates of Mesotrypsin
The specificity constant kcat/Km is often used to compare different substrates of a given enzyme and to identify substrates that meet kinetic criteria to be classified as “good” substrates for an enzyme. Specific protein substrates of proteases are typically recognized with a Km in the high nanomolar to low micromolar range, a catalytic rate constant in the range of 0.1–50 s−1, and a kcat/Km in the range of 104 to 107 (e.g. see Table 2 in Ref. 15 and Table 1 in Ref. 37; also see Refs. 38 and 39). The kinetic constants kcat, Km, and kcat/Km for cleavage of the human Kunitz domains by mesotrypsin are summarized in Table 2. Like APPI (15), APLP2, bikunin-KD2, and HAI2-KD1 meet kinetic criteria to be considered as good protein substrates of mesotrypsin and represent candidate physiological targets of mesotrypsin. TFPI1-KD2 reveals a similarly high kcat/Km ratio, but individual values for kcat and Km, and particularly the slower rate of catalytic turnover, classify this Kunitz domain as more a temporary inhibitor than a good substrate of mesotrypsin.
TABLE 2.
Kinetics of Kunitz domain inhibition of and hydrolysis by mesotrypsin
| Kunitz domain | kcat | Ki | kcat/Km |
|---|---|---|---|
| s−1 | m | s−1 m−1 | |
| BPTIa | 1.9 × 10−4 | 1.35 × 10−5 | 1.4 × 101 |
| APPIb | 4.2 × 10−2 | 1.36 × 10−7 | 3.1 × 105 |
| APLP2-KD | 1.1 × 10−1 | 6.33 × 10−7 | 1.7 × 105 |
| Bikunin-KD2 | 4.1 × 10−2 | 1.38 × 10−7 | 3.0 × 105 |
| HAI2-KD1 | 4.4 × 10−2 | 3.79 × 10−7 | 1.2 × 105 |
| TFPI1-KD1 | ∼1.2 × 10−1 | ∼7.5 × 10−5 | ∼1.6 × 103 |
| TFPI1-KD2 | 5.8 × 10−3 | 4.26 × 10−8 | 1.4 × 105 |
| TFPI2-KD1 | 2.6 × 10−2 | 2.75 × 10−6 | 9.4 × 103 |
A Loop of Canonical-like Conformation Found within Human E-selectin Is Not a Specific Substrate of Mesotrypsin
We previously found that the reactive site of APPI, specifically targeted for cleavage by mesotrypsin, is recognized by mesotrypsin only in the context of the three-dimensional structure of the Kunitz domain because a linear peptide mimicking the sequence of the APPI canonical loop was incapable of inhibiting mesotrypsin (15). We hypothesized that stabilization of substrate cleavage sites in a canonical-like conformation may be necessary for molecular recognition by mesotrypsin, a concept known as “conformational specificity” (40). Our finding above that multiple human Kunitz domains are good mesotrypsin substrates demonstrates successful application of this conformational criterion in the prediction of mesotrypsin substrates. We next wondered whether taking conformational specificity into account could help us to identify additional mesotrypsin substrates and proteolytic sites beyond the classic families of canonical protease inhibitors. As candidate substrates, we considered a set of protein structures previously identified by Jackson and Russell (41), featuring exposed loops with conformational resemblance to the P2–P′2 canonical loop of BPTI, and identified the Arg84-Gln85 bond of E-selectin, a cell surface glycoprotein, as a potential mesotrypsin cleavage site. Docking the E-selectin lectin domain (PDB entry 1ESL) (42) with mesotrypsin indicated that this bond could be accommodated in the mesotrypsin active site with minimal adjustment (Fig. 4A).
FIGURE 4.
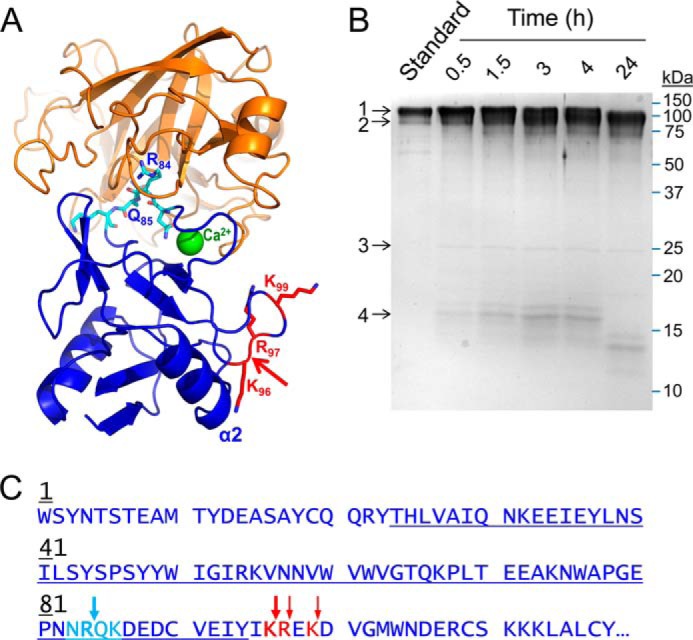
Proteolysis of recombinant human E-selectin by mesotrypsin. A, the crystal structure of mesotrypsin (orange; PDB entry 2R9P, chain B) docked with the C-type lectin domain of human E-selectin (blue; PDB entry 1ESL) in which the lectin residues 83–86 (cyan), which feature a canonical-like backbone conformation, are positioned in the mesotrypsin active site cleft. The molecular surfaces are roughly sterically compatible. The actual mesotrypsin cleavage sites in E-selectin that were subsequently identified experimentally are shown in red, and the primary initial nick site is indicated by the red arrow. In the absence of major conformational changes in E-selectin, binding of mesotrypsin to this cleavage site would be sterically blocked by the adjacent α2 helix. B, Coomassie-stained 15% SDS-polyacrylamide gel shows gradual cleavage of 5 μm recombinant human E-selectin (band 1) by 0.25 μm mesotrypsin (band 3) over a 24-h time course to generate a large C-terminal fragment (band 2) and a smaller N-terminal fragment (band 4), which was subsequently further degraded. The first lane shows a 1-μg undigested E-selectin run as a standard; subsequent lanes show time point samples collected at times indicated above the gel. C, the amino acid sequence of the C-type lectin domain of human E-selectin is shown; this domain encompasses residues 1–118 of the 589-residue mature secreted glycoprotein. The loop of canonical-like conformation is colored cyan, with the Arg84-Gln85 bond hypothesized to be a potential mesotrypsin cleavage site indicated by a cyan arrow. The actual mesotrypsin cleavage sites identified by N-terminal sequencing of the large cleavage fragment are indicated by red arrows. The blue underlining indicates the sequence coverage obtained from in-gel chymotrypsin digestion and nanoLC-MS/MS analysis of the 16-kDa cleavage fragment.
To experimentally test the prediction that the Arg84-Gln85 bond of E-selectin may be a specific mesotrypsin cleavage site, we carried out time course incubations of recombinant E-selectin with active mesotrypsin and analyzed time point samples by SDS-PAGE. Initial reactions using a low molar ratio of enzyme to substrate (1:1000 or 1:100, as in the studies with Kunitz domains) showed no evidence of proteolysis in time courses up to 24 h, indicating that E-selectin is not an efficiently cleaved mesotrypsin substrate. To ascertain whether the Arg84-Gln85 bond would ultimately be attacked by mesotrypsin as an initial nick site, albeit slowly, we ran time courses with a higher molar ratio of enzyme to substrate (1:20). We found evidence for slow cleavage over 4 h to produce a small fragment of ∼16 kDa and a larger fragment that was not well resolved from the intact protein; the 16-kDa fragment was further degraded over 24 h (Fig. 4B).
To identify the initial site of cleavage, reaction samples were resolved by SDS-PAGE and electroblotted, and the bands of interest were excised and subjected to N-terminal sequencing. The 16 kDa band produced the sequence Trp-Ser-Tyr-X-Thr-Ser expected for the N terminus of the mature secreted E-selectin protein, whereas for the larger fragment, the most abundant sequence detected was Arg-Glu-Lys-Asp-Val identifying the Lys96-Arg97 cleavage site, followed by Glu-Lys-Asp-Val-Gly identifying the Arg97-Glu98 cleavage site and Asp-Val-Gly-Met-Trp identifying the Lys99-Asp100 cleavage site (Fig. 4C). When the gel band containing the 16-kDa fragment was further analyzed by nanoLC-MS/MS, we observed peptides offering complete coverage of the E-selectin sequence spanning residues Thr24–Tyr94; this region encompasses the hypothesized Arg84-Gln85 cleavage site (Fig. 4C). Thus, it was determined definitively that limited proteolysis of E-selectin by mesotrypsin targets Lys96-Arg97, and not Arg84-Gln85, as the primary initial nick site.
Of interest, the Lys96-Arg97 site is not found in a canonical-like conformation in the E-selectin crystal structure, and an attempt to position this peptide bond in the mesotrypsin active site resulted in numerous intermolecular bumps involving multiple loops and the α2 helix of E-selectin. We conclude that mesotrypsin cleavage of Lys96-Arg97 requires substantial conformational change of E-selectin and that the preference of mesotrypsin for cleavage of this site over other accessible candidate sites is determined by elements of specificity yet to be identified. Importantly, the very slow rate at which this bond is cleaved and the persistence of the higher molecular weight fragment without further cleavage over 24 h (Fig. 4B) are consistent with the idea that mesotrypsin is a poor general trypsin that cannot effectively degrade a random native protein like E-selectin. Instead, mesotrypsin employs a complex combination of sequence and conformational specificity determinants to efficiently recognize and cleave specific substrates, such as APP, APLP2, bikunin, and HAI2.
Crystal Structures of Mesotrypsin Bound to Bikunin and HAI2 Kunitz Domains Reveal Recognition of Canonical Loop
To gain insight into structural aspects of substrate recognition by mesotrypsin, we cocrystallized the catalytically inactive mesotrypsin-S195A mutant with bikunin-KD2 and with HAI2-KD1 and solved the x-ray structures of these complexes. Data collection and refinement statistics are summarized in Table 3. We had previously found that mesotrypsin targets the reactive site bonds within the canonical inhibitory loops of BPTI (22) and APPI (15, 23) and hypothesized that for bikunin-KD2 and HAI2-KD1 as well, the conformation of the canonical loop and exposure of the favorable P1 Arg residue would similarly drive recognition of the canonical loop. As anticipated, the new structures feature the canonical binding loops of bikunin-KD2 and HAI2-KD1 positioned in the mesotrypsin active site, with the reactive site bond, Arg15-Ala16 (BPTI numbering scheme), oriented appropriately for cleavage, as described in greater detail for each complex below.
TABLE 3.
Crystallographic data collection and refinement statistics
| Mesotrypsin-S195A·bikunin-KD2 | Mesotrypsin-S195A·HAI2-KD1 | |
|---|---|---|
| PDB ID | 4U30 | 4U32 |
| Complexes per asymmetric unit | 4 | 1 |
| Space group | P3 | P212121 |
| Unit cell (Å) | 164.0, 164.0, 81.0, 90, 90, 120 | 37.2, 70.2, 123.0, 90, 90, 90 |
| Resolution (Å) | 2.4 | 1.65 |
| Unique reflections | 80,120 | 36,369 |
| Completeness (%) | 99.9 (99.9)a | 92.0 (88.1) |
| Multiplicity | 6.0 (6.2) | 9.5 (8.6) |
| Mean I/σ | 12.24 (4.88) | 21.71 (3.69) |
| Rmerge | 0.133 (0.420) | 0.092 (0.464) |
| Rcryst/Rfree (%) | 18.8/22.3 | 18.0/21.3 |
| Average B-factor (Å2) | 29 | 13.0 |
| Protein atoms | 8476 | 2400 |
| Water molecules | 291 | 222 |
| Root mean square deviation, bonds (Å) | 0.019 | 0.022 |
| Root mean square deviation, angles (degrees) | 1.928 | 2.136 |
| φ,ψ angle distributionb | ||
| Favored regions (%) | 96 | 98 |
| Allowed regions (%) | 4 | 2 |
| Outliers (%) | 0 | 0 |
a Values in parentheses are for the highest resolution shell.
b Ramachandran distributions are reported as defined by the PDB validation server/MolProbity.
Mesotrypsin was cocrystallized with bikunin-KD2 in space group P3 with four copies of the heterodimeric complex in the asymmetric unit and was refined to 2.4 Å resolution; an overview of the structure represented by the chain A-chain X complex is shown in Fig. 5A. The uncomplexed structure of intact bikunin, composed of two Kunitz domains, has been reported previously (43), and bikunin-KD2 in the new complex structure aligns very closely to the uncomplexed form (PDB entry 1BIK) with a root mean square deviation of 0.535 Å (Fig. 5B). Examining the superposition of the two structures, it is apparent that binding of intact bikunin to mesotrypsin would require structural adjustment to avoid a steric clash between the β-strands of bikunin-KD1 and a stretch of mesotrypsin loop C, involving mesotrypsin residues 92–97 (Fig. 5B). In the unbound structure, the binding loop of bikunin-KD2 deviated from the characteristic canonical conformation in the vicinity of primed-side residues Ala16 and Phe17; the peptide linkage between these residues was flipped relative to that seen in BPTI, and the P′2 side chain of Phe17 pointed toward the hydrophobic core of the Kunitz domain (43). In the complex with mesotrypsin, these residues have shifted to more closely approximate the standard canonical conformation (Fig. 5C). The P1 residue Arg15 is inserted deeply into the S1 specificity pocket to interact with mesotrypsin residue Asp189, the backbone amide of P′2 residue Phe17 forms a hydrogen bond with the carbonyl oxygen of mesotrypsin Phe41, and the Phe17 side chain packs parallel to the side chain of mesotrypsin Arg193 with a separation of ∼4 Å (Fig. 5, C and D). The bikunin-KD2 Arg15-Ala16 reactive site bond is positioned as expected for cleavage relative to catalytic triad residues Ser195 (in our structure Ala195), His57, and Asp102 (Fig. 5D).
FIGURE 5.

Structure of the mesotrypsin·bikunin-KD2 complex. A, structural overview shows mesotrypsin in a light gray schematic representation bound to bikunin-KD2 in green. Catalytic triad residues of the mesotrypsin active site are colored red, and Arg15 of the bikunin reactive site, positioned in the S1 specificity pocket of mesotrypsin, is rendered in a stick representation. B, the mesotrypsin·bikunin-HAI2-KD1 structure is superposed with that of intact bikunin (PDB entry 1BIK). With the canonical loop of bikunin-KD2 bound in the mesotrypsin active site, the β-strands of bikunin-KD1 clash with residues 92–97 of mesotrypsin loop C. C, mesotrypsin-bound bikunin-KD2 (green), showing P3–P4′ residues in standard orientation with nonprimed residues on the left and primed residues on the right, reveals conformational changes relative to unbound bikunin (tan) in the region of the P′1 and P′2 residues Ala16 and Phe17. Mesotrypsin is colored gray and rendered with a semitransparent surface. D, the bikunin-KD2 Arg15-Ala16 reactive site bond is positioned as would be expected for cleavage in the mesotrypsin active site by the mesotrypsin catalytic triad of Ser195 (in our structure mutated to Ala), His57, and Asp102. A black dashed line highlighted with a gold arrow illustrates a hydrogen bond formed between bikunin Phe17 nitrogen and mesotrypsin Phe41 oxygen.
The structures of HAI2 and its constitutent domains have not been previously reported. Mesotrypsin was cocrystallized with HAI2-KD1 in space group P212121 with a single heterodimeric complex in the asymmetric unit. In the solved structure, refined to 1.65 Å resolution, HAI2-KD1 displays the typical pear-shaped Kunitz domain fold, characterized by N- and C-terminal helical regions at the base, a central antiparallel β-turn, and a binding loop of canonical conformation at the top (Fig. 6A). Our electron density maps also revealed HAI2-KD1 to be glycosylated on Asn24 (Asn57 in the full-length HAI2 precursor protein), and we built in a single N-acetylglucosamine residue to account for this clear density in our maps. As seen in other members of this fold family, the compact structure of HAI2-KD1 is stabilized by three conserved disulfide bonds. As in the case of the mesotrypsin·bikunin-KD2 complex, the side chain of Arg15 occupies the S1 specificity pocket, forming a salt bridge with Asp189, and the Arg15-Ala16 reactive site bond is positioned as expected for cleavage by the catalytic Ser195 (in our structure Ala195) and other catalytic triad residues His57 and Asp102 (Fig. 6B).
FIGURE 6.
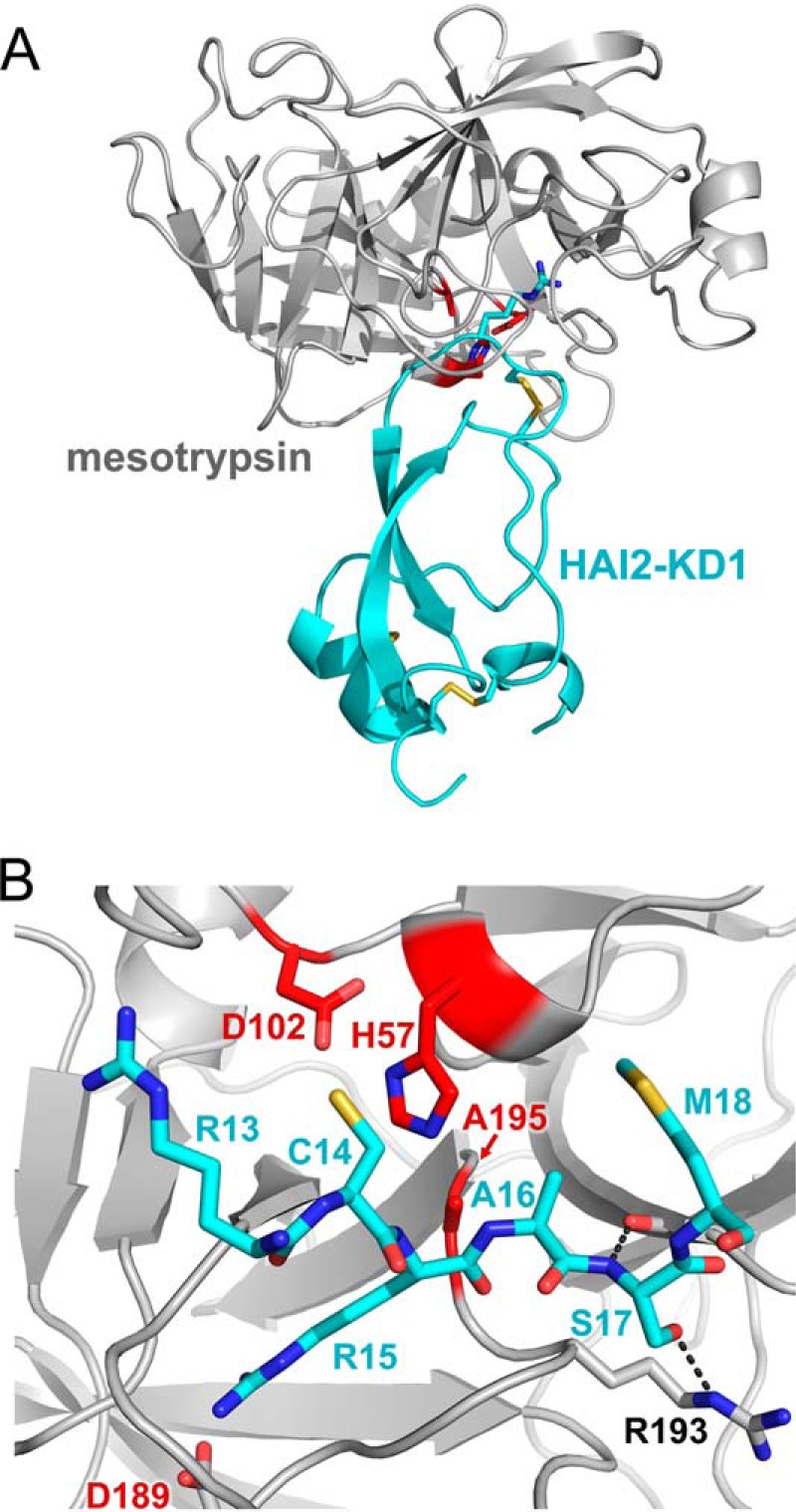
Structure of the mesotrypsin·HAI2-KD1 complex. A, structural overview shows mesotrypsin in a light gray schematic representation bound to HAI2-KD1 in cyan. Catalytic triad residues of the mesotrypsin active site are colored red, and Arg15 of the HAI2-KD1 reactive site, positioned in the S1 specificity pocket of mesotrypsin, is rendered in a stick representation. B, mesotrypsin active site occupied by HAI2-KD1 P3–P′3 residues is shown in standard orientation, with nonprimed residues on the left and primed residues on the right. The HAI2-KD1 Arg15-Ala16 reactive site bond is positioned as would be expected for cleavage by the mesotrypsin catalytic triad of Ser195 (in our structure mutated to Ala), His57, and Asp102. Black dashed lines illustrate hydrogen bonds formed between HAI2-KD1 Ser17 nitrogen and mesotrypsin Phe41 oxygen and between HAI2-KD1 Ser17 Oγ and mesotrypsin Arg193 Nϵ.
Although our two structures represent enzyme complexes with good substrates by kinetic criteria, and not mesotrypsin inhibitors, the orientations of reactive site bonds are indistinguishable from those observed in canonical inhibitor complexes with other trypsins, such as the complex of bovine trypsin with BPTI (PDB entry 2FTL) (44) (Fig. 7A). This observation is consistent with other evidence that such protease·inhibitor complexes represent ideal Michaelis complexes (21). Serine proteases inhibited by canonical inhibitors may undergo rapid acylation, the first step of catalytic cleavage, but progress of the reaction is blocked when the primed-side leaving group residues fail to dissociate from the active site, held in place by a combination of interactions with the enzyme and with the inhibitor scaffold (21, 45, 46). This leads to a thermodynamic equilibrium in which religation of the peptide bond is strongly favored, explaining why the intact form of the inhibitor is observed crystallographically in enzyme·inhibitor complexes.
FIGURE 7.
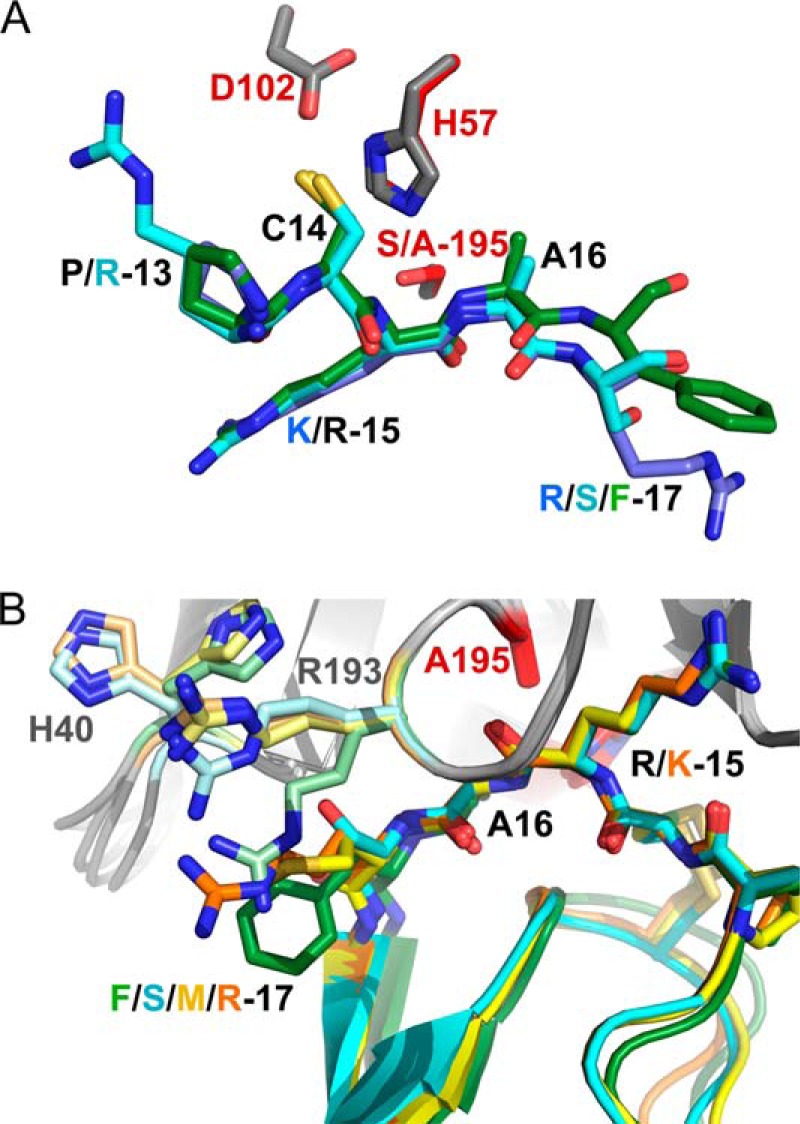
Comparison of mesotrypsin·bikunin-KD2 and mesotrypsin·HAI2-KD1 complexes with other Kunitz domain complexes. A, orientation of canonical loop P3-P′3 residues of bikunin-KD2 (green) and HAI2-KD1 (cyan) relative to mesotrypsin catalytic triad residues (gray) superpose closely with orientations seen in the archetypal serine protease/canonical inhibitor interaction as represented by BPTI (blue) bound to bovine trypsin (catalytic triad residues in red; PDB entry 2FTL). Structures are shown in the standard orientation, with nonprimed residues on the left and primed residues on the right. B, different P′2 residues among Kunitz domains induce distinct conformations for mesotrypsin residues on the primed side of the active site. Bikunin-KD2, HAI2-KD1, APPI, and BPTI are shown in green, cyan, yellow, and orange, respectively, with corresponding side chain conformations of mesotrypsin Arg193 and His40 shown in pale green, pale cyan, pale yellow, and pale orange, respectively. Orientation is similar to that shown in Figs. 5A and 6A, with primed residues on the right and nonprimed residues on the left.
The accelerated cleavage of canonical inhibitors by mesotrypsin, compared with other trypsins, has been hypothesized to result from a weakening of favorable interactions and introduction of unfavorable interactions between mesotrypsin and the primed-side residues of the canonical inhibitor binding loop, facilitating dissociation of the primed-side leaving group residues from the active site (2, 22, 47). The two new structures are reasonably consistent with this mechanism, because favorable primed-side interactions in each structure are minimal. The mesotrypsin·bikunin-KD2 structure reveals only a single hydrogen bond between the mesotrypsin Phe41 carbonyl oxygen and amide nitrogen of P′2 residue Phe17 (Fig. 5D). The proximity of the bikunin Phe17 side chain to mesotrypsin Arg193 (Fig. 5, C and D) suggests a potentially stabilizing cation-π interaction (48); however, affinity measurements and mathematical modeling that show a deleterious effect of P′2 aromatic residues on mesotrypsin affinity (see below) indicate that this interaction is probably not significantly stabilizing. In the mesotrypsin·HAI2-KD1 structure, two hydrogen bonds contribute to primed-side interactions at the intermolecular interface: one between the mesotrypsin Phe41 carbonyl oxygen and the amide nitrogen of HAI2-KD1 P′2 residue Ser17 and a second between the Ser17 Oγ and Nϵ of mesotrypsin Arg193 (Fig. 6B).
In previous structural studies of mesotrypsin bound to BPTI and APPI (22, 23, 49), we have noted conformational changes in the mesotrypsin primed-side residues Arg193 and His40, relative to the positions of these residues when the primed-side subsites are unoccupied (as revealed by the mesotrypsin·benzamidine structure 1H4W (12)). We have interpreted the conformational changes as deriving from deleterious steric and electrostatic conflicts between mesotrypsin Arg193, which in other trypsins is a conserved Gly residue, and the P′2 residue of a bound Kunitz domain protein. The clash results in upward displacement of Arg193 into a cleft on the surface of mesotrypsin, between the two β-barrel domains. In mutagenesis studies, we have found that the P′2 residue is a critical determinant of mesotrypsin affinity, because bulky or charged residues are strongly disfavored at this position (49). Interestingly, the two new structures each show novel conformations of Arg193 distinct from those observed previously (Fig. 7B). In the complex with HAI2-KD1, which possesses Ser17 in the P′2 position, Arg193 is pushed upward to a slightly lesser extent than seen in complexes with BPTI and APPI (Fig. 7B) and is positioned to allow formation of a hydrogen bond between the Ser17 side chain and Arg· Nϵ (Fig. 6B). In the complex with bikunin-KD2, Arg193 assumes a position pointed more downward, packing parallel to P′2 residue Phe17 (Fig. 7B), and the guanidinium group is displaced by only about 1.5 Å from its position in the mesotrypsin·benzamidine complex. Surprisingly, the positions assumed by mesotrypsin primed-side residues in the structures do not correlate as expected with the susceptibility of the Kunitz domains to cleavage. For example, BPTI and APPI binding result in nearly identical conformations of Arg193 (Fig. 7B), but APPI is a good substrate, whereas BPTI is a slowly cleaved poor substrate (Table 2). Conversely, APPI, bikunin-KD2, and HAI2-KD1 binding induce a spectrum of dissimilar conformations in Arg193 (Fig. 7B), but all are good substrates of mesotrypsin with very similar kcat and Km values (Table 2). Based on the new data, we now hypothesize that the ability of mesotrypsin to accelerate cleavage of canonical inhibitors may stem from increased protein dynamics of Arg193 and surrounding residues that favor dissociation of the primed-side leaving group upon cleavage; these dynamics are only hinted at by the distribution of side chain conformations in our static structures.
Mathematical Modeling Provides Insights into Determinants of Kunitz Domain Cleavage
The new kinetics studies and crystal structures reported here can be combined with results from our previous work, featuring structures and kinetics for cleavage of many BPTI and APPI mutants (23, 49), from which we wish to extract significant correlations that link sequence or structural features to affinity and susceptibility to cleavage. Initially, we examined the cleavage sites in the available Kunitz domain structures for solvent accessibility and for flexibility as evidenced by B-factors, two structural descriptors that we have previously identified as being important in proteolysis (50, 51). However, given the close structural similarity among Kunitz domains, we did not find correlative differences in these descriptors in our data set.
Next, we examined the primary sequence surrounding the reactive site bond for correlations with binding affinity and cleavage rates. Our earlier work has identified P1 and P′2 residues as major mesotrypsin specificity determinants through experiments analyzing BPTI and APPI variants with mutations at these positions (23, 49). To assess the degree to which sequence differences at these two positions alone may account for differential mesotrypsin binding and proteolytic susceptibility among Kunitz domains, we developed a single linear regression model in which the only variables considered were the identities of P1 and P′2 residues. We further made the simplifying assumption that the impact of the residues at the two positions would be additive. Regression analysis requires that the same substitution must be present at least twice in the data set to allow detection of correlations. After applying this filter, the remaining data set included APPI (and mutants), BPTI (and mutants), bikunin-KD2, and TFPI1-KD2. APLP2, HAI2-KD1, TFPI1-KD1, and TFPI2-KD1 were excluded from analysis because they possessed unique P′2 residues (Val, Ser, Ile, and Leu, respectively) without a second appearance among our data set. Next, we identified the largest possible subset of Kunitz domains that could be effectively described by a single linear regression model (Equations 2 and 3). APPI, bikunin-KD2, and TFPI1-KD2 were used to construct a consistent model that explains 96% of the data variability in kcat (R2 = 0.96) and 98% of the data variability in Km (R2 = 0.98). Thus, the differences in behavior toward mesotrypsin of APPI, bikunin-KD2, and TFPI1-KD2 are seemingly well explained by sequence differences in the vicinity of the scissile bond.
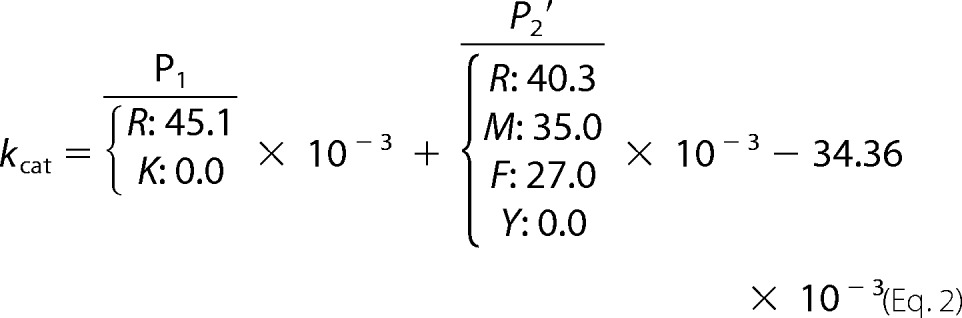 |
 |
Whereas the human Kunitz domains APPI, bikunin-KD2, and TFPI1-KD2 show similar cleavage behavior and are well described by our simple linear model, BPTI and its variants are cleaved much more slowly than the human Kunitz domains (Table 2) (23, 49). The considerably enhanced stability of BPTI to mesotrypsin cleavage, compared with other Kunitz domains, appears to be an intrinsic property of the BPTI scaffold, as we have noted previously (23). The mechanistic explanation for this enhanced stability is not obvious from the very similar three-dimensional structures of the Kunitz domains, and it is presumably attributable to nonconserved sequence elements that are yet to be identified.
DISCUSSION
In this study, we have used a hypothesis-based approach incorporating both cleavage site sequence and conformational elements to identify three new specific substrates of mesotrypsin. By using a conformational criterion, that the cleavage site peptide backbone should be found in the canonical conformation characteristic of canonical serine protease inhibitors, we identified an initial set of putative mesotrypsin substrates. Of the six representative human Kunitz domains from this set that we then produced and assessed experimentally as candidate mesotrypsin substrates, three (APLP2, bikunin-KD2, and HAI2-KD1) met our kinetic criteria for good substrates: kcat/Km > 1 × 105 and kcat > 1 × 10−2. We would anticipate that the comparable “hit” rate among a non-conformationally selected panel of candidate substrates would be far lower than the 50% that we found in our set of human Kunitz domains. In support of this assumption, it is intriguing that the 535-residue extracellular domain of E-selectin was highly resistant to mesotrypsin proteolysis. Within the sequence of E-selectin, there are numerous Lys and Arg residues that could have been, but were not, mesotrypsin cleavage sites. Examining the sequence more closely, we find the motif Cys-Arg-Ala-Val at residues 304–307 within the third Sushi domain of E-selectin, identical to the P2–P′2 sequence of the APLP2 canonical loop, which we found to be an excellent mesotrypsin substrate. This same sequence in the context of E-selectin showed no evidence of cleavage in our nanoLC-MS/MS analyses, even in samples that had been incubated for 24 h with a 1:20 molar ratio of mesotrypsin/E-selectin. We hypothesize that differences in conformational presentation of the sequence may account for the differences in mesotrypsin's ability to recognize and cleave this sequence motif.
Initially, we hypothesized that E-selectin might represent a candidate mesotrypsin substrate because crystal structures of E-selectin show that residues 83–86 within the lectin domain are found in a canonical-like conformation with Arg in the P1 position (41). However, in the results described here, we found no evidence for mesotrypsin cleavage at this site. In providing a negative example, this study may offer clues to help us interpret the phenomenon of conformational substrate selection by mesotrypsin. One potentially important difference between the canonical loops of Kunitz domains and that found in E-selectin relates to backbone flexibility. In the context of the Kunitz domain fold, the canonical binding loop is rigidly structured, with mobility limited by the compact packing of the protein, by a disulfide bond that links the P2 residue to the scaffold, and by hydrogen bonds that tether the backbone of P′1 and P′3 residues to the scaffold. This conformational stability of the inhibitory loop is believed to contribute to the unusually high affinity interactions of canonical inhibitors toward cognate serine proteases (18), and mutations that confer increased flexibility on the canonical loop also diminish affinity toward target proteases (44). By contrast, the canonical-like loop of E-selectin is minimally restrained by intramolecular contacts, and very high B-factors in this region of E-selectin crystal structures suggest that the loop is highly flexible (42). Although the canonical-like conformation of residues 83–86 is conserved in most structures of E-selectin and the structurally similar P-selectin, a structure of P-selectin bound to a peptide ligand (PDB entry 1G1S) shows dramatic remodeling of the loop with some backbone atoms displaced by as much as 12 Å (52), further highlighting the flexibility in the loop. We now hypothesize that the relative rigidity of the canonical loop found in Kunitz domain inhibitors, but not in selectins, is important for substrate recognition by mesotrypsin.
Although the human Kunitz domains as a group show similar cleavage behavior that can be well described by a simple linear model, BPTI and its variants are cleaved much more slowly than the human Kunitz domains. It may be that coevolution of mesotrypsin with the human Kunitz domains has tuned its ability to effectively target this group of substrates for proteolysis, in contrast with the non-coevolved Kunitz domain BPTI. A similar phenomenon has been demonstrated for human caspase 3, which was found to cleave its natural substrates orders of magnitude more efficiently than it cleaves non-coevolved E. coli proteins (39). The kinetic efficiency of cleavage of the human Kunitz domains by mesotrypsin, in contrast with BPTI, argues in favor of the potential physiological relevance of these cleavage events in vivo.
In vivo cleavage of the specific substrates that we have identified in our study is anticipated to contribute to the physiological and pathological activities of mesotrypsin. APP and the highly homologous APLP2 are both ubiquitously expressed transmembrane proteins with large extracellular regions composed of multiple distinct domains. Splice isoforms of APP and APLP2 that contain the Kunitz protease inhibitor domain can function as anticoagulants by potently inhibiting factor XIa and other proteases of the coagulation cascade (53–55); in the brain, these inhibitors have been shown to play an important role in regulating cerebral thrombosis (56). Because the trypsinogen 4 splice isoform of mesotrypsinogen is also highly expressed in the brain (3, 4), it may be that cleavage of APP/APLP2 Kunitz domains by mesotrypsin plays an opposing role in regulating thrombosis. Importantly, we previously demonstrated that cleavage of the APP Kunitz domain by mesotrypsin weakened its affinity for factor XIa and for trypsin by ∼100-fold (15), and we anticipate that the inhibitory potential of APLP2 will be similarly compromised by mesotrypsin cleavage. Outside of the brain, we and others have found that mesotrypsin is expressed in some epithelial cancers, where it contributes to tumor progression and metastasis (8–10, 57, 58); this effect could be mediated in part by mesotrypsin cleavage of APP/APLP2. Thrombin generation and coagulation are known to facilitate cancer progression (59–61), and inactivation of the protease inhibitor function of APP and APLP2 by mesotrypsin could contribute to the overproduction of thrombin in the tumor microenvironment.
Bikunin is a serine protease inhibitor composed of two Kunitz domains that is present in amniotic fluid, blood, and urine; in blood, it also exists as part of the inter-α-inhibitor complex (62, 63). Although the true physiological function of bikunin remains unclear, it is a weak inhibitor of many trypsin-like serine proteases (35), is more effective than other plasma protease inhibitors for inhibiting cell surface-bound plasmin (64), and also appears to have protease-independent signaling activities mediated through an unidentified receptor (62, 63). Both plasmin-inhibitory and signaling activities may contribute to potent anti-invasive and anti-metastatic activities that have been demonstrated in cell culture models and in vivo (64, 65). Cleavage of bikunin-KD2 by mesotrypsin would be expected to greatly diminish its inhibitory capability toward plasmin and its related anti-metastatic function; it is unclear whether cleavage may also interfere with bikunin's protease-independent signaling activities.
HAI2, also known as placental bikunin, is another compound inhibitor composed of two Kunitz domains (66). It potently inhibits many serine proteases with subnanomolar affinities (66) and is believed to be a physiological inhibitor of hepatocyte growth factor (HGF) activator (67) as well as hepsin (68) and matriptase (69, 70), two type II transmembrane serine proteases that, like HGF activator, can convert latent pro-hepatocyte growth factor/scatter factor (pro-HGF/SF) into the two-chain active signaling heterodimer. Additionally, HAI2 is a physiological inhibitor of prostasin, a glycosylphosphatidylinositol-anchored protease involved in regulation of matriptase activation and shedding (71, 72). HAI2-KD1, shown here to be a good substrate for mesotrypsin, has been found to be the domain responsible for inhibition of HGF activator (67). Activated HGF/SF binds to its receptor tyrosine kinase MET to induce dimerization and initiate phosphorylation cascades leading to comprehensive cellular changes that, in the deregulated context of cancer, drive malignant transformation and progression (73). HAI2 has been found to be a natural tumor suppressor in renal cell carcinoma (74), breast cancer (75, 76), and prostate cancer (77, 78), the loss of which leads to tumor growth and progression attributable at least in part to increased MET signaling. We have found mesotrypsin to be up-regulated with progression in prostate cancers and to contribute to invasion and metastasis (10), and it is highly plausible that these activities of mesotrypsin may be mediated at least in part through cleavage and inactivation of HAI2, with resulting increases in HGF/SF activation and MET signaling.
In sum, we have used a combination of sequence and conformational information to predict candidate substrates of mesotrypsin and have experimentally validated three new mesotrypsin targets, APLP2, bikunin, and HAI2, that are cleaved with kinetic parameters characteristic of physiologically relevant substrates. Each of these three substrates functions as a physiological inhibitor of serine proteases known to contribute to tumor progression and metastasis, and thus their cleavage and inactivation by mesotrypsin offers plausible mechanisms by which mesotrypsin can promote tumor progression. Further studies using tumor models and analysis of human biospecimens will be required to establish the specific significance of each of these mesotrypsin substrates in vivo.
More generally, our study provides an example of how sequence and structural information can be integrated in the prediction of protease substrates, but it also points to potential difficulties in the proteome-scale implementation of predictive algorithms for identifying protease substrates. We and others have examined structural features of limited proteolysis sites and made efforts toward implementation of such predictive algorithms with some success (39, 50, 51, 79–81). However, at least in the case of mesotrypsin, sequence, conformation, and straightforward structural descriptors such as solvent accessibility and protrusion, although important, appear to be insufficient to accurately distinguish among some candidate substrates. Our data suggest that complex local interactions and dynamics in the vicinity of putative cleavage sites, which are not obvious from examination of static structures, may have profound effects on substrate preference.
This work was supported, in whole or in part, by National Institutes of Health Grant R01CA154387 (to E. S. R.). Proteomics analysis was conducted at the Medical Genome Facility Proteomics Core, which is supported in part by Mayo Clinic Cancer Center Support Grant P30 CA15083. Diffraction data were measured at beamlines X25 and X29 of the National Synchrotron Light Source, which is supported by the Offices of Biological and Environmental Research and of Basic Energy Sciences of the United States Department of Energy and the National Center for Research Resources of the National Institutes of Health.
The atomic coordinates and structure factors (codes 4U30 and 4U32) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- BPTI
- bovine pancreatic trypsin inhibitor
- APLP2
- amyloid precursor-like protein 2
- HAI2
- hepatocyte growth factor activator inhibitor type 2
- APP
- amyloid precursor protein
- APPI
- APP Kunitz protease inhibitor domain
- TFPI1 and TFPI2
- tissue factor pathway inhibitor-1 and -2, respectively
- APLP2-KD
- APLP2 Kunitz domain
- bikunin-KD2
- bikunin Kunitz domain 2
- HAI1-KD1 and -KD2
- hepatocyte growth factor activator inhibitor type 1 Kunitz domain 1 and 2, respectively
- HAI2-KD1 and -KD2
- HAI2 Kunitz domain 1 and 2, respectively
- TFPI1-KD1 and -KD2
- TFPI1 Kunitz domain 1 and 2, respectively
- TFPI2-KD1
- TFPI2 Kunitz domain 1
- Z
- benzoxycarbonyl
- pNA
- p-nitroanilide
- nanoLC-MS/MS
- nanoflow liquid chromatography tandem mass spectrometry
- HGF
- hepatocyte growth factor
- SF
- scatter factor
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PDB
- Protein Data Bank.
REFERENCES
- 1. Rinderknecht H., Renner I. G., Abramson S. B., Carmack C. (1984) Mesotrypsin: a new inhibitor-resistant protease from a zymogen in human pancreatic tissue and fluid. Gastroenterology 86, 681–692 [PubMed] [Google Scholar]
- 2. Salameh M. A., Radisky E. S. (2013) Biochemical and structural insights into mesotrypsin: an unusual human trypsin. Int. J. Biochem. Mol. Biol. 4, 129–139 [PMC free article] [PubMed] [Google Scholar]
- 3. Tóth J., Siklódi E., Medveczky P., Gallatz K., Németh P., Szilágyi L., Gráf L., Palkovits M. (2007) Regional distribution of human trypsinogen 4 in human brain at mRNA and protein level. Neurochem. Res. 32, 1423–1433 [DOI] [PubMed] [Google Scholar]
- 4. Wiegand U., Corbach S., Minn A., Kang J., Müller-Hill B. (1993) Cloning of the cDNA encoding human brain trypsinogen and characterization of its product. Gene 136, 167–175 [DOI] [PubMed] [Google Scholar]
- 5. Nakanishi J., Yamamoto M., Koyama J., Sato J., Hibino T. (2010) Keratinocytes synthesize enteropeptidase and multiple forms of trypsinogen during terminal differentiation. J. Invest. Dermatol. 130, 944–952 [DOI] [PubMed] [Google Scholar]
- 6. Miyai M., Matsumoto Y., Yamanishi H., Yamamoto-Tanaka M., Tsuboi R., Hibino T. (2014) Keratinocyte-Specific Mesotrypsin Contributes to the Desquamation Process via Kallikrein Activation and LEKTI Degradation. J. Invest. Dermatol. 134, 1665–1674 [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto-Tanaka M., Motoyama A., Miyai M., Matsunaga Y., Matsuda J., Tsuboi R., Hibino T. (2014) Mesotrypsin and caspase-14 participate in prosaposin processing: potential relevance to epidermal permeability barrier formation. J. Biol. Chem. 289, 20026–20038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hockla A., Radisky D. C., Radisky E. S. (2010) Mesotrypsin promotes malignant growth of breast cancer cells through shedding of CD109. Breast Cancer Res. Treat. 124, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang G., Cao F., Ren G., Gao D., Bhakta V., Zhang Y., Cao H., Dong Z., Zang W., Zhang S., Wong H. H., Hiley C., Crnogorac-Jurcevic T., Lemoine N. R., Wang Y. (2010) PRSS3 promotes tumour growth and metastasis of human pancreatic cancer. Gut 59, 1535–1544 [DOI] [PubMed] [Google Scholar]
- 10. Hockla A., Miller E., Salameh M. A., Copland J. A., Radisky D. C., Radisky E. S. (2012) PRSS3/mesotrypsin is a therapeutic target for metastatic prostate cancer. Mol. Cancer Res. 10, 1555–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Radisky E. S. (2013) PRSS3/mesotrypsin in prostate cancer progression: implications for translational medicine. Asian J. Androl. 15, 439–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katona G., Berglund G. I., Hajdu J., Gráf L., Szilágyi L. (2002) Crystal structure reveals basis for the inhibitor resistance of human brain trypsin. J. Mol. Biol. 315, 1209–1218 [DOI] [PubMed] [Google Scholar]
- 13. Nyaruhucha C. N., Kito M., Fukuoka S. I. (1997) Identification and expression of the cDNA-encoding human mesotrypsin(ogen), an isoform of trypsin with inhibitor resistance. J. Biol. Chem. 272, 10573–10578 [DOI] [PubMed] [Google Scholar]
- 14. Szmola R., Kukor Z., Sahin-Tóth M. (2003) Human mesotrypsin is a unique digestive protease specialized for the degradation of trypsin inhibitors. J. Biol. Chem. 278, 48580–48589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salameh M. A., Robinson J. L., Navaneetham D., Sinha D., Madden B. J., Walsh P. N., Radisky E. S. (2010) The amyloid precursor protein/protease nexin 2 Kunitz inhibitor domain is a highly specific substrate of mesotrypsin. J. Biol. Chem. 285, 1939–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rawlings N. D., Barrett A. J., Bateman A. (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ascenzi P., Bocedi A., Bolognesi M., Spallarossa A., Coletta M., De Cristofaro R., Menegatti E. (2003) The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor): a milestone protein. Curr. Protein Pept. Sci. 4, 231–251 [DOI] [PubMed] [Google Scholar]
- 18. Bode W., Huber R. (1992) Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 204, 433–451 [DOI] [PubMed] [Google Scholar]
- 19. Krowarsch D., Cierpicki T., Jelen F., Otlewski J. (2003) Canonical protein inhibitors of serine proteases. Cell. Mol. Life Sci. 60, 2427–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laskowski M., Jr., Kato I. (1980) Protein inhibitors of proteinases. Annu. Rev. Biochem. 49, 593–626 [DOI] [PubMed] [Google Scholar]
- 21. Radisky E. S., Koshland D. E., Jr. (2002) A clogged gutter mechanism for protease inhibitors. Proc. Natl. Acad. Sci. U.S.A. 99, 10316–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salameh M. A., Soares A. S., Hockla A., Radisky E. S. (2008) Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J. Biol. Chem. 283, 4115–4123 [DOI] [PubMed] [Google Scholar]
- 23. Salameh M. A., Soares A. S., Navaneetham D., Sinha D., Walsh P. N., Radisky E. S. (2010) Determinants of affinity and proteolytic stability in interactions of Kunitz family protease inhibitors with mesotrypsin. J. Biol. Chem. 285, 36884–36896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chase T., Jr., Shaw E. (1967) p-Nitrophenyl-p′-guanidinobenzoate HCl: a new active site titrant for trypsin. Biochem. Biophys. Res. Commun. 29, 508–514 [DOI] [PubMed] [Google Scholar]
- 25. Navaneetham D., Sinha D., Walsh P. N. (2010) Mechanisms and specificity of factor XIa and trypsin inhibition by protease nexin 2 and basic pancreatic trypsin inhibitor. J Biochem. 148, 467–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Navaneetham D., Jin L., Pandey P., Strickler J. E., Babine R. E., Abdel-Meguid S. S., Walsh P. N. (2005) Structural and mutational analyses of the molecular interactions between the catalytic domain of factor XIa and the Kunitz protease inhibitor domain of protease nexin 2. J. Biol. Chem. 280, 36165–36175 [DOI] [PubMed] [Google Scholar]
- 27. DelMar E. G., Largman C., Brodrick J. W., Geokas M. C. (1979) A sensitive new substrate for chymotrypsin. Anal. Biochem. 99, 316–320 [DOI] [PubMed] [Google Scholar]
- 28. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 29. Vagin A. A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 30, 1022–1025 [Google Scholar]
- 30. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32. Read R. J., Adams P. D., Arendall W. B., 3rd, Brunger A. T., Emsley P., Joosten R. P., Kleywegt G. J., Krissinel E. B., Lütteke T., Otwinowski Z., Perrakis A., Richardson J. S., Sheffler W. H., Smith J. L., Tickle I. J., Vriend G., Zwart P. H. (2011) A new generation of crystallographic validation tools for the protein data bank. Structure 19, 1395–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 34. Kirchhofer D., Peek M., Li W., Stamos J., Eigenbrot C., Kadkhodayan S., Elliott J. M., Corpuz R. T., Lazarus R. A., Moran P. (2003) Tissue expression, protease specificity, and Kunitz domain functions of hepatocyte growth factor activator inhibitor-1B (HAI-1B), a new splice variant of HAI-1. J. Biol. Chem. 278, 36341–36349 [DOI] [PubMed] [Google Scholar]
- 35. Potempa J., Kwon K., Chawla R., Travis J. (1989) Inter-α-trypsin inhibitor. Inhibition spectrum of native and derived forms. J. Biol. Chem. 264, 15109–15114 [PubMed] [Google Scholar]
- 36. Cornish-Bowden A. (1995) Fundamentals of Enzyme Kinetics, pp 105–108, Portland Press, London [Google Scholar]
- 37. Radisky E. S., King D. S., Kwan G., Koshland D. E., Jr. (2003) The role of the protein core in the inhibitory power of the classic serine protease inhibitor, chymotrypsin inhibitor 2. Biochemistry 42, 6484–6492 [DOI] [PubMed] [Google Scholar]
- 38. Agard N. J., Mahrus S., Trinidad J. C., Lynn A., Burlingame A. L., Wells J. A. (2012) Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc. Natl. Acad. Sci. U.S.A. 109, 1913–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Timmer J. C., Zhu W., Pop C., Regan T., Snipas S. J., Eroshkin A. M., Riedl S. J., Salvesen G. S. (2009) Structural and kinetic determinants of protease substrates. Nat. Struct. Mol. Biol. 16, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wright H. T. (1977) Secondary and conformational specificities of trypsin and chymotrypsin. Eur. J. Biochem. 73, 567–578 [DOI] [PubMed] [Google Scholar]
- 41. Jackson R. M., Russell R. B. (2000) The serine protease inhibitor canonical loop conformation: examples found in extracellular hydrolases, toxins, cytokines and viral proteins. J. Mol. Biol. 296, 325–334 [DOI] [PubMed] [Google Scholar]
- 42. Graves B. J., Crowther R. L., Chandran C., Rumberger J. M., Li S., Huang K. S., Presky D. H., Familletti P. C., Wolitzky B. A., Burns D. K. (1994) Insight into E-selectin/ligand interaction from the crystal structure and mutagenesis of the lec/EGF domains. Nature 367, 532–538 [DOI] [PubMed] [Google Scholar]
- 43. Xu Y., Carr P. D., Guss J. M., Ollis D. L. (1998) The crystal structure of bikunin from the inter-α-inhibitor complex: a serine protease inhibitor with two Kunitz domains. J. Mol. Biol. 276, 955–966 [DOI] [PubMed] [Google Scholar]
- 44. Hanson W. M., Domek G. J., Horvath M. P., Goldenberg D. P. (2007) Rigidification of a flexible protease inhibitor variant upon binding to trypsin. J. Mol. Biol. 366, 230–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Radisky E. S., Kwan G., Karen Lu C. J., Koshland D. E., Jr. (2004) Binding, proteolytic, and crystallographic analyses of mutations at the protease-inhibitor interface of the subtilisin BPN′/chymotrypsin inhibitor 2 complex. Biochemistry 43, 13648–13656 [DOI] [PubMed] [Google Scholar]
- 46. Zakharova E., Horvath M. P., Goldenberg D. P. (2009) Structure of a serine protease poised to resynthesize a peptide bond. Proc. Natl. Acad. Sci. U.S.A. 106, 11034–11039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Salameh M. A., Soares A. S., Alloy A., Radisky E. S. (2012) Presence versus absence of hydrogen bond donor Tyr-39 influences interactions of cationic trypsin and mesotrypsin with protein protease inhibitors. Protein Sci. 21, 1103–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cohen M., Reichmann D., Neuvirth H., Schreiber G. (2008) Similar chemistry, but different bond preferences in inter versus intra-protein interactions. Proteins 72, 741–753 [DOI] [PubMed] [Google Scholar]
- 49. Salameh M. A., Soares A. S., Hockla A., Radisky D. C., Radisky E. S. (2011) The P(2)′ residue is a key determinant of mesotrypsin specificity: engineering a high-affinity inhibitor with anticancer activity. Biochem. J. 440, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kazanov M. D., Igarashi Y., Eroshkin A. M., Cieplak P., Ratnikov B., Zhang Y., Li Z., Godzik A., Osterman A. L., Smith J. W. (2011) Structural determinants of limited proteolysis. J. Proteome Res. 10, 3642–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Belushkin A. A., Vinogradov D. V., Gelfand M. S., Osterman A. L., Cieplak P., Kazanov M. D. (2014) Sequence-derived structural features driving proteolytic processing. Proteomics 14, 42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Somers W. S., Tang J., Shaw G. D., Camphausen R. T. (2000) Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell 103, 467–479 [DOI] [PubMed] [Google Scholar]
- 53. Van Nostrand W. E., Wagner S. L., Farrow J. S., Cunningham D. D. (1990) Immunopurification and protease inhibitory properties of protease nexin-2/amyloid β-protein precursor. J. Biol. Chem. 265, 9591–9594 [PubMed] [Google Scholar]
- 54. Wagner S. L., Siegel R. S., Vedvick T. S., Raschke W. C., Van Nostrand W. E. (1992) High level expression, purification, and characterization of the Kunitz-type protease inhibitor domain of protease nexin-2/amyloid β-protein precursor. Biochem. Biophys. Res. Commun. 186, 1138–1145 [DOI] [PubMed] [Google Scholar]
- 55. Van Nostrand W. E., Schmaier A. H., Neiditch B. R., Siegel R. S., Raschke W. C., Sisodia S. S., Wagner S. L. (1994) Expression, purification, and characterization of the Kunitz-type proteinase inhibitor domain of the amyloid β-protein precursor-like protein-2. Biochim. Biophys. Acta 1209, 165–170 [DOI] [PubMed] [Google Scholar]
- 56. Xu F., Previti M. L., Nieman M. T., Davis J., Schmaier A. H., Van Nostrand W. E. (2009) AβPP/APLP2 family of Kunitz serine proteinase inhibitors regulate cerebral thrombosis. J. Neurosci. 29, 5666–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Diederichs S., Bulk E., Steffen B., Ji P., Tickenbrock L., Lang K., Zänker K. S., Metzger R., Schneider P. M., Gerke V., Thomas M., Berdel W. E., Serve H., Müller-Tidow C. (2004) S100 family members and trypsinogens are predictors of distant metastasis and survival in early-stage non-small cell lung cancer. Cancer Res. 64, 5564–5569 [DOI] [PubMed] [Google Scholar]
- 58. Han S., Lee C. W., Trevino J. G., Hughes S. J., Sarosi G. A., Jr. (2013) Autocrine extra-pancreatic trypsin 3 secretion promotes cell proliferation and survival in esophageal adenocarcinoma. PLoS One 8, e76667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nierodzik M. L., Karpatkin S. (2006) Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 10, 355–362 [DOI] [PubMed] [Google Scholar]
- 60. Ruf W., Mueller B. M. (2006) Thrombin generation and the pathogenesis of cancer. Semin. Thromb. Hemost. 32, 61–68 [DOI] [PubMed] [Google Scholar]
- 61. Gil-Bernabé A. M., Lucotti S., Muschel R. J. (2013) Coagulation and metastasis: what does the experimental literature tell us? Br. J. Haematol. 162, 433–441 [DOI] [PubMed] [Google Scholar]
- 62. Fries E., Blom A. M. (2000) Bikunin: not just a plasma proteinase inhibitor. Int. J. Biochem. Cell Biol. 32, 125–137 [DOI] [PubMed] [Google Scholar]
- 63. Kobayashi H., Suzuki M., Hirashima Y., Terao T. (2003) The protease inhibitor bikunin, a novel anti-metastatic agent. Biol. Chem. 384, 749–754 [DOI] [PubMed] [Google Scholar]
- 64. Kobayashi H., Shinohara H., Takeuchi K., Itoh M., Fujie M., Saitoh M., Terao T. (1994) Inhibition of the soluble and the tumor cell receptor-bound plasmin by urinary trypsin inhibitor and subsequent effects on tumor cell invasion and metastasis. Cancer Res. 54, 844–849 [PubMed] [Google Scholar]
- 65. Yagyu T., Kobayashi H., Matsuzaki H., Wakahara K., Kondo T., Kurita N., Sekino H., Inagaki K. (2006) Enhanced spontaneous metastasis in bikunin-deficient mice. Int. J. Cancer 118, 2322–2328 [DOI] [PubMed] [Google Scholar]
- 66. Delaria K. A., Muller D. K., Marlor C. W., Brown J. E., Das R. C., Roczniak S. O., Tamburini P. P. (1997) Characterization of placental bikunin, a novel human serine protease inhibitor. J. Biol. Chem. 272, 12209–12214 [DOI] [PubMed] [Google Scholar]
- 67. Qin L., Denda K., Shimomura T., Kawaguchi T., Kitamura N. (1998) Functional characterization of Kunitz domains in hepatocyte growth factor activator inhibitor type 2. FEBS Lett. 436, 111–114 [DOI] [PubMed] [Google Scholar]
- 68. Kirchhofer D., Peek M., Lipari M. T., Billeci K., Fan B., Moran P. (2005) Hepsin activates pro-hepatocyte growth factor and is inhibited by hepatocyte growth factor activator inhibitor-1B (HAI-1B) and HAI-2. FEBS Lett. 579, 1945–1950 [DOI] [PubMed] [Google Scholar]
- 69. Szabo R., Hobson J. P., List K., Molinolo A., Lin C. Y., Bugge T. H. (2008) Potent inhibition and global co-localization implicate the transmembrane Kunitz-type serine protease inhibitor hepatocyte growth factor activator inhibitor-2 in the regulation of epithelial matriptase activity. J. Biol. Chem. 283, 29495–29504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Szabo R., Hobson J. P., Christoph K., Kosa P., List K., Bugge T. H. (2009) Regulation of cell surface protease matriptase by HAI2 is essential for placental development, neural tube closure and embryonic survival in mice. Development 136, 2653–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Friis S., Sales K. U., Schafer J. M., Vogel L. K., Kataoka H., Bugge T. H. (2014) The protease inhibitor HAI-2, but not HAI-1, regulates matriptase activation and shedding through prostasin. J. Biol. Chem. 289, 22319–22332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Friis S., Uzzun Sales K., Godiksen S., Peters D. E., Lin C. Y., Vogel L. K., Bugge T. H. (2013) A matriptase-prostasin reciprocal zymogen activation complex with unique features: prostasin as a non-enzymatic co-factor for matriptase activation. J. Biol. Chem. 288, 19028–19039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gherardi E., Birchmeier W., Birchmeier C., Vande Woude G. (2012) Targeting MET in cancer: rationale and progress. Nat. Rev. Cancer 12, 89–103 [DOI] [PubMed] [Google Scholar]
- 74. Morris M. R., Gentle D., Abdulrahman M., Maina E. N., Gupta K., Banks R. E., Wiesener M. S., Kishida T., Yao M., Teh B., Latif F., Maher E. R. (2005) Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 65, 4598–4606 [DOI] [PubMed] [Google Scholar]
- 75. Parr C., Jiang W. G. (2006) Hepatocyte growth factor activation inhibitors (HAI-1 and HAI-2) regulate HGF-induced invasion of human breast cancer cells. Int. J. Cancer 119, 1176–1183 [DOI] [PubMed] [Google Scholar]
- 76. Parr C., Watkins G., Mansel R. E., Jiang W. G. (2004) The hepatocyte growth factor regulatory factors in human breast cancer. Clin. Cancer Res. 10, 202–211 [DOI] [PubMed] [Google Scholar]
- 77. Tsai C. H., Teng C. H., Tu Y. T., Cheng T. S., Wu S. R., Ko C. J., Shyu H. Y., Lan S. W., Huang H. P., Tzeng S. F., Johnson M. D., Lin C. Y., Hsiao P. W., Lee M. S. (2014) HAI-2 suppresses the invasive growth and metastasis of prostate cancer through regulation of matriptase. Oncogene 33, 4643–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bergum C., List K. (2010) Loss of the matriptase inhibitor HAI-2 during prostate cancer progression. Prostate 70, 1422–1428 [DOI] [PubMed] [Google Scholar]
- 79. Barkan D. T., Hostetter D. R., Mahrus S., Pieper U., Wells J. A., Craik C. S., Sali A. (2010) Prediction of protease substrates using sequence and structure features. Bioinformatics 26, 1714–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Song J., Tan H., Perry A. J., Akutsu T., Webb G. I., Whisstock J. C., Pike R. N. (2012) PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One 7, e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hubbard S. J., Beynon R. J., Thornton J. M. (1998) Assessment of conformational parameters as predictors of limited proteolytic sites in native protein structures. Protein Eng. 11, 349–359 [DOI] [PubMed] [Google Scholar]
- 82. Gouet P., Courcelle E., Stuart D. I., Metoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]
- 83. Petersen L. C., Bjørn S. E., Olsen O. H., Nordfang O., Norris F., Norris K. (1996) Inhibitory properties of separate recombinant Kunitz-type-protease-inhibitor domains from tissue-factor-pathway inhibitor. Eur. J. Biochem. 235, 310–316 [DOI] [PubMed] [Google Scholar]
- 84. Chand H. S., Schmidt A. E., Bajaj S. P., Kisiel W. (2004) Structure-function analysis of the reactive site in the first Kunitz-type domain of human tissue factor pathway inhibitor-2. J. Biol. Chem. 279, 17500–17507 [DOI] [PubMed] [Google Scholar]
- 85. Nagy A., Trexler M., Patthy L. (2003) Expression, purification and characterization of the second Kunitz-type protease inhibitor domain of the human WFIKKN protein. Eur. J. Biochem. 270, 2101–2107 [DOI] [PubMed] [Google Scholar]