

Background: The inability to up-regulate “regeneration-associated genes” contributes to the failure of axonal regeneration after injury.
Results: Expression of activated CREB and cAMP up-regulation co-regulate AP1-dependent regeneration-associated gene expression and enhance axon growth.
Conclusion: Targeting transcription factor activity and the associated second messengers facilitates regenerative growth.
Significance: This transcription factor plus small molecule approach may be an efficient means to stimulate axon regeneration.
Keywords: AP1 Transcription Factor (AP-1), cAMP Response Element-binding Protein (CREB), Cyclic AMP (cAMP), Regeneration, Transcription, Regeneration-associated Gene
Abstract
To regenerate damaged axons, neurons must express a cassette of regeneration-associated genes (RAGs) that increases intrinsic growth capacity and confers resistance to extrinsic inhibitory cues. Here we show that dibutyrl-cAMP or forskolin combined with constitutive-active CREB are superior to either agent alone in driving neurite growth on permissive and inhibitory substrates. Of the RAGs examined, only arginase 1 (Arg1) expression correlated with the increased neurite growth induced by the cAMP/CREB combination, both of which were AP1-dependent. This suggests that cAMP-induced AP1 activity is necessary and interacts with CREB to drive expression of RAGs relevant for regeneration and demonstrates that combining a small molecule (cAMP) with an activated transcription factor (CREB) stimulates the gene expression necessary to enhance axonal regeneration.
Introduction
The capacity of peripheral axons of sensory neurons to regenerate after injury is highly dependent on the transcriptional response initiated by axon damage (1). This response includes the induction of regeneration-associated genes (RAGs)2 (2–4) such as the activating transcription factor 3 (ATF3), arginase 1 (Arg1), galanin (Gal), and interleukin 6 (IL6). Moreover, although CNS-projecting axons do not regenerate after injury, the induction of RAGs by peripheral axon injury in sensory neurons facilitates the growth of subsequently lesioned CNS-projecting axons of the same neurons (5). The increase of cAMP levels after peripheral axon injury is thought to initiate regeneration-associated transcription (6); however, some studies indicate that cAMP up-regulation alone is less effective than peripheral nerve injury in driving axon regeneration (3, 7, 8). This suggests that regeneration-associated transcription requires additional biochemical effectors of peripheral nerve injury for optimal efficacy. Among the different transcription factors involved in axon regeneration (9, 10), the cAMP-responsive element binding protein (CREB) has received significant attention given the ability of cAMP analogues to facilitate regeneration of some CNS axons in vivo and to promote neurite growth in the presence of myelin-derived inhibitors in vitro (6, 11, 12). Furthermore, expression of a constitutive-active CREB fusion protein allows dorsal root ganglion (DRG) neurons to overcome myelin-derived inhibitors of axonal growth in vitro and central axon regeneration of sensory neurons following a dorsal column lesion in vivo (13); however, the specific contribution of cAMP-signaling and CREB to regeneration-associated transcription remains unclear.
To address the roles of cAMP and CREB in promoting axon growth and regeneration-associated transcription, we targeted CREB activity by expressing constitutive-active (CREB-CA) and dominant-negative (CREB-DN) CREB variants in DRG neurons. We show that CREB-CA and dibutyryl-cAMP (Bt2cAMP) have both common and distinct targets among the RAGs that we investigated. Of note, CREB-CA and Bt2cAMP synergized to enhance neurite growth in an AP1-dependent fashion on inhibitory substrates, which mirrored the expression pattern of Arg1. Our findings indicate that maximum axon growth requires both CREB and cAMP-regulated AP1 activity. These results further reveal the complexity of regeneration-associated transcription and suggest that a combinatorial strategy affecting both CREB and AP1 activation may be optimal to trigger the regenerative phenotype.
EXPERIMENTAL PROCEDURES
Use of animals conformed to the Burke-Cornell IACUC guidelines under approved protocols.
Primary Neuron and Cell Culture
Mouse cortical neurons were cultured from E14.5 embryos as previously described and used for experiments 1 day later (14). Mouse DRG neurons were cultured from E12.5 embryos as previously described and plated in Neurobasal/B27 media supplemented with 50 ng/ml NGF (EMD Millipore) + 10 μm 5-fluorodeoxyuridine. Media were changed at 4 days in vitro, and cultures were used at 7 days in vitro when nearly all Schwann cells were killed. CHO cells stably expressing myelin-associated glycoprotein (MAG) or the empty pSJL vector were cultured as previously described (15). All cell culture media components were obtained from Invitrogen unless otherwise noted.
Promoter-Reporter Assays
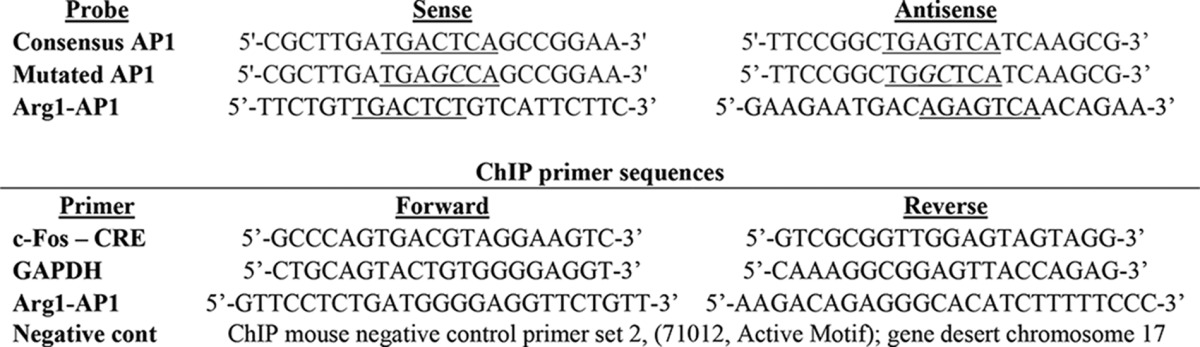
DRG neurons were transfected with firefly luciferase promoter-reporter constructs containing 4782, 3291, 2781, 1405, 809, and 112 bp of the Arg1 promoter (16), a mutated Arg1–112-bp promoter, or consensus AP1 promoter plus an HSV-TK-renilla luciferase plasmid transfection control. 0.8 μg of reporter plasmid + 0.04 μg of TK-renilla were complexed with Lipofectamine LTX and Plus reagent (1:2; μg of DNA:μl of Lipofectamine). The neurons were then treated with Bt2cAMP (2 mm; N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate; Sigma) and then assayed 24 h later using a dual-luciferase kit (Promega). Data are expressed as the ratio of firefly:renilla luminescence normalized to control.
The putative AP1 binding site of the Arg1–112-bp promoter was mutated to a sequence with diminished AP1 binding (from 5′-tgaCTct-3′ to 5′-tgaGCct-3′) (17) by site-directed mutagenesis using the QuikChange II kit (Agilent Technologies).
Real Time RT-PCR
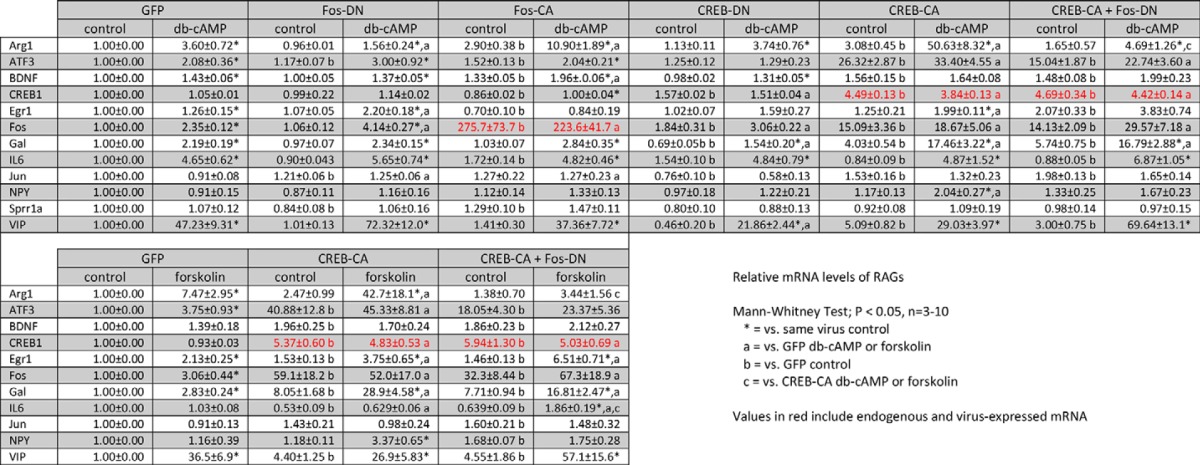
RNA was harvested with TriReagent (Sigma), column-purified, and treated with DNase (Directzol kit, Zymo Research). cDNA libraries were generated (iScript, Bio-Rad) from which gene expression was assayed by real time PCR using TaqMan FAM-labeled probes (Invitrogen) with VIC-labeled β-actin or GAPDH included in duplex as endogenous controls. For Arg1, the endogenous controls were run in parallel. Relative expression levels of each gene are summarized in Table 1. The probes used were: Arg1, Mm00475988_m1; ATF3, Mm00476032_m1; BDNF, Mm04230607_s1; CREB1, Mm00501607_m1; Egr1, Mm00656724_m1; Fos, Mm00487425_m1; Gal, Mm00439056_m1; IL6, Mm00446190_m1; Jun, Mm00495062_s1; JunD, Mm00498088_s1; NPY, Mm03048253_m1; Sprr1a, Mm01962902_s1; VIP, Mm00660234_m1.
TABLE 1.
mRNA expression of RAGs
EMSA
Nuclear proteins were prepared from DRG neuron cultures after treatment with Bt2cAMP (NE-PER kit; Pierce). 50 fmol of 5′ IRDye700-labeled double-stranded DNA probes were combined with 5 μg of nuclear protein, incubated at room temperature for 20 min, and resolved on a gradient acrylamide mini gel (Bio-Rad) at 100 V for 2 h. For supershift experiments, 5 μg of each antibody was added to the nuclear protein/probe mixture before incubation. The gels were imaged using the LiCor gel imaging system (LiCor). The antibodies used were: rabbit IgG (12-370, Millipore), c-fos (SC-253X, Santa Cruz Biotechnology), c-Jun (SC-45X, Santa Cruz), JunD (SC-74X, Santa Cruz), and FosB (SC-28213X, Santa Cruz). Probe sequences are listed in Table 2.
TABLE 2.
EMSA probe sequences (consensus or putative binding site underlined)
ChIP
After treatments, cultures were fixed in 1% formalin (Sigma) in PBS for 9 min at room temperature and then quenched with 125 mm glycine in PBS for 5 min. Cells were collected in ice-cold harvesting buffer (100 mm Tris-HCl, 10 mm DTT, pH 9.4), pelleted, and lysed. The resulting chromatin was sheared by bath sonication (Bioruptor, Diagenode) and immunoprecipitated with 3 μg of antibody using the low cell number ChIP kit (Diagenode). Enrichment of promoter regions was probed using SYBR-Green real time PCR (Invitrogen). The antibodies used were as with the EMSA supershift assays, CREB (17-600; Millipore) and acetyl-H3 (06-599; Millipore). The primer sequences are listed in Table 2.
Lentiviral Vector Generation
The coding sequences of modified CREB and Fos proteins were cloned into the LentiLox 3.7 backbone under the control of the synapsin I promoter, restricting expression to neurons. The viruses were produced by the University of Iowa Gene Transfer Vector Core (Iowa City, IA). Viruses were added at a multiplicity of infection of 5 directly to culture media at 4 days in vitro, and the neurons were treated or trypsinized at 7 days in vitro.
MAG Inhibition Assays
Untreated or lentivirus-transduced neurons were trypsinized and replated upon MAG- and R2-CHO cell monolayers in clear-bottom 96-well plates at ∼150 neurons/well. Bt2cAMP (final 2 mm) or forskolin (final 10 μm) was added 1 h later. 24 h later the co-cultures were fixed with 4% paraformaldehyde and then processed for fluorescence immunocytochemistry with an anti-βIII-tubulin antibody (1:500, rabbit monoclonal; Epitomics). Neurons were imaged using a flash cytometer (Trophos) that captures images of entire wells of the 96-well plate. The total neurite length/neuron was automatically measured using the neurite outgrowth application in Metamorph (Molecular Devices). Neurite length data are expressed as total neurite length per neuron. Representative higher magnification images were taken with an inverted Motic AE31 epifluorescence microscope with a 20× objective.
Statistical Analysis
Data were plotted as the mean ± S.E. unless otherwise indicated from at least three independent experiments from separate primary cultures. The statistical tests used are indicated in the figure legends. For mRNA expression, the data are plotted as box and whiskers to show the range, median, and mean obtained between independent experiments.
RESULTS
Increasing neuronal cAMP levels can promote neurite growth and reverse growth cone repulsion to inhibitory cues in vitro (3, 11, 18–20). To assess neurite growth, we replated primary DRG neurons upon CHO cell monolayers stably expressing either MAG or a control vector (R2) in media lacking NGF to limit basal neurite growth. Neurons plated on MAG-CHO cells exhibited significant inhibition of neurite growth compared with those on R2-CHO cells, consistent with the inhibitory properties of MAG as a component of CNS myelin (Fig. 1, A–C) (15). The addition of Bt2cAMP, a cell-permeable cAMP analog, increased neurite length on both cell types, indicating that Bt2cAMP stimulates neurite growth on both inhibitory and permissive substrates (Fig. 1, A–C).
FIGURE 1.

Coregulation of neurite growth and RAG transcription by active CREB and Bt2cAMP. A–C, Bt2cAMP promotes neurite growth on inhibitory (MAG-CHO) and permissive (R2-CHO) substrates. A, representative fluorescence micrographs of βIII-tubulin-labeled DRG neurons on CHO cells; Bt2cAMP (db-cAMP; 2 mm). Quantitation of total neurite length/neuron on MAG-CHO (B) or R2-CHO cells (C) is shown. *, p < 0.05 versus control, one-way ANOVA with Newman-Keuls post test. D, modulation of CREB-mediated transcriptional activity. Expression of dominant-negative (CREB-DN) or constitutive-active (CREB-CA) forms of CREB inhibited or potentiated the Bt2cAMP-induced expression of a CRE-driven luciferase reporter, respectively. *, p < 0.01; n.s. = not significant, two-way ANOVA with Bonferroni post test, n = 6–14. E–G, CREB synergizes with Bt2cAMP to enhance neurite growth on MAG. DRG neurons transduced with CREB-DN or CREB-CA were replated on CHO cells. E, fluorescence micrographs and (F and G) quantitation show that Bt2cAMP increased neurite growth in CREB-independent fashion, although CREB-CA was sufficient to increase basal neurite growth. CREB-CA showed synergy with Bt2cAMP to further enhance neurite growth on MAG-CHO and was additive on R2-CHO cell substrates. *, p < 0.0001; n.s. = p > 0.05; +, p < 0.0001 versus GFP control, two-way ANOVA with Bonferroni post test; n = 972–1984 neurons. H, mRNA expression profile of candidate RAGs. A heat map was generated from mRNA expression levels of the indicated RAGS from DRG neurons expressing GFP or CREB-CA and then treated with Bt2cAMP. The expression levels of each gene were then correlated to axon length on MAG-CHO cells for each manipulation. The correlation coefficient and whether each gene was induced by Bt2cAMP or blocked by CREB-DN are listed. VIP, vasoactive-intestinal peptide. *, p < 0.05 Pearson's test. For Induced by Bt2cAMP? column, Y = p < 0.05 GFP control versus GFP + Bt2cAMP. For the Blocked by CREB-DN column, Y = p < 0.05 CREB-DN control versus CREB-DN+Bt2cAMP, Mann-Whitney test; n = 4–10. 2 mm Bt2cAMP was used in all panels unless otherwise noted. Scale bars = 50 μm.
The transcription-dependent axon growth effects of cAMP have been attributed to the activation of CREB (13). To assess the role of CREB in neurite growth, we expressed a constitutive-active CREB fusion protein (VP16CREB; referred to as CREB-CA), a dominant-negative CREB protein (ACREB; referred to as CREB-DN), or GFP control using lentiviruses to manipulate CREB transcriptional activity (21, 22). At a multiplicity of infection of 5, this yielded 100% transduction efficiency. In DRG neurons, the expression of CREB-CA further increased Bt2cAMP-induced CRE reporter activity, whereas CREB-DN blocked CRE activity induced by Bt2cAMP (Fig. 1D). DRG neurons transduced with these viruses were replated upon the CHO cell monolayers to assess neurite growth. Consistent with previous studies, CREB-CA enhanced basal neurite growth on both MAG- and R2-CHO cells (Fig. 1, E–G) (13). Interestingly, CREB-CA showed marked synergy with Bt2cAMP, causing a greater than additive increase in neurite growth on MAG-CHO cells. Surprisingly, we found that the Bt2cAMP-mediated neurite growth persisted in neurons expressing CREB-DN (Fig. 1, E–G). These results indicate that although activation of CREB-dependent gene expression can by itself promote neurite growth, it is not necessary for cAMP-triggered growth, which signifies the existence of an alternative growth-promoting pathway downstream of cAMP.
To examine the specific contribution of CREB and its upstream second messenger cAMP to RAG expression, we measured the transcript levels of several candidate RAGs (2, 3, 9, 10, 23–27) in neurons expressing either GFP, CREB-CA, or CREB-DN in the presence or absence of Bt2cAMP (Fig. 1H). Consistent with previous reports, Bt2cAMP increased the expression levels of a subset of these RAGs in GFP-transduced neurons (Fig. 1H); however, the behavior of these Bt2cAMP-responsive genes in neurons expressing either CREB-CA or CREB-DN were markedly different. We observed several distinct gene expression patterns. Of note are the following. 1) Vasoactive-intestinal peptide (VIP) was highly and maximally induced (∼47-fold) by Bt2cAMP. 2) ATF3 behaved as a canonical CREB target; CREB-DN prevented ATF3 induction by Bt2cAMP, whereas CREB-CA strongly induced its expression regardless of Bt2cAMP treatment. 3) IL6 transcription is completely CREB-independent; neither CREB-DN nor CREB-CA altered the induction of IL6 by Bt2cAMP. 4) Arg1 and Gal were up-regulated by Bt2cAMP in a CREB-independent manner (i.e. CREB-DN did not block their induction), although CREB-DN significantly decreased basal and, thus, Bt2cAMP-induced levels of Gal. CREB-CA alone also up-regulated their basal expression, but this was greatly increased in neurons that were also treated with Bt2cAMP (∼17–50-fold). The expression levels of each gene are summarized as a heat map (Fig. 1H) and in Table 1.
We next correlated RAG expression levels to neurite length on MAG-CHO cells under each CREB manipulation to determine the genes associated with neurite growth. By this analysis, Arg1 and Gal were the Bt2cAMP-induced genes with the highest correlation between expression levels and neurite length (R2 = 0.954 and 0.958, respectively; Fig. 1H). Because Gal expression was attenuated by CREB-DN, we focused on the transcriptional regulation of Arg1, which was unaltered by CREB-DN, to further explore the synergy between Bt2cAMP and CREB-CA in driving specific RAG expression.
To identify the promoter regions critical for Bt2cAMP responsiveness, we used a series of Arg1 promoter-luciferase reporter constructs with progressively shorter promoter regions (−4.8 kb, −3.3 kb, −2.8 kb, −1.4 kb, −0.8 kb, and −112 bp). Luciferase activity was equally up-regulated by Bt2cAMP (2 mm for 24 h) with all constructs, indicating that the minimum-essential promoter region resided in the initial 112 bp (Fig. 2A). To determine whether CREB binds to this region, we assessed the occupancy of CREB by ChIP from neurons treated with Bt2cAMP (2 mm) for 2 h, a time point we have previously shown as maximal CREB activation (14). The initial 112-bp region of the Arg1 promoter was not enriched by CREB ChIP, indicating that CREB does not directly bind this minimal Arg1 promoter (Fig. 2C). By contrast, the CRE region of the Fos promoter, an established CREB target gene, was significantly enriched (28).
FIGURE 2.

Arg1 transcription is mediated by AP1. A, promoter-reporter constructs containing different lengths of the Arg1 promoter were equally activated by Bt2cAMP in DRG neurons, indicating that the initial 112 bp contains the minimum essential promoter. p < 0.05, Bt2cAMP versus control for each construct, Mann-Whitney test, n = 4. B, top, the initial 112 bp of the Arg1 promoter contains putative CREB and AP1 binding sites. Bottom, site-directed mutagenesis of the putative Arg1-AP1 binding site. C, chromatin from cortical neurons treated with Bt2cAMP was subjected to ChIP with a CREB antibody. CREB occupancy in the initial 112 bp of the Arg1 promoter was unchanged by Bt2cAMP, whereas occupancy was increased at the c-fos promoter, a CREB target gene. *, p < 0.05; n.s. = p > 0.05, Mann-Whitney test, n = 4. D, mutation of the putative Arg1-AP1 site abrogates promoter activation by Bt2cAMP (db-cAMP). *, p < 0.01; two-way ANOVA with Bonferroni post test, n = 3. E, AP1 nuclear protein complexes bind to the Arg1 promoter. Nuclear proteins from Bt2cAMP-treated DRG (left and middle panels) and cortical (cort) neurons (right panel) were assayed by EMSA with an AP1 consensus (asterisk) or putative Arg1-AP1 (arrowheads) binding site probes. Bt2cAMP induced prominent mobility shifts of both probes. Cross competition with a 180-fold excess of unlabeled AP1 probe eliminated binding of the Arg1 probe, indicating that the same nuclear protein complexes bind to both probes. Representative gels from 3 independent experiments are shown. F, AP1 subunit supershift. Inclusion of a JunD antibody induced a prominent supershift, whereas c-Fos and c-Jun antibodies decreased Bt2cAMP-induced binding to the consensus AP1 EMSA probe. G, AP1 subunit occupancy at the Arg1 promoter. Chromatin from DRG neurons treated with Bt2cAMP was subjected to ChIP with antibodies against the indicated AP1 subunits. Basal JunD occupancy was decreased and c-Jun occupancy was increased by Bt2cAMP. Open bars show no enrichment of a gene desert site by these antibodies as a negative control. p values are as indicated; Student's t test; n = 3. H, Bt2cAMP does not change c-Jun or JunD expression levels. DRG neurons were treated with Bt2cAMP and assayed for c-Jun and JunD expression at the indicated times. Two-way ANOVA, n = 4. I, modulation of AP1 transcriptional activity. Dominant negative (Fos-DN) and constitutive active (Fos-CA) AP1 variants were expressed in DRG neurons. Treatment with Bt2cAMP induced expression of a consensus AP1-luciferase reporter, which was abrogated by Fos-DN and potentiated by Fos-CA. *, p < 0.05; +, p < 0.05 versus GFP control; one-way ANOVA with Bonferroni post test within group, n = 4–6. J, Arg1 transcription is AP1-dependent and -sufficient. Expression of Fos-DN blocked the Bt2cAMP-mediated up-regulation of Arg1 mRNA in DRG neurons, whereas Fos-CA increased basal and potentiated Bt2cAMP-induced Arg1 transcription. Parentheses indicate mean -fold induction over GFP control. *, p < 0.05 versus control for same virus condition; +, p < 0.0001 versus GFP control; p values are indicated for other comparisons, Mann-Whitney test, n = 10. 2 mm Bt2cAMP was used for all experiments.
To determine which other transcription factor might bind to this region, we searched for binding sites using the Transfac 7.0 public database and identified a putative AP1 binding site (Fig. 2B, top panel). To assess its necessity, we mutated two nucleotides within this site of the Arg1–112-bp construct (Fig. 2B, lower panel), which attenuated Bt2cAMP-induced reporter activity, indicating that the site was essential for promoter activation (Fig. 2D). We then used an EMSA with a consensus AP1 site probe to establish that Bt2cAMP (2 mm for 24 h) increased AP1 DNA binding activity of nuclear proteins from DRG neurons (Fig. 2E, left panel). Similarly, Bt2cAMP increased binding to a probe containing the putative Arg1-AP1 site (Fig. 2E, middle panel). Bt2cAMP also induced a mobility shift of the Arg1-AP1 probe with nuclear proteins from cortical neurons, indicating that binding to this site is regulated by Bt2cAMP in multiple neuron types (Fig. 2E, right panel). As expected, an excess of an unlabeled AP1 probe eliminated the binding of both labeled probes, indicating that the Bt2cAMP-induced mobility shifts were specific to the AP1 probe sequence and that the same nuclear protein complexes may bind both the Arg1 and consensus AP1 probes (Fig. 2E). Conversely, an excess of an unlabeled probe with a mutated AP1 sequence did not eliminate binding to the labeled probes (data not shown). To identify the AP1 subunits that are active in DRG neurons, we used antibodies against c-fos, FosB, JunD, and c-jun to supershift the consensus AP1 probe. The JunD antibody exhibited a prominent supershift, whereas the c-fos and c-jun antibodies attenuated binding of nuclear protein complexes to the probe, indicating that these subunits may be active after Bt2cAMP treatment (Fig. 2F).
We then assessed the occupancy of c-fos, c-jun, and JunD at the Arg1 promoter using ChIP analysis. Under basal conditions, the occupancy of only JunD was evident; treatment with Bt2cAMP released JunD while simultaneously inducing c-jun occupancy (Fig. 2G). These antibodies did not enrich chromatin from a gene desert used as a negative control. To determine whether the changes in AP1 activity were due to the transcriptional regulation of AP1 subunits, we measured the expression of Jun and JunD at 2, 8, and 24 h and found no change associated with Bt2cAMP exposure (Fig. 2H).
We next addressed the sufficiency and necessity of AP1 activity in driving Arg1 transcription by directly modulating AP1 activity using lentiviruses to express dominant-negative (AFos; referred to as Fos-DN) or constitutive-active (FosVP16; referred to as Fos-CA) variants of the Fos AP1 subunit (29, 30). The expression of Fos-DN blocked Bt2cAMP-induced expression of an AP1 response element-driven luciferase reporter, whereas Fos-CA increased basal and Bt2cAMP-induced reporter activity (Fig. 2I). Importantly, Arg1 mRNA levels mirrored this pattern, as Fos-DN attenuated Arg1 induction by Bt2cAMP, whereas Fos-CA increased basal Arg1 levels and potentiated Bt2cAMP-mediated Arg1 expression (Fig. 2J). These data indicate that AP1 activity is a necessary mediator of Bt2cAMP-induced Arg1 transcription.
We next asked whether CREB-CA and Bt2cAMP induced Arg1 expression by activating AP1. The expression of CREB-CA increased AP1 reporter activity, indicating that it may directly drive AP1-mediated transcription (Fig. 3A). To determine whether AP1 activity was necessary, we co-expressed CREB-CA and Fos-DN to drive CREB activity while blocking AP1. This reversed the synergy between CREB-CA and Bt2cAMP, indicating that AP1 activity was necessary for Arg1 induction and that CREB gene products may interact with AP1 to further drive RAG transcription (Fig. 3B). As AP1 subunits can be CREB target genes, we measured the expression levels of two prominent subunits, Fos (Fig. 3C) and Jun (Fig. 3D), in neurons transduced with CREB-CA. The expression of Fos was markedly induced by CREB-CA (15-fold), whereas Jun levels were modestly, but significantly, up-regulated (1.5-fold). These data suggest that CREB-CA may increase AP1 activity by increasing the expression of AP1 subunits. To further probe the role of AP1 in RAG transcription, we measured the expression levels of all 12 RAGs under each manipulation of CREB and AP1 to see which other genes were also AP1-dependent (Fig. 3E, left panels, and Table 1). Interestingly, only Arg1 showed AP1 dependence for both Bt2cAMP and CREB-CA-induced expression (Table 1).
FIGURE 3.

CREB-CA mediated Arg1 expression is AP1-dependent. A, expression of CREB-CA in DRG neurons increases basal and Bt2cAMP (db-cAMP)-induced AP1 reporter expression. *, p < 0.05 versus control for same virus condition; +, p < 0.05 versus GFP control, Mann-Whitney test, n = 6. B, the synergistic expression of Arg1 by CREB-CA and Bt2cAMP is AP1-dependent. The synergy between CREB-CA and Bt2cAMP was abrogated by co-expression of Fos-DN to block AP1-dependent transcription. * p < 0.05 versus control for same virus condition, p values are indicated for other comparisons, Mann-Whitney test, n = 10. Gray bars are re-plotted from Fig. 2 for comparison. C and D, CREB-CA increases the expression of c-Fos (C) and c-Jun (D) subunits of AP1. *, p < 0.05 versus control for same virus condition; p values are indicated for other comparisons, Mann-Whitney test, n = 4. E, expression profile of candidate RAGs. Heat maps were generated of RAG expression levels from DRG neurons transduced with lentiviruses delivering the indicated modified transcription factors and then treated with Bt2cAMP (2 mm) or forskolin (10 μm) for 24 h. Nearly all RAGs responsive to Bt2cAMP responded similarly to forskolin, indicating that cAMP mediates the effects of both agents.
Because Bt2cAMP is a cell membrane permeant analog of cAMP that requires hydroylzation of the butyrate side chains for activity (31), we tested whether off-target actions of this compound contributed to our gene expression changes. We used forskolin (10 μm), an activator of cellular adenylate cyclase, to generate intracellular cAMP in neurons transduced with GFP, CREB-CA, and CREB-CA + Fos-DN (Fig. 3E, right panel, and Table 1). Interestingly, only IL6 expression deviated from that observed with Bt2cAMP, as IL6 levels were not induced by forskolin (Fig. 3). This congruence indicates that the gene expression effects and interaction of CREB-CA with Bt2cAMP are indeed cAMP-dependent.
We next assessed whether the manipulation of CREB and AP1 altered neurite growth of DRG neurons. Blocking AP1 activity with Fos-DN inhibited Bt2cAMP-mediated neurite growth on both MAG- and R2-CHO cells while driving AP1 activity with Fos-CA increased neurite growth in combination with Bt2cAMP only on MAG (Fig. 4, A–C); however, both basal and Bt2cAMP-induced neurite growth was greater in CREB-CA-expressing neurons. This effect of CREB-CA was reversed in cells co-transduced with CREB-CA and Fos-DN, indicating that AP1 mediated these effects of CREB activation (Fig. 4, A–C). This necessity of AP1 was also evident on the permissive R2-CHO cells, although the effect of CREB-CA was additive. These data indicate that these manipulations drive both intrinsic growth and counteract the extrinsic inhibitory signals from MAG (Fig. 4, A–C). These experiments excluded NGF from the replating media to limit basal neurite growth from the trophic effects of NGF; thus, expression of Fos-CA and CREB-CA may simply reconstitute some of the transcriptional effects of NGF. We repeated these experiments with a low concentration of NGF (1 ng/ml) to determine whether neurite growth was still affected by manipulation of CREB and AP1 activity. The inclusion of NGF increased both basal and Bt2cAMP-mediated neurite growth on MAG- and R2-CHO cells; however, blocking AP1 with Fos-DN only partially abrogated the effect of Bt2cAMP on MAG-CHO cells, indicating that NGF can activate AP1-independent pathways that act with Bt2cAMP to promote neurite growth (Fig. 4, D and E). Despite the presence of NGF, the AP1-dependent interaction between CREB-CA and Bt2cAMP persisted, indicating that the benefit derived from further activating CREB required AP1 activity (Fig. 4, D and E). Finally, we used forskolin (10 μm) to verify that the neurite growth effects of Bt2cAMP were due to the elevation of intracellular cAMP levels. Similar to gene expression, the neurite growth-promoting effect and interaction with CREB-CA mirrored that of Bt2cAMP (Fig. 4, F and G); however, the magnitude of growth was greater with forskolin, indicating that it may generate higher local levels of free cAMP than the Bt2cAMP analog.
FIGURE 4.

Enhancement of neurite growth of by Bt2cAMP and CREB is AP1-dependent. DRG neurons transduced with lentiviruses delivering the indicated modified transcription factors or GFP were replated on MAG- and R2-CHO cell monolayers and then treated with Bt2cAMP (db-cAMP, 2 mm) ± NGF (1 ng/ml) or forskolin (10 μm) without NGF for 24 h. A, representative fluorescence micrographs of βIII-tubulin-labeled DRG neurons on MAG (no NGF). Scale bar = 50 μm. B–G, quantitation of mean neurite length per neuron. Neurons in B, C, D, and E were treated with Bt2cAMP, whereas F and G were treated with forskolin. Bars in gray are re-plotted from Fig. 1 for comparison. *, p < 0.01, two-way ANOVA with Bonferroni post test, n = 403–1984 neurons from 3–5 independent experiments.
To evaluate the relationship between the expression levels of the candidate RAGs and neurite growth on MAG-CHO cells with each manipulation of AP1 and CREB activity, we normalized the Bt2cAMP-induced expression levels of each gene and the corresponding neurite length data, assigning the levels obtained with GFP as the baseline (0) and the manipulation with highest gene expression/neurite growth as the maximum (100) and asked whether gene expression correlated with neurite length (Fig. 5, A and B). We found significant correlation between these factors only for Arg1 (R2 = 0.9699, p = 0.0003, two-tailed Pearson test; Fig. 5A), indicating that the transcriptional program regulating Arg1 and possibly other RAGs not queried in our study may be the most relevant to neurite growth in DRG neurons. These findings also suggest that Arg1 expression may be a good biomarker for the activation of this program. Together, these data indicate that these RAGs may not be maximally up-regulated by Bt2cAMP alone and that further activation of transcription pathways downstream of cAMP (CREB and AP1) concurrent with cAMP up-regulation may provide superior RAG expression and neurite growth (Fig. 5C).
FIGURE 5.

AP1 activity dictates the axon growth-related RAG expression profile. A and B, gene expression and axon growth correlation for RAGs. A, representative plot of Arg1 normalized Bt2cAMP-induced expression levels (y axis) versus axon growth on MAG-CHO cells (x axis) of DRG neurons with the indicated manipulations. Data were normalized between 0 and 100, where the value for GFP = 0 and the maximum response = 100. B, correlation coefficients of all RAGs. When all manipulations were considered, only the profile of Arg1 expression was significantly correlated with axon growth (p = 0.0003, Pearson's test), indicating that the CREB-sufficient but AP1-dependent transcriptome is relevant for promoting axon growth. C, scheme for the transcriptional regulation of cAMP-induced RAG expression. Bt2cAMP activates the transcription of Arg1 in a CREB-sufficient but AP1-dependent manner. Activated CREB synergizes with cAMP-induced AP1 activity to drive higher levels of Arg1 transcription as well as neurite growth (red arrows).
DISCUSSION
Analysis of the peripheral nerve injury-induced transcriptome has identified individual genes and pathways that contribute to axon regeneration. These transcriptional responses function to increase intrinsic growth capacity and confer resilience to inhibitory cues, allowing neurons to regenerate damaged peripheral nervous system and CNS axons. The convergent nature of these responses stresses the importance of engaging these pathways sufficiently upstream of the terminal effectors (2, 3, 9, 10, 32–34). Although cAMP-activated pathways are important components of this machinery, a number of studies show that cAMP elevation alone may insufficiently drive RAG transcription and is only effective at improving regeneration in some settings (3, 6, 8, 12, 20, 35). This complexity is further exemplified by our disparate results regarding the efficacy of constitutive-active and dominant-negative versions of CREB and Fos to influence Bt2cAMP induction of RAGs and promote or interfere with Bt2cAMP-dependent neurite growth. In our studies we found that exposure to Bt2cAMP promoted neurite growth in DRG neurons, and this ability was further potentiated by the expression of a CREB-CA variant in the same cells. Moreover, our experiments identified the transcription factor AP1 as a likely candidate to synergize with CREB to enhance neurite growth on inhibitory substrates through the regulation of specific RAGs, such as Arg1 (Fig. 5C).
Although the expression of CREB-CA was sufficient to drive Arg1 transcription and neurite growth, we could not block the effects of Bt2cAMP by inhibiting CREB activity with CREB-DN (Fig. 1H and Table 1). Instead, we found that both the Bt2cAMP- and CREB-CA-mediated transcription of Arg1 was AP1-dependent in DRG neurons, indicating that both manipulations act by inducing AP1 activity (Figs. 2J and 3B). Together, this suggests that Bt2cAMP activates both AP1 and CREB in parallel and that CREB-activated gene products likely interact with AP1 to further increase the expression of certain RAGs, including Arg1. Although we focused on an AP1 site in the initial 112 bp of the Arg1 promoter (Fig. A–G), other putative AP1 binding sites have been described further upstream and are functional in non-neuronal cell types; whether these sites act cooperatively in DRG neurons remains to be determined (36, 37). Both Bt2cAMP and CREB-CA increased AP1-mediated transcriptional activity in our study (Figs. 2I and 3A). Multiple mechanisms can increase AP1 activity, including the transcriptional up-regulation of AP1 subunits, changes in subunit composition, and subunit phosphorylation (38). In our study both Bt2cAMP and, to a greater extent, CREB-CA increased Fos mRNA levels (Fig. 3C), whereas only CREB-CA increased Jun mRNA (Fig. 3D). Although the incorporation of Fos subunits can increase AP1 DNA binding and transcriptional activity, we did not find changes in Fos occupancy at the Arg1 promoter after Bt2cAMP treatment in control cells within the detection limits of our assay (Fig. 2G); however, along with the induction of Fos by Bt2cAMP, the inhibition Arg1 expression by Fos-DN and the effect of the c-Fos antibody on AP1 EMSA probe binding suggest a role for Fos in regulating Arg1 transcription (Fig. 2, F and J). With CREB-CA, the high levels of Fos expression and Jun induction may indeed drive the AP1-dependent transcription, including that of Arg1 (Fig. 3, C and D). AP1 transcriptional activity is enhanced by the phosphorylation of the N-terminal transactivation domain of Jun by c-Jun-N-terminal kinases (JNK) (39). This may be a point of AP1 regulation by cAMP, as cross-talk between cAMP-activated pathways (both protein kinase A and EPAC-mediated) and MAPK/JNK pathways has been described in other cell types and suggests the importance of the non-transcriptional actions of cAMP (40). We found a concurrent increase in Jun and decrease in JunD occupancy at the Arg1 promoter after Bt2cAMP treatment (Fig. 2G), that was not associated with changes in mRNA levels of either subunit (Fig. 2H). JunD is an atypical member of the AP1 family that can act as both a transcriptional activator and repressor (41). Together our data suggest that JunD binding to the Arg1 promoter in the basal state may interfere with CREB-CA-driven AP1-dependent transcription; upon Bt2cAMP treatment, the activation of AP1 may release JunD to enable strong transcription of AP1 targets downstream of CREB-CA.
Importantly, AP1 was necessary for Bt2cAMP-mediated neurite growth on both permissive and inhibitory substrates (Fig. 4, B and C), which is consistent with the prominent role for AP1 in mediating peripheral axon regeneration in vivo. AP1 activity is up-regulated in DRG neurons after peripheral nerve injury and remains elevated until target reinnervation (42), whereas targeted deletion of c-Jun in neurons impedes regeneration after axotomy (43), indicating that AP1 is necessary to maintain RAG expression to facilitate regeneration. On a network level, AP1 subunits have been identified as central hubs of injury-induced gene transcription in DRG mRNA profiling studies (3, 9, 32). Although other regeneration-associated AP1 targets have been identified, our data suggest that up-regulating any single gene product, including Arg1, is unlikely to harness the full effect of activating upstream transcription factors to drive the regenerative “program.” This is evidenced by the inability of S-(2-boronoethyl)-l-cysteine (BEC), a pharmacological inhibitor of Arg1 enzymatic activity, to block the neurite growth effect of CREB-CA and Bt2cAMP (data not shown). As Arg1 has been previously shown to play important roles in nervous system repair by promoting polyamine synthesis, inhibiting nitric oxide production, and regulating protein translation, the lack of necessity for Arg1 does not preclude an important role for Arg1 in axonal regeneration but suggests that other genes and pathways likely compensate for the loss of Arg1 activity.
An important finding in this study is that the expression of Arg1, Gal, and NPY were further increased by directly activating CREB and/or AP1 transcriptional activity in conjunction with Bt2cAMP (Fig. 3H and Table 1). This suggests that the activation of either transcription factor by Bt2cAMP is insufficient to drive maximal RAG expression and neurite growth or, alternatively, that Bt2cAMP triggers an inhibitory feedback loop such as the inducible cAMP early repressor (ICER) that can be overcome by the constitutive-active transcription factor variants (30). Notably, endogenous CREB mRNA levels were not altered by Bt2cAMP, and total levels of CREB transcripts (both from endogenous CREB and CREB-CA) were increased to 5-fold over GFP-transduced neurons by CREB-CA expression. This could indicate that the endogenous levels of CREB may be limiting in mediating RAG transcription in response to Bt2cAMP. By contrast, the combined Fos levels were 275-fold increased in transduced Fos-CA neurons (Fig. 3E and Table 1), indicating that high Fos expression alone is insufficient to recapitulate the CREB-CA effect. Together this suggests that other direct CREB-CA target genes are required in addition to the activation of AP1.
In the context of peripheral nerve injury, many pathways are simultaneously activated within neurons and in surrounding non-neuronal cells. These include the retrograde transport of protein complexes from inured axons, including transcription factors (i.e. STAT3) and activators of injury signaling (i.e. JNK and ERK) that ultimately lead to transcriptional changes at the soma (9, 44–46). Additionally, perturbation of the normal injury response of macrophages and Schwann cells can occlude axon regeneration and the peripheral lesion conditioning effect (47–50). Some of these factors, such macrophage-released oncomodulin, work in synergy with cAMP to promote both peripheral nervous system and CNS axon regeneration (50, 51). It is likely that these effectors converge with cAMP-mediated signaling and gene transcription to drive axonal regeneration.
After injury, CNS axons are exposed to factors that inhibit regeneration, including myelin proteins such as MAG. Although this may alter the transcriptional landscape of the associated neurons, previous CNS axon injury of DRG neurons does not preclude the induction of RAGs after peripheral axon injury, suggesting that the injury-induced transcriptional programs are not repressed by inhibitory substrates (27). In the context of our studies, we expect that similar transcriptional changes occur in the neurons replated onto MAG-CHO cells as those plated upon laminin.
Our findings suggest that efforts to promote axonal regeneration by increasing neuronal cAMP levels alone may yield limited success. Rather, we identify a CREB-mediated, AP1-dependent transcriptional “module” that functions in concert with cAMP to promote robust neurite growth under inhibitory and permissive conditions. Although our data reaffirm the importance of CREB for axonal regeneration, they indicate that cAMP in conjunction with CREB activation is necessary to maximally activate the transcriptional program that recapitulates the regenerative phenotype. This interdependence between CREB and cAMP suggests that a strategy targeting more than a single transcription pathway (i.e. both CREB and AP1) may be more efficient than the genetic activation of a single transcriptional factor. This idea is consistent with the observed superior efficacy of multimodal combinatorial approaches utilizing cAMP elevation in combination with neurotrophic factors to promote regeneration (52–54). Additionally, as Arg1 mRNA expression is regulated in a CREB-sufficient and AP1-dependent fashion, Arg1 levels may serve as a relevant biomarker for pharmacological or genetic manipulations aimed at promoting regenerative gene transcription.
Acknowledgments
We thank Dr. Sidney M. Morris for the Arg1 promoter constructs, Dr. Wilfredo Mellado for technical assistance, and Dr. Chris Henderson for careful reading of the manuscript. The Instituto de Neurociencias is a Centre of Excellence Severo Ochoa.
This work was supported, in whole or in part, by National Institutes of Health Grant NR010797 (to D. W.). This work was also supported by grants from the Dr. Miriam and Sheldon G. Adelson Foundation (to R. R.), The Center for Research Excellence in Spinal Cord Injury (Grant DOHC019772; to R. R.), and the Burke Medical Research Institute Foundation (to D. W.). The Barco laboratory is supported by Grants SAF2011-22855 from the Spanish Ministry of Science and Innovation and Prometeo/2012/005 from the Generalitat Valenciana.
This work is dedicated to the memory of Dr. Marie T. Filbin. Her groundbreaking work set the foundation for these studies. “If we see farther, it is by standing on the shoulders of giants.”
- RAG
- regeneration-associated gene
- CREB
- cAMP-responsive element binding protein
- AP1
- activator protein 1
- Arg1
- arginase 1
- Gal
- galanin
- DRG
- dorsal root ganglion
- CA
- constitutive-active
- DN
- dominant-negative
- Bt2cAMP
- dibutyryl cyclic AMP
- MAG
- myelin-associated glycoprotein
- Fos-DN
- dominant-negative Fos
- ATF3
- activating transcription factor 3
- ANOVA
- analysis of variance.
REFERENCES
- 1. Smith D. S., Skene J. H. (1997) A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 17, 646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cao Z., Gao Y., Bryson J. B., Hou J., Chaudhry N., Siddiq M., Martinez J., Spencer T., Carmel J., Hart R. B., Filbin M. T. (2006) The cytokine interleukin-6 is sufficient but not necessary to mimic the peripheral conditioning lesion effect on axonal growth. J. Neurosci. 26, 5565–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blesch A., Lu P., Tsukada S., Alto L. T., Roet K., Coppola G., Geschwind D., Tuszynski M. H. (2012) Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to camp-mediated effects. Exp. Neurol. 235, 162–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lesiak A., Pelz C., Ando H., Zhu M., Davare M., Lambert T. J., Hansen K. F., Obrietan K., Appleyard S. M., Impey S., Wayman G. A. (2013) A genome-wide screen of CREB occupancy identifies the RhoA inhibitors Par6C and Rnd3 as regulators of BDNF-induced synaptogenesis. PloS ONE 8, e64658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neumann S., Woolf C. J. (1999) Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91 [DOI] [PubMed] [Google Scholar]
- 6. Qiu J., Cai D., Dai H., McAtee M., Hoffman P. N., Bregman B. S., Filbin M. T. (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34, 895–903 [DOI] [PubMed] [Google Scholar]
- 7. Udina E., Furey M., Busch S., Silver J., Gordon T., Fouad K. (2008) Electrical stimulation of intact peripheral sensory axons in rats promotes outgrowth of their central projections. Exp. Neurol. 210, 238–247 [DOI] [PubMed] [Google Scholar]
- 8. Costa L. M., Pereira J. E., Filipe V. M., Magalhães L. G., Couto P. A., Gonzalo-Orden J. M., Raimondo S., Geuna S., Maurício A. C., Nikulina E., Filbin M. T., Varejão A. S. (2013) Rolipram promotes functional recovery after contusive thoracic spinal cord injury in rats. Behav. Brain Res. 243, 66–73 [DOI] [PubMed] [Google Scholar]
- 9. Ben-Yaakov K., Dagan S. Y., Segal-Ruder Y., Shalem O., Vuppalanchi D., Willis D. E., Yudin D., Rishal I., Rother F., Bader M., Blesch A., Pilpel Y., Twiss J. L., Fainzilber M. (2012) Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 31, 1350–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Michaelevski I., Segal-Ruder Y., Rozenbaum M., Medzihradszky K. F., Shalem O., Coppola G., Horn-Saban S., Ben-Yaakov K., Dagan S. Y., Rishal I., Geschwind D. H., Pilpel Y., Burlingame A. L., Fainzilber M. (2010) Signaling to transcription networks in the neuronal retrograde injury response. Sci. Signal. 3, ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cai D., Shen Y., De Bellard M., Tang S., Filbin M. T. (1999) Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron 22, 89–101 [DOI] [PubMed] [Google Scholar]
- 12. Neumann S., Bradke F., Tessier-Lavigne M., Basbaum A. I. (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34, 885–893 [DOI] [PubMed] [Google Scholar]
- 13. Gao Y., Deng K., Hou J., Bryson J. B., Barco A., Nikulina E., Spencer T., Mellado W., Kandel E. R., Filbin M. T. (2004) Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44, 609–621 [DOI] [PubMed] [Google Scholar]
- 14. Ma T. C., Campana A., Lange P. S., Lee H. H., Banerjee K., Bryson J. B., Mahishi L., Alam S., Giger R. J., Barnes S., Morris S. M., Jr., Willis D. E., Twiss J. L., Filbin M. T., Ratan R. R. (2010) A large-scale chemical screen for regulators of the arginase 1 promoter identifies the soy isoflavone daidzeinas a clinically approved small molecule that can promote neuronal protection or regeneration via a cAMP-independent pathway. J. Neurosci. 30, 739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mukhopadhyay G., Doherty P., Walsh F. S., Crocker P. R., Filbin M. T. (1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13, 757–767 [DOI] [PubMed] [Google Scholar]
- 16. Gray M. J., Poljakovic M., Kepka-Lenhart D., Morris S. M., Jr. (2005) Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPβ. Gene 353, 98–106 [DOI] [PubMed] [Google Scholar]
- 17. Jessen B. A., Qin Q., Rice R. H. (2000) Functional AP1 and CRE response elements in the human keratinocyte transglutaminase promoter mediating Whn suppression. Gene 254, 77–85 [DOI] [PubMed] [Google Scholar]
- 18. Song H., Ming G., He Z., Lehmann M., McKerracher L., Tessier-Lavigne M., Poo M. (1998) Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science 281, 1515–1518 [DOI] [PubMed] [Google Scholar]
- 19. Andersen P. L., Webber C. A., Kimura K. A., Schreyer D. J. (2000) Cyclic AMP prevents an increase in GAP-43 but promotes neurite growth in cultured adult rat dorsal root ganglion neurons. Exp. Neurol. 166, 153–165 [DOI] [PubMed] [Google Scholar]
- 20. Li Y., Irwin N., Yin Y., Lanser M., Benowitz L. I. (2003) Axon regeneration in goldfish and rat retinal ganglion cells: differential responsiveness to carbohydrates and cAMP. J. Neurosci. 23, 7830–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barco A., Alarcon J. M., Kandel E. R. (2002) Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108, 689–703 [DOI] [PubMed] [Google Scholar]
- 22. Ahn S., Olive M., Aggarwal S., Krylov D., Ginty D. D., Vinson C. (1998) A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol. Cell. Biol. 18, 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cai D., Deng K., Mellado W., Lee J., Ratan R. R., Filbin M. T. (2002) Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 35, 711–719 [DOI] [PubMed] [Google Scholar]
- 24. Anouar Y., Lee H. W., Eiden L. E. (1999) Both inducible and constitutive activator protein-1-like transcription factors are used for transcriptional activation of the galanin gene by different first and second messenger pathways. Mol. Pharmacol. 56, 162–169 [DOI] [PubMed] [Google Scholar]
- 25. Boeshore K. L., Schreiber R. C., Vaccariello S. A., Sachs H. H., Salazar R., Lee J., Ratan R. R., Leahy P., Zigmond R. E. (2004) Novel changes in gene expression following axotomy of a sympathetic ganglion: a microarray analysis. J. Neurobiol. 59, 216–235 [DOI] [PubMed] [Google Scholar]
- 26. Seijffers R., Allchorne A. J., Woolf C. J. (2006) The transcription factor ATF-3 promotes neurite outgrowth. Mol. Cell Neurosci. 32, 143–154 [DOI] [PubMed] [Google Scholar]
- 27. Ylera B., Ertürk A., Hellal F., Nadrigny F., Hurtado A., Tahirovic S., Oudega M., Kirchhoff F., Bradke F. (2009) Chronically CNS-injured adult sensory neurons gain regenerative competence upon a lesion of their peripheral axon. Curr. Biol. 19, 930–936 [DOI] [PubMed] [Google Scholar]
- 28. Sheng M., McFadden G., Greenberg M. E. (1990) Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron 4, 571–582 [DOI] [PubMed] [Google Scholar]
- 29. Olive M., Krylov D., Echlin D. R., Gardner K., Taparowsky E., Vinson C. (1997) A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J. Biol. Chem. 272, 18586–18594 [DOI] [PubMed] [Google Scholar]
- 30. Benito E., Valor L. M., Jimenez-Minchan M., Huber W., Barco A. (2011) cAMP response element-binding protein is a primary hub of activity-driven neuronal gene expression. J. Neurosci. 31, 18237–18250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwede F., Maronde E., Genieser H., Jastorff B. (2000) Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther. 87, 199–226 [DOI] [PubMed] [Google Scholar]
- 32. Ma C. H., Omura T., Cobos E. J., Latrémolière A., Ghasemlou N., Brenner G. J., van Veen E., Barrett L., Sawada T., Gao F., Coppola G., Gertler F., Costigan M., Geschwind D., Woolf C. J. (2011) Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J. Clin. Invest. 121, 4332–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gumy L. F., Yeo G. S., Tung Y. C., Zivraj K. H., Willis D., Coppola G., Lam B. Y., Twiss J. L., Holt C. E., Fawcett J. W. (2011) Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17, 85–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Costigan M., Befort K., Karchewski L., Griffin R. S., D'Urso D., Allchorne A., Sitarski J., Mannion J. W., Pratt R. E., Woolf C. J. (2002) Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nikulina E., Tidwell J. L., Dai H. N., Bregman B. S., Filbin M. T. (2004) The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc. Natl. Acad. Sci. U.S.A. 101, 8786–8790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharda D. R., Yu S., Ray M., Squadrito M. L., De Palma M., Wynn T. A., Morris S. M., Jr., Hankey P. A. (2011) Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J. Immunol. 187, 2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu W., Chandrasekharan U. M., Bandyopadhyay S., Morris S. M., Jr., DiCorleto P. E., Kashyap V. S. (2010) Thrombin induces endothelial arginase through AP-1 activation. Am. J. Physiol. Cell Physiol. 298, C952–C960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Angel P., Karin M. (1991) The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072, 129–157 [DOI] [PubMed] [Google Scholar]
- 39. Smeal T., Binetruy B., Mercola D. A., Birrer M., Karin M. (1991) Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 354, 494–496 [DOI] [PubMed] [Google Scholar]
- 40. Eid A. H., Chotani M. A., Mitra S., Miller T. J., Flavahan N. A. (2008) Cyclic AMP acts through Rap1 and JNK signaling to increase expression of cutaneous smooth muscle α2C-adrenoceptors. Am. J. Physiol. Heart Circ. Physiol. 295, H266–H272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hernandez J. M., Floyd D. H., Weilbaecher K. N., Green P. L., Boris-Lawrie K. (2008) Multiple facets of junD gene expression are atypical among AP-1 family members. Oncogene 27, 4757–4767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kenney A. M., Kocsis J. D. (1998) Peripheral axotomy induces long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and junD in adult rat dorsal root ganglia in vivo. J. Neurosci. 18, 1318–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raivich G., Bohatschek M., Da Costa C., Iwata O., Galiano M., Hristova M., Nateri A. S., Makwana M., Riera-Sans L., Wolfer D. P., Lipp H. P., Aguzzi A., Wagner E. F., Behrens A. (2004) The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43, 57–67 [DOI] [PubMed] [Google Scholar]
- 44. Shin J. E., Cho Y., Beirowski B., Milbrandt J., Cavalli V., DiAntonio A. (2012) Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 74, 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cavalli V., Kujala P., Klumperman J., Goldstein L. S. (2005) Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 168, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hanz S., Perlson E., Willis D., Zheng J. Q., Massarwa R., Huerta J. J., Koltzenburg M., Kohler M., van-Minnen J., Twiss J. L., Fainzilber M. (2003) Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 40, 1095–1104 [DOI] [PubMed] [Google Scholar]
- 47. Arthur-Farraj P. J., Latouche M., Wilton D. K., Quintes S., Chabrol E., Banerjee A., Woodhoo A., Jenkins B., Rahman M., Turmaine M., Wicher G. K., Mitter R., Greensmith L., Behrens A., Raivich G., Mirsky R., Jessen K. R. (2012) c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75, 633–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niemi J. P., DeFrancesco-Lisowitz A., Roldán-Hernández L., Lindborg J. A., Mandell D., Zigmond R. E. (2013) A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J. Neurosci. 33, 16236–16248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fontana X., Hristova M., Da Costa C., Patodia S., Thei L., Makwana M., Spencer-Dene B., Latouche M., Mirsky R., Jessen K. R., Klein R., Raivich G., Behrens A. (2012) c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 198, 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kwon M. J., Kim J., Shin H., Jeong S. R., Kang Y. M., Choi J. Y., Hwang D. H., Kim B. G. (2013) Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J. Neurosci. 33, 15095–15108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yin Y., Cui Q., Gilbert H. Y., Yang Y., Yang Z., Berlinicke C., Li Z., Zaverucha-do-Valle C., He H., Petkova V., Zack D. J., Benowitz L. I. (2009) Oncomodulin links inflammation to optic nerve regeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 19587–19592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kadoya K., Tsukada S., Lu P., Coppola G., Geschwind D., Filbin M. T., Blesch A., Tuszynski M. H. (2009) Combined intrinsic and extrinsic neuronal mechanisms facilitate bridging axonal regeneration one year after spinal cord injury. Neuron 64, 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu P., Yang H., Jones L. L., Filbin M. T., Tuszynski M. H. (2004) Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J. Neurosci. 24, 6402–6409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pearse D. D., Pereira F. C., Marcillo A. E., Bates M. L., Berrocal Y. A., Filbin M. T., Bunge M. B. (2004) cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med. 10, 610–616 [DOI] [PubMed] [Google Scholar]