
Fig. 1. Loss-of-function IFNGR2 alleles associated with MSMD.
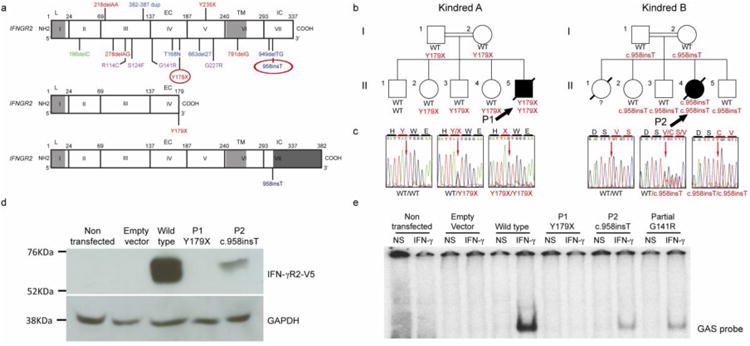
a) Schematic representation of the human IFNGR2 gene and corresponding protein with all previously described MSMD-associated mutations: in blue, mutations causing AR complete IFN-γR2 deficiency with detectable surface expression; in red, AR IFN-γR2 deficiency with no receptor expression; in purple, AR partial IFN-γR2 deficiency, and in green, AD IFN-γR2 deficiency. The different domains of the protein are indicated as L (leader peptide), EC (extracellular domain), TM (transmembrane domain) and IC (intracellular domain). Below this diagram, the consequences of the two mutations described here are shown: in the middle panel, a short IFN-γR2 caused by the creation of a premature stop codon (Y179X) and at the bottom, a longer predicted protein caused by the c.958insT variant. The extra portion of the protein due to the frame shift is indicated at dark gray at the end of the diagram.
b) Familial segregation of the two mutations (Y179X and c.958insT) found in IFNGR2. The index case (P1 and P2) of each pedigree is indicated with solid symbols and an arrow. Each kindred is designated by a capital letter (A-B), and each generation by a Roman numeral (I-II).
c) Electropherograms of a wild-type homozygous individual, a heterozygous carrier and a homozygous carrier are shown in this panel, in which the position of the mutant form is indicated with an arrow.
d) Overexpression of the WT and mutant alleles of IFNGR2 in IFN-γR2-deficient SV40-fibroblasts, revealing a lack of expression of Y179X and the residual expression of 958insT. The results presented are from one experiment representative of the three independent experiments carried out.
e) EMSA was carried out after IFN-γ stimulation of the same transfected cells as in D. A complete absence of DNA binding was observed for the nonsense mutation. The frameshift insertion showed a partial DNA binding, which was comparable to a previously reported partial deficient mutation. The results shown are representative of five independent experiments.