

Abstract
The nonheme diiron enzyme cyanobacterial aldehyde deformylating oxygenase, cADO, catalyzes the highly unusual deformylation of aliphatic aldehydes to alkanes and formate. We have determined crystal structures for the enzyme with a long-chain water-soluble aldehyde and medium-chain carboxylic acid bound to the active site. These structures delineate a hydrophobic channel that connects the solvent with the deeply buried active site and reveal a mode of substrate binding that is different from previously determined structures with long-chain fatty acids bound. The structures also identify a water channel leading to the active site that could facilitate the entry of protons required in the reaction. NMR studies examining 1-[13C]-octanal binding to cADO indicate that the enzyme binds the aldehyde form rather than the hydrated form. Lastly, the fortuitous cocrystallization of the metal-free form of the protein with aldehyde bound has revealed protein conformation changes that are involved in binding iron.
The biosynthesis of aliphatic hydrocarbons has recently garnered much interest as drop in replacements for conventional transportation fuels.1−6 In Nature, various organisms such as plants,7 insects,8 birds,9 and microbes5 are known to produce long-chain alkanes. These alkanes are derived from fatty acyl-CoA esters in a two-step biosynthetic pathway that involves a fatty-acyl-CoA reductase (FAR) that first converts the fatty acyl-CoA ester to the corresponding aldehyde.10−13 Next, an aldehyde decarbonylase (AD) cleaves the C1–C2 (aldehyde carbon−α-carbon) bond of the aldehyde to generate an alkane and, depending upon the organism, CO,14,15 CO2,8,16 or HCO2H.17,18
Although alkanes are very simple molecules, their production from aldehydes is a chemically difficult transformation,19 and the AD enzymes as a group remain very poorly understood.20 This is due in large part to the fact that most of these enzymes are integral membrane proteins that have proved hard to purify and express in recombinant form. Recently, however, a soluble version of AD was discovered in cyanobacteria,5 which is referred to as either cADO21 (cyanobacterial aldehyde deformylating oxygenase) or in earlier reports as cAD (cyanobacterial aldehyde decarbonylase).5,17,18,22,23 Although the physiological role of cADO in cyanobacteria remains unknown, the amenability of the enzyme has stimulated significant interest in understanding its mechanism and its use in engineering new pathways to generate alkane-based biofuels.
cADO is a nonheme di-iron protein that catalyzes the conversion of aldehydes to alkanes and formate in a reaction that requires O2 and reducing equivalents, as shown in Scheme 1.17,18,22,23
Scheme 1. Deformylation Reaction of Aldehydes Catalyzed by cADO.
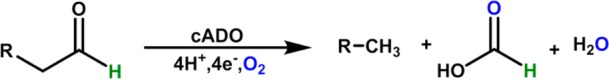
Labeling studies have shown that the aldehyde proton is retained in formate17,18 and that one of the oxygen atoms derives from molecular oxygen,22 whereas the proton in the product alkane derives from the solvent.17,18 Spectroscopic studies point to the initial formation of a diferric intermediate in the cADO catalyzed reaction.24 Addition of a further electron to this complex is proposed to lead to its breakdown and scission of the C1–C2 bond. A radical mechanism for C1–C2 bond cleavage is supported by the observed ring-opening of cyclopropyl aldehydes and oxiranyl aldehydes designed to act as “radical clocks” during deformylation by cADO.25,26
The crystal structure of the enzyme reveals it to possess an α-helical structure with the di-iron center housed in a ferritin-like four-helix bundle at the core of the protein.5,27 The iron atoms are coordinated by two histidine and four carboxylate residues, as is seen in other di-iron oxygenases, such as methane monoxygenase, ribonucleotide reductase, stearoyl-ACP-Δ9-desaturase, and toluene-4-monooxygenase.28−34 All cADO structures solved so far include an aliphatic fatty acid that is bound in a hydrophobic channel, with the carboxylate group providing a bridging ligand to the two iron atoms.27,35 The fatty acid presumably mimics the binding of the aldehyde substrate; however, it is unclear how the substrate gains access to the active site because the hydrophobic channel is completely enclosed by the protein.
Here, we address the question of how substrates and metal ions gain access to the active site of cADO. We have solved crystal structures of cADO with a long-chain, water-soluble aldehyde bound and a medium-chain carboxylic bound that reveal a new mode of substrate binding and protein conformational changes involved in binding iron. We have also undertaken NMR studies to establish whether the enzyme binds the aldehyde or its hydrated form, which predominates in solution.
Results and Discussion
NMR Studies of Substrate Binding to cADO
In aqueous solution, aldehydes exist in equilibrium with the hydrated (gem-diol) forms, which predominate. Mechanistic studies have assumed that it is the aldehyde form that is reactive toward deformylation. However, it is unclear which form initially binds to the enzyme, as it would be plausible for the enzyme to bind the hydrated aldehyde and for dehydration to occur at the active site; this could be catalyzed by the Lewis acidic diferric form of the enzyme. The gem-diol and carbonyl forms of the aldehyde carbon resonate at very different frequencies in the 13C NMR spectrum, making NMR an excellent technique to probe which form binds to the enzyme.
We examined the binding of 1-[13C]-octanal to cADO (Figure 1). These experiments were conducted on the diferric form of the enzyme in the absence of an auxiliary reducing system so that turnover could not occur. 1-[13C]-octanal exhibits peaks at 208 ppm due to the carbonyl form and at 92 ppm due to the diol form. Addition of increasing concentrations of cADO to a 1 mM solution of octanal in assay buffer resulted in a progressive reduction in intensity for the peak at 208 ppm. At 125 μM cADO, the 208 ppm peak could no longer be detected, whereas the peak at 92 ppm retained significant intensity. The loss of the 208 peak indicates that the enzyme binds the aldehyde form. The peak is lost due to broadening caused by chemical exchange with the enzyme-bound form on the NMR time scale; some broadening may also be due to paramagnetic relaxation associated with the iron center. Note that the unbound aldehyde remains in chemical equilibrium with the hydrated form but this equilibrium is slow on the NMR time scale so that the peak at 92 ppm is not broadened. Further increasing the enzyme concentration to be equimolar with the substrate resulted in almost complete loss of the 92 ppm peak as well. This is due to the re-establishment of the aldehyde-hydrate equilibrium in solution as more enzyme is added and available to bind the aldehyde form. Eventually all the substrate is converted to the aldehyde form and becomes bound to the enzyme.
Figure 1.

Binding of 1-[13C]-octanal to cADO monitored by 13C NMR. Octanal (1 mM) was titrated with increasing concentrations of cADO and the peaks correspond to the carbonyl (208 ppm) and hydrate forms (92 ppm) were monitored. At 125 μM cADO, the carbonyl peak is completely lost indicating that the enzyme selectively binds this form.
Synthesis and Activity of a Water-Soluble Long-Chain Aldehyde Substrate
Previously solved crystal structures of cADO all feature a long-chain fatty acid bound that was presumed to occupy the binding site of the aldehyde substrate.5,27,35 Recent studies have identified a mixture of palmitic, stearic, and oleic acids as being bound,35 which most likely are derived from E. coli during purification. Furthermore, it appears that the fatty acids are necessary for crystallization as even extensively lipid-depleted enzyme crystallizes with fatty acids bound.
We aimed to obtain a structure of the enzyme with an aldehyde bound, which would be more mechanistically informative. Preliminary screens with short-chain aldehydes in the crystallization buffer failed to obtained crystals of cADO and the cocrystallization of the enzyme with long-chain aldehydes, such as octadecanal, proved infeasible due to the very low solubility of these compounds. Therefore, we synthesized a water-soluble mimic of icosanal, 11-(2-(2-ethoxyethoxy)ethoxy)undecanal (1) that incorporates three oxygen atoms in the carbon chain (Scheme 2). This compound was straightforwardly synthesized by coupling silyl-protected 11-bromoundecanol to 2-(2-ethoxyethoxy)ethanol followed by deprotection and oxidation of the alcohol to the aldehyde, as described in the Supporting Information.
Scheme 2. Structures of 11-(2-(2-Ethoxyethoxy)ethoxy)undecanal, 1, and trans-2-Nonylcyclopropane-1-carboxylic Acid, 2.
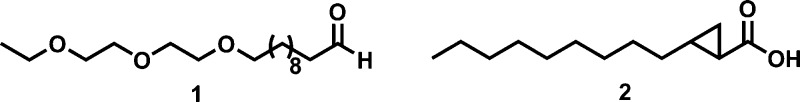
Compound 1 was soluble at millimolar concentrations under the buffer conditions typically used to assay cADO.18,25 We compared the activity of cADO in catalyzing the deformylation of compound 1 (500 μM) and octadecanal (500 μM + 4% DMSO to improve solubility) using previously described assay conditions with NADH and PMS as the auxiliary reducing system.25,26 cADO deformylated 1 to produce 1-(2-(2-ethoxyethoxy)ethoxy)decane at a rate 0.049 ± 0.002 min–1, a rate only ∼2-fold slower than the deformylation of octadecanal to heptadecane under similar conditions.
Crystallization of cADO with Substrate Analogs Bound
During the course of this investigation, we solved the structure of cADO with the following substrate analogs bound: stearate (i.e., enzyme as isolated from E. coli), PDB ID 4TW3; with compound 1 bound, PDB ID 4PGK; and with trans-2-nonylcyclopropane-1-carboxylate, 2, bound (Scheme 2), PDB ID 4PG1. We also solved the structure of a mutant enzyme, cADO-L194A, with 1 bound, PDB ID 4PGI. These structures are discussed individually in detail in the following. In each case, the protein crystallized in the space group, P43212, and the structures were solved by molecular replacement to resolutions ranging from 1.6 to 2.2 Å, with refinement statistics given in Table 1.
Table 1. Data Collection and Refinement Statistics for Crystal Structures.
| data set | cADO (stearate) | cADO-(1) | cADOL194A-(1) | cADO-(2) |
|---|---|---|---|---|
| PDB ID | 4TW3 | 4PGK | 4PGI | 4PG1 |
| space group | P43212 | P43212 | P43212 | P43212 |
| unit cell | a = b = 77.21; c = 116.57 | a = b = 77.34; c = 116.96 | a = b = 77.07; c = 117.26 | a = b = 77.12; c = 116.20 |
| α = β = γ = 90 | α = β = γ = 90 | α = β = γ = 90 | α = β = γ = 90 | |
| wavelength (Å) | 0.97872 | 0.97856 | 0.97872 | 1.0781 |
| resolution (Å) | 1.60 (1.60–1.63) | 2.17 (2.17–2.21) | 2.08 (2.08–2.12) | 1.70 (1.70–1.73) |
| Rsym (%) | 6.6 (71.9) | 8.5 (92.3) | 8.7 (57.0) | 7.3 (86.4) |
| {I/σI} | 20 (3) | 20 (5) | 20 (3) | 20 (3) |
| completeness (%) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 99.9 (100.0) |
| redundancy | 14.4 (14.3) | 14.4 (14.7) | 14.3 (14.6) | 14.2 (14.3) |
| Refinement Statistics | ||||
| resolution (Å) | 1.60 (38.61–1.60) | 2.17 (22.13–2.17) | 2.08 (22.48–2.08) | 1.70 (36.60–1.70) |
| R-factor (%) | 17.3 | 19.4 | 19.9 | 18.6 |
| Rfree (%) | 18.9 | 21.3 | 22.8 | 20.5 |
| protein atoms | 1815 | 1907 | 1886 | 1783 |
| water molecules | 306 | 227 | 148 | 204 |
| unique reflections rmsd | 47 171 | 19 412 | 21 862 | 39 033 |
| bonds | 0.010 | 0.010 | 0.010 | 0.010 |
| angles | 0.89 | 0.97 | 0.91 | 0.85 |
| MolProbity score | 0.95 | 1.19 | 1.26 | 0.64 |
| clash score | 1.91 | 2.37 | 4.47 | 0.28 |
Structure of cADO with Stearate Bound
To facilitate our analysis of the enzyme, we redetermined the structure of cADO with stearate bound at 1.60 Å resolution (PDB ID 4TW3). The enzyme comprises 8 α-helices (Figure 2A), of which helices 1 (blue), 2 (cyan), 4 (green), and 5 (yellow-green) make up the canonical antiparallel 4-helix bundle that provides the coordinating protein ligands to the di-iron center. Helix 3 (cyan–green), which is kinked in the middle, crosses over helix 1 before connecting through a short loop to helix 4. Helices 6 (yellow), 7 (orange), and 8 (red) form a subdomain in which helices 6 and 7 pack against helices 1, 3, and 4, whereas the shorter helix 8 packs at an angle of ∼45 across the interface of helices 6 and 7. Close inspection of the structure revealed a shallow hydrophobic cavity on the surface composed of side chains from helices 6, 7, and 8. This region of the surface is notably devoid of crystallographically resolved water molecules, as illustrated in Figure 2B. Although crystallographic symmetry places identical hydrophobic patches from neighboring protein molecules in close proximity, there are no intermolecular van der Waals contacts between hydrophobic residues of different proteins and the protein in solution is a monomer.
Figure 2.
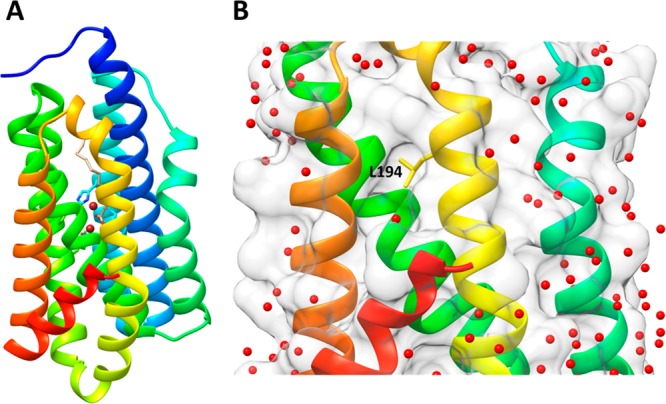
Structure of cADO with stearate bound (PDB ID 4TW3). (A) Ribbon diagram of cADO with helices colored from the N-terminus: 1 (blue), 2 (cyan), 3 (cyan-green), 4 (green), 5 (yellow–green), 6 (yellow), 7 (orange), and 8 (red); the iron atoms are brown and stearate is shown in gray. (B) The region containing the surface of helices 6, 7, and 8 shows a narrow hydrophobic cavity that is devoid of crystallographically resolved water molecules; L194 is shown in stick form at the center of the hydrophobic cavity.
At the center of the hydrophobic cavity is L194, which is located on helix 6 at the interface between the aqueous protein exterior and hydrophobic protein interior. Interestingly, this residue appeared highly mobile as evidenced by the poor electron density and high temperature factors of L194 side chain carbon atoms. This degree of side chain mobility is not observed for surrounding side chains in the hydrophobic patch or buried hydrophobic side chains elsewhere in the protein.
These observations suggested that L194 might serve as a gateway for substrate entry. Therefore, to assess the extent to which L194 might constrict access to the substrate-binding channel, we mutated this residue to alanine. However, this mutant, referred to as cADO-L194A, had kinetic properties very similar to the wild-type enzyme (data not shown) suggesting that L194 does not play a kinetically significant role in limiting substrate access to the active site.
Structure of cADO with 1 Bound
We were next able to solve the structures of cADO and cADO-L194A with 1 bound at 2.17 and 2.08 Å resolution, respectively (PDB IDs 4PGK and 4PGI, respectively). These structures were essentially identical, differing only in minor alterations to side chain conformations of residues in the immediate vicinity of the mutation. However, because the L194A mutant structure is at higher resolution, we have used this structure in our discussion of 1 binding to cADO in complex as described in the following.
The initial inspection of the electron density 2F0 – Fc map contoured at 1σ (Figure 3 and Supporting Information Figure S10) for the complex of cADO-L194A with 1 revealed several interesting features. First, where the substrate was expected to bind, there was T-shaped region of electron density extending from the metal center with one fork of the T occupying the cavity previously observed to bind stearic acid. The other fork exited the protein structure adjacent to L194 (A194 in the mutant cADO), supporting the view that this is the substrate entry point. The overall shape and the relatively poor definition of this electron density made it apparent that 1 must bind to the enzyme in two quite different, and somewhat mobile, conformations. A second area of ambiguity concerned the electron density of helix 5, which appeared partially unfolded between residues 154 and 165. Notably, this region encompasses E157 and H160, which supply ligands to Fe2. Lastly, the electron density for the metal ions in the structure refined to an occupancy of only ∼60%, which is considerably lower than that of any fatty acid-containing cADO structure.
Figure 3.
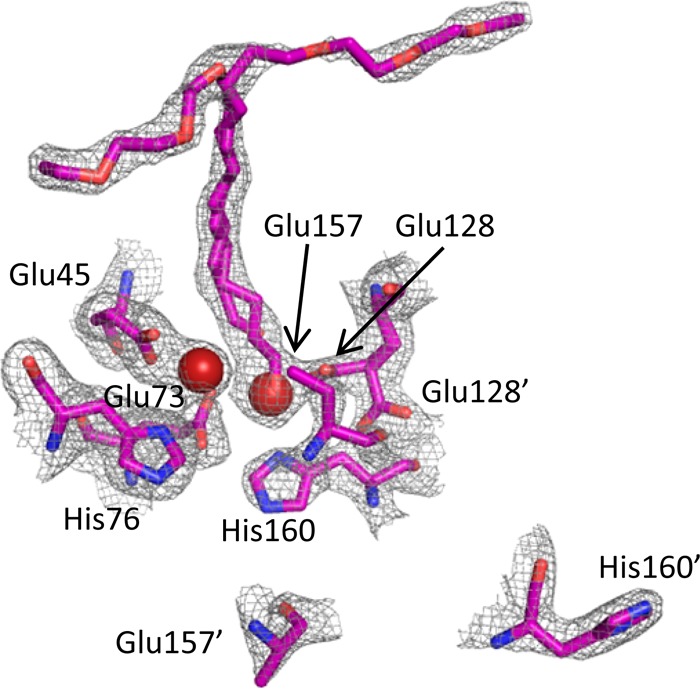
Electron density 2F0 – Fc map contoured at 1σ for cADO-L194A (PDB ID 4PGI) complexed with 1. The substrate can be modeled in two conformations to fit the T-shaped region of electron density, suggesting that at least two conformations of the substrate binding are possible. The electron density map also indicates that the region encompassing E157 and H160 also exists in two conformations: one in which these residues coordinate the metal; the other in which helix 5 is partially unfolded. The carboxylate group of E157 is disordered in both conformations and has therefore not been modeled in the structure.
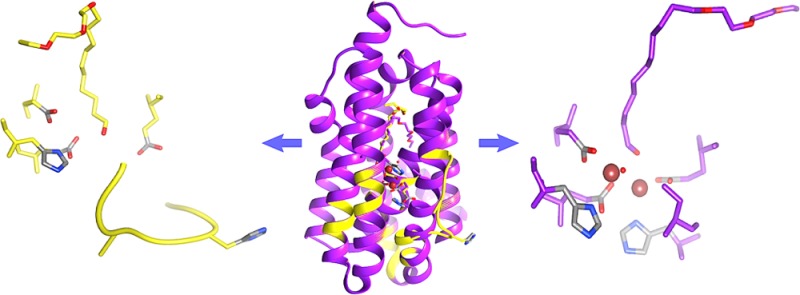
Taken together, these observations led to the conclusion that (at least) two structural forms of cADO were represented within the crystal. Although there is some ambiguity associated with interpreting electron density maps in which residues and ligands appear to adopt more than one conformation, we consider that, as discussed below, the most reasonable interpretation is that one form represents the metal-containing protein in which the substrate enters the protein at L194, whereas the other represents the metal-free protein in which helix 5 is partially unfolded and the substrate binds in a similar conformation to stearate. These conformations are shown in Figure 4. To support this conclusion, each conformation was fitted and refined independently. The resulting electron density maps produced density for the alternate conformation in both the Fo–Fc maps (contoured at 3σ) and in the 2Fo–Fc maps (contoured at 1σ). A figure showing the alternate positions of His160 and Glu157 overlaid with a map calculated from the refined B conformation of the helix is available in the Supporting Information (Figure S10). The low metal content of the crystals is not surprising because, as purified from E. coli, only 30–35% of the active sites contain metal, which we have previously characterized as being mainly iron and zinc,18 and no additional divalent metal ions were present in the crystallization buffer.
Figure 4.

Structure of metal-bound (purple) and metal-free (yellow) cADO-L194A in complex with 1 (PDB ID 4PGI). (A) Overlay of the two conformations of cADO; metal-bound form in purple; metal-free form in yellow, in which helix 5 is partially unfolded between the residues 154 and 165. (B) Details of the active site geometry for the metal-bound form. (C) Detail of the substrate entry channel showing conformational changes in L194 and F130 with the structure of the stearate-bound form of cADO superimposed for comparison. (D) Details of the active site geometry in the metal-free form in which the substrate intrudes into the metal-binding site. The carboxylate group of E157 is disordered in both conformations and has therefore not been modeled in the structure.
Structure of Metal-Bound cADO
In this structure, the coordinating ligands surrounding the two metal atoms adopt a similar geometry to that seen in the structures of the cADO/stearate complex and other dinuclear metalloenzymes that possess the canonical ‘4-carboxylate, 2-histidine’ binding motif. As shown in Figure 4B, for Fe1, the carboxylate of E45 from helix 1 provides a bidentate ligand, whereas E73 and H76 from helix 2 provide monodentate ligands. Similarly, for Fe2, E128 from helix 4 provides a bidentate ligand, and H160 from helix 5 provides a monodentate ligand. The carboxylate of E157, which should provide the final protein ligand, was not resolved in the electron density maps. This suggests that it is conformationally labile and because of this we do not include it in the model. The fifth coordination site to Fe1 is occupied by water, whereas it is the aldehyde oxygen that supplies the fifth ligand to Fe2, with an Fe–O distance of 2.5 Å.
In the metal-bound form of cADO, there is insufficient space available within the hydrophobic cavity for compound 1 to bind in the same conformation as stearate. We therefore modeled the aldehyde chain to fit the electron density that exits the protein between helices 6 and 7 (Figure 4B). To accommodate 1, the side chain of L194 is displaced by ∼2 Å, (measured with respect to Cγ) compared to the structure of the stearate-binding enzyme. V197 on the consecutive helical turn accommodates the rotomeric shift of L194. The phenyl ring of F130 undergoes a χ2 rotation of 50 degrees so the face of the ring now lines the channel that terminates at the protein exterior as illustrated in Figure 4C. Attempts to crystallize the enzyme in the fully metalated form by including Fe2+ or Zn2+ ions in the crystallization buffer together with compound 1 were unsuccessful.
Structure of Metal-Free cADO
The cocrystallization of the metal-free form of the enzyme was unexpected and has provided insight into the pathway by which metals gain access to the active site of cADO. Several attempts to crystallize apo-cADO in the presence of 1 failed to yield crystals, presumably due to the conformational lability of the protein. Therefore, we conclude that the fortuitous presence of an approximately 50:50 mixture of apo- and metallo-enzyme, as isolated from E. coli, allowed the apoenzyme to cocrystallize with the metallo-enzyme and thereby be trapped for crystallographic analysis.
In the apo enzyme, residues 154–165, which in the metalated enzyme are contained within helix 5 directly adjacent to the diiron center, unfold to form a solvent-exposed loop (Figure 4D). This region of the protein includes E157 and H160, which coordinate Fe2. A less dramatic change in the conformation of the other ligand to Fe2, E128, is also seen; small changes in the positions of Y52 and Q123 also occur, which adopt alternate hydrogen bonding arrangements within the protein. In contrast, the positions of the ligands to Fe1 are very little changed; interactions with surrounding residues appear to maintain their metal-binding conformations in the absence of metal.
The conformation of helix 5 does not appear to be influenced by crystal packing effects in either conformation. None of the residues in helix 5 make contacts with other symmetry related protein molecules within the crystal lattice. The side-chain of the closest residue Tyr163 is makes the closest approach, 4.4 Å. Helical residues N-terminal to Y163 are completely solvent exposed whereas residues C-terminal to Y163 are greater than 5.3 Å from any symmetry related molecule.
Helix 5 is one of the two more solvent exposed helices housing the di-iron center and its partial unfolding suggests a mechanism by which metal ions can enter the active site. The metal-free structures of other structurally related di-iron enzymes have been described previously, notably Class I ribonucleotide reductase R2 subunit (RNR R2)36 and methane monooxygenase hydroxylase subunit (MMOH).37 In both these enzymes, the tertiary structure remains intact and the metal-chelating ligands adopt conformations that are close to their metal-bound conformations. In contrast, in cADO, one metal binding site remains intact, but the localized unfolding of helix 5 between residues 154 and 165 completely dissembles the second metal-binding site. Interestingly, the corresponding helices in RNR R2 and MMOH are similarly the more solvent exposed of the helices housing the di-iron center.37,38 cADO is smaller than either RNR R2 or MMOH and so may require metal coordination to stabilize the folded structure, whereas for RNR and MMO more extensive tertiary interactions with other helices in the protein presumably stabilize the metal-free structure. This suggests that, although RNR R2 and MMOH remain folded in the absence of metals, the same localized unfolding may provide a mechanism for metalation in these and other enzymes of this structural class.
In the metal-free structure, the electron density due to the substrate, 1, extends into the region that would be occupied by Fe2 (Figures 4D and 3). It is possible that the intrusion of the substrate into the metal-binding site may have helped stabilize the open conformation of helix 5, allowing this conformation to be trapped in the crystal. The additional space available to the substrate in the metal-free structure, allows the tail end of 1 to be accommodated completely within the protein in a conformation very similar to that seen for cADO with stearate bound.
Although we chose to model the substrate in two conformations associated with metal-bound and metal-free forms of cADO, we do not rule out the possibility that the substrate may bind in other orientations. The conformational mobility of the long aliphatic chain, combined with the lack of any hydrogen-bonding residues in the substrate-binding channel that might serve as “anchor points” for the substrate, suggest that many energetically similar configurations may be possible. In particular, it is possible to model the substrate in a nonproductive binding mode in which the aliphatic chain stretches across the top of the T-shaped channel, with the aldehyde carbonyl extended into the solvent rather than binding to the metal.
Structure of cADO with trans-2-Nonylcyclopropane-1-Carboxylate (2) Bound
Because the electron density map associated with 1 bound to cADO left some ambiguity as to its mode of binding, we also attempted to cocrystallize cADO with other aldehydes. We were able to obtain highly diffracting crystals in the presence of trans-2-nonylcyclopropane-1-carbaldehyde, which allowed the structure to be solved at 1.7 Å resolution, PDB ID 4PG1.
During refinement it became apparent that the aldehyde carbon had oxidized to the carboxylic acid, to give compound 2 (Scheme 2), a finding confirmed by chromatographic analysis of the mother liquor. This is not especially surprising given that aldehydes are sensitive to oxidation by oxygen in the presence of metal ions, and no attempt was made to rigorously exclude oxygen from the crystallization trays. Nevertheless, the structure is instructive because it reveals two molecules of 2 bound to the protein (Figure 5), which clearly delineate the entry point for the substrate. The high quality of the electron density map allowed the cyclopropyl ring to be clearly resolved, removing any ambiguity about the identity and orientation of the ligands. Interestingly, although the aldehyde of 2 was synthesized in racemic form, it is the 1R,2R enantiomer of 2 that cocrystallizes with the enzyme in both positions.
Figure 5.

Structure of cADO in complex with cyclopropyl carboxylic acid, 2 (PDB ID 4PG1). (A) The structure of cADO shows two molecules of 2 bound to the enzyme; one molecule directly coordinates to the di-iron center by the carboxyl group, other molecule assumes a “reverse” orientation where the carboxyl group extends into the solvent. (B) Overlay of structures of cADO with stearate bound (tan) and 2 bound (orange) illustrating large scale movement of helix 8 (for clarity the stearate molecule has been omitted from the figure).
As shown in Figure 5, one molecule of 2 is coordinated to the metal center through the carboxylate group, the other molecule binds to the protein in the substrate entry channel in a “reverse” orientation and extends into the solvent, with the carboxylate group making a hydrogen bond with the guanidine group of R191 and last three carbon atoms of 2 contacting L194. As seen with 1, access to the protein interior is provided through a displacement of the L194 side chain to accommodate the aliphatic tail of 2. The binding of 2 also results in a large-scale movement of helix 8 (Figure 5B), which pivots about its N-terminus resulting in a displacement of the C-terminus of the helix of ∼4.3 Å with respect to the stearate-bound structure. This movement is accompanied by the formation of additional hydrophobic contacts between 2 and residues in helix 8.
The structure of cADO with 2 bound also provided insight into another aspect of the reaction—the pathway by which the proton that is introduced into the product alkane is shuttled between the solvent and the interior of the protein. A chain of water molecules is seen in the structure that enters the protein between helices 1 and 3 close to the metal center (Figure 6). These waters are stabilized by hydrogen bonds to Q108, E47, N51, and Q123 and could potentially form a proton relay to the solvent. The three closest potential proton donors, a water molecule, the side-chain of Q123 and an iron-bound water/hydroxide, are each about 4.8 Å from the α-carbon of 2. Although this is too far for them to act as proton donors, one may imagine that in the catalytically active enzyme relatively small movements in the active site may bring any of these donors within range. Furthermore, recently published solvent isotope effect measurements have provided evidence that the proton is most likely transferred from the iron-bound water molecule.39
Figure 6.
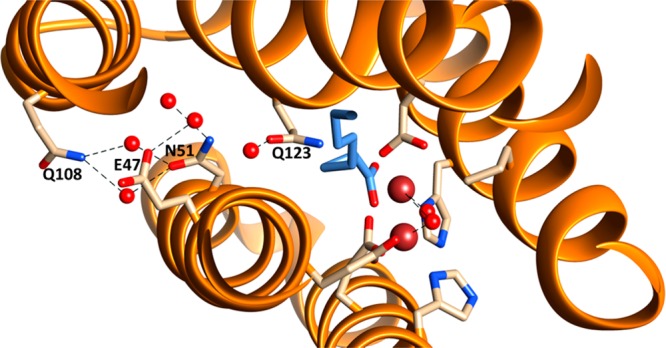
Water molecules within the structure of cADO with 2 bound (PDB ID 4PG1). A chain of water molecules were resolved, which enter the enzyme in between helices 1 and 3 and are hydrogen bonded to Q108, E47, N51, and Q123. The channel potentially provides a conduit to supply the proton necessary for alkane formation.
The structures of cADO with 1 and 2 bound have illuminated the likely entry point for the substrate, which was not apparent from previous structures with stearate or palmitate bound. The structures also demonstrate that for long-chain aldehydes several modes of binding are possible and that for short-chain aldehydes more than one molecule can bind at a time. These observations suggest that nonproductive modes of substrate binding are possible and may contribute to the low rates of turnover measured for cADO. For example, substrate inhibition might occur with short aldehydes if a second substrate molecule is bound in the channel preventing product release. Likewise, if a long-chain aldehyde bound across the top of the ‘T’-shaped substrate channel, instead of in the channel leading to the metal center, this would effectively inhibit the enzyme complex. This suggests that protein engineering could be used to occlude the “dead-end” branch of the substrate-binding channel and possibly improve catalytic efficiency.
A final intriguing observation is that the entry point for the aldehyde is in the center of a hydrophobic depression on the enzyme’s surface that is flanked by several positively charged residues. This suggests that cADO may interact with either another protein for the delivery of these very hydrophobic molecules, or possibly the cell membrane. No interaction partners for cADO have been identified and the enzyme does not appear to be membrane associated; however, it seems unlikely that cytosolic long-chain aldehydes would be the physiological substrates for cADO because of their extremely low solubilities. Indeed, it is unclear why some species of cyanobacteria biosynthesize alkanes, and it remains possible that the physiological substrates for cADO are other hydrophobic molecules.
Conclusion
The deformylation reaction catalyzed by cADO is chemically complex and unlike that of other nonheme iron oxygenases in that O2 is completely reduced to water during the catalytic cycle. This reaction requires that, in addition to the aldehyde substrate and O2, further electrons and a proton be delivered to the active site during the reaction.17,18,22,23 cADO deformylates aldehydes extremely slowly,17,18,21,23,35 with kcat for even the fastest substrates not exceeding ∼1 min–1. We had considered that the improved solubility of 1 and its product might result in faster turnover; however, the sluggish rate at which 1 is deformylated indicates that the solubility of the substrate is not a limiting factor in the rate of turnover.
A key step in understanding both the mechanism of the enzyme and its possible function in vivo is to understand how the substrates gain access to the active site. The NMR studies presented here indicate that the enzyme selectively binds the aldehyde form from solution, rather than the hydrate. The selectivity may arise because the hydrated form is too hydrophilic to enter the hydrophobic substrate-binding channel. The crystal structures of cADO with substrate analogs bound have identified local protein conformational changes that allow iron to enter the active site of cADO and a pathway by which the proton required in the reaction may enter the active site. Notably, the substrate-binding channel identified in these structures is different from that inferred for previous structures containing fatty acids. These observations should facilitate protein engineering to improve the efficiency of the enzyme.
Methods
Materials
Phenazine methosulfate (PMS) and ferrous ammonium sulfate were from Sigma-Aldrich; NADH was obtained from Acros Organics; 1-[13C]-octanal was obtained from Cambridge Isotope Laboratories.
Synthesis of 11-(2-(2-Ethoxyethoxy)ethoxy)undecanal, 1, and trans-2-Nonylcyclopropane-1-Carbaldehyde (Aldehyde of Compound 2)
Details of the synthesis and characterization of compound 1 and the aldehyde of compound 2 are described in the Supporting Information.
Enzyme Purification and Reconstitution
The purification of recombinant P marinus cADO from Escherichia coli was performed as described previously.18,23 Activity assays of cADO with compound 1 were performed as described previously.25,26
Preparation of Samples for NMR
NMR samples of the diferric form of cADO were prepared in 50 mM potassium phosphate buffer (pH 7.2) and 20 mM CHAPS (500 μL volume). 1-[13C]-octanal was dissolved in d6-DMSO for a stock concentration of 100 mM and added to the proteins solution to give the desired final concentration. Spectra were acquired using a Varian VNMRS 700 MHz spectrometer with a 5 mm PFG AutoX broadband probe at 4 °C using 2000 scans.
Crystallization and Structure Determination
For crystallization of cADO or cADOL194A with 1, the compound was added directly to a solution of 396 μM cADO in 50 mM HEPES, pH 7.4 for a final concentration of 10 mM 1. Samples were incubated for 16 h at 4 °C immediately prior to crystallization screening. The protein samples used for these experiments did not undergo removal/replacement of endogenous metals and cADO retained the metal content as isolated from E. coli. The crystals of cADO and cADOL194A with 1, from which the highest resolution data were collected and were crystallized against 20% PEG 3350, 0.3 M NaCl and 20% PEG 3350, 0.2 M sodium tartrate, respectively, with drops containing equal volumes of protein sample and precipitant.
For crystallization of cADO with 2, the sample was prepared by adding 2.0 equiv of Fe(II) in the form of ferrous ammonium sulfate to 400 μM apo cADO in 50 mM HEPES buffer, pH 7.4 with 2% glycerol under a microaerobic environment. Protein solutions were oxidized for 1 h under atmospheric conditions at 4 °C where they acquired a slight yellow color and metal oxidation was verified by UV absorbance at 360 nm, which is characteristic of a iron(III)-oxo species. Stock solutions of 100 mM of the aldehyde of 2 in DMSO were prepared and added to diferric cADO to give final concentrations of 2 mM or 4 mM of the aldehyde of 2. Diferric cADO solutions containing of the aldehyde of 2 were incubated for 16 h at 4 °C immediately prior to crystallization. The crystals of cADO with 2 mM and 4 mM 2 from which the highest resolution data were collected and were crystallized in 35% PEG 400, 0.1 M Tris pH 8.5, and 30% PEG 550MME, 0.1 M Tris, 0.1 M ammonium acetate pH 8.5, respectively.
Crystals were cryoprotected in liquid N2 in their mother liquor for data collection. Data were collected at the Advanced Photon Source (APS) (LS-CAT Beamlines 21-D, 21-F, and 21-G) at Argonne National Laboratory on a MarCCD (Mar USA, Evanston, IL) at wavelengths of 1.0781, 0.97872, and 0.97857 Å, respectively, at −180 °C. Data were processed and scaled with HKL2000.40 All proteins for this study crystallized in space group P43212 with unit cell parameters as listed in Supporting Information Table S1. All proteins contain a monomer in the asymmetric unit.
Phases were initially determined by molecular replacement with a native structure of cADO having all ligands and water molecules removed (PDB ID: 2OC5) using Phaser41 in the CCP4i program suite.42 Smiles for the compounds were created using Molinspiration WebME Editor 3.81 (Dr. Peter Ertl, http://www.molinspiration/docu/webme/index.html) while restraints were produced using mogul=qm option in grade.43,44 Protein models were refined by rigid body refinement and restrained refinement using Buster.44 Side chains were visualized and adjusted using Coot45 with 2F0–Fc and F0–Fc electron density maps from Buster. Structures were validated using Molprobity.46 All residues are in allowed regions of the Ramachandran plot. Data refinement statistics are given in Table 1.
Acknowledgments
This research was supported in part by grants from the National Science Foundation, CHE 1152055, CBET 1336636, the National Institutes of Health, GM 093088, and the European Union, FP-7 256808. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). We thank Dr. Jennifer Meagher for her assistance with crystallization and Dr. David Smith for his help with data collection at LS-CAT.
Supporting Information Available
The procedures for the synthesis and characterization of 11-(2-(2-ethoxyethoxy)ethoxy)undecanal (1) and trans-2-nonylcyclopropane-1-carbaldehyde (2). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates of the protein structures discussed in this manuscript have been deposited in the Protein Data Bank with IDs 4TW3, 4PGK, 4PGI, and 4PG1.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Connor M. R.; Atsumi S. (2010) Synthetic biology guides biofuel production. J. Biomed. Biotechnol. 541698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasling J. D. (2010) Manufacturing molecules through metabolic engineering. Science 330, 1355–1358. [DOI] [PubMed] [Google Scholar]
- Steen E. J.; Kang Y. S.; Bokinsky G.; Hu Z. H.; Schirmer A.; McClure A.; del Cardayre S. B.; Keasling J. D. (2010) Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463, 559–562. [DOI] [PubMed] [Google Scholar]
- Clarke N. D. (2010) Protein engineering for bioenergy and biomass-based chemicals. Curr. Opin. Struct. Biol. 20, 527–532. [DOI] [PubMed] [Google Scholar]
- Schirmer A.; Rude M. A.; Li X. Z.; Popova E.; del Cardayre S. B. (2010) Microbial biosynthesis of alkanes. Science 329, 559–562. [DOI] [PubMed] [Google Scholar]
- Ghim C. M.; Kim T.; Mitchell R. J.; Lee S. K. (2010) Synthetic biology for biofuels: Building designer microbes from the scratch. Biotechnol. Bioprocess. Eng. 15, 11–21. [Google Scholar]
- Aarts M. G. M.; Keijzer C. J.; Stiekema W. J.; Pereira A. (1995) Molecular characterization of the CER1 gene of arabidopsis involved in epicuticular wax biosynthesis and pollen fertility. Plant Cell 7, 2115–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed J. R.; Vanderwel D.; Choi S. W.; Pomonis J. G.; Reitz R. C.; Blomquist G. J. (1994) Unusual mechanism of hydrocarbon formation in the housefly cytochrome-P450 converts aldehyde to the sex-pheromone component (Z)-9-tricosene and CO2. Proc. Natl. Acad. Sci. U.S.A. 91, 10000–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheesbrough T. M.; Kolattukudy P. E. (1988) Microsomal preparation from an animal tissue catalyzes release of carbon-monoxide from a fatty aldehyde to generate an alkane. J. Biol. Chem. 263, 2738–2743. [PubMed] [Google Scholar]
- Bourdenx B.; Bernard A.; Domergue F.; Pascal S.; Leger A.; Roby D.; Pervent M.; Vile D.; Haslam R. P.; Napier J. A.; Lessire R.; Joubes J. (2011) Overexpression of arabidopsis ECERIFERUM1 promotes wax very-long-chain alkane biosynthesis and influences plant response to biotic and abiotic stresses. Plant Physiol. 156, 29–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland O.; Zheng H. Q.; Hepworth S. R.; Lam P.; Jetter R.; Kunst L. (2006) CER4 encodes an alcohol-forming fatty acyl-coenzyme A reductase involved in cuticular wax production in Arabidopsis. Plant Physiol. 142, 866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Kolattukudy P. E. (1995) Solubilization and purification of aldehyde-generating fatty Acyl-Coa reductase from green-alga Botryococcus braunii. FEBS Lett. 370, 15–18. [DOI] [PubMed] [Google Scholar]
- Lin F.; Das D.; Lin X. N.; Marsh E. N. G. (2013) Aldehyde-forming fatty acyl-CoA reductase from cyanobacteria: Expression, purification, and characterization of the recombinant enzyme. FEBS J. 280, 4773–4781. [DOI] [PubMed] [Google Scholar]
- Cheesbrough T. M.; Kolattukudy P. E. (1984) Alkane biosynthesis by decarbonylation of aldehydes catalyzed by a particulate preparation from Pisum sativum. Proc. Natl. Acad. Sci. U.S.A. 81, 6613–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis M.; Kolattukudy P. E. (1992) A Cobalt-porphyrin enzyme converts a fatty aldehyde to a hydrocarbon and Co. Proc. Natl. Acad. Sci. U.S.A. 89, 5306–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qui Y.; Tittiger C.; Wicker-Thomas C.; Le Goff G.; Young S.; Wajnberg E.; Fricaux T.; Taquet N.; Blomquist G. J.; Feyereisen R. (2012) An insect-specific P450 oxidative decarbonylase for cuticular hydrocarbon biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 109, 14858–14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warui D. M.; Li N.; Norgaard H.; Krebs C.; Bollinger J. M.; Booker S. J. (2011) Detection of formate, rather than carbon monoxide, as the stoichiometric coproduct in conversion of fatty aldehydes to alkanes by a cyanobacterial aldehyde decarbonylase. J. Am. Chem. Soc. 133, 3316–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D.; Eser B. E.; Han J.; Sciore A.; Marsh E. N. G. (2011) Oxygen-independent decarbonylation of aldehydes by cyano-bacterial aldehyde decarbonylase: A new reaction of di-iron enzymes. Angew. Chem., Int. Ed. 50, 7148–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buist P. H. (2007) Exotic biomodification of fatty acids. Nat. Prod. Rep. 24, 1110–1127. [DOI] [PubMed] [Google Scholar]
- Marsh E. N. G.; Waugh M. W. (2013) Aldehyde decarbonylases: Enigmatic enzymes of hydrocarbon biosynthesis. ACS Catal. 3, 2515–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Chang W.-C.; Warui D. M.; Booker S. J.; Krebs C.; Bollinger J. M. (2012) Evidence for only oxygenative cleavage of aldehydes to alk(a/e)nes and formate by cyanobacterial “aldehyde decarbonylase”. Biochemistry 51, 7908–7916. [DOI] [PubMed] [Google Scholar]
- Li N.; Norgaard H.; Warui D. M.; Booker S. J.; Krebs C.; Bollinger J. M. (2011) Conversion of fatty aldehydes to alka(e)nes and foramte by a cyanobacterial aldehyde decarbonylase: Crypric redox by an unusual dimetal oxygenase. J. Am. Chem. Soc. 133, 7148–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser B. E.; Das D.; Han J.; Jones P. R.; Marsh E. N. G. (2011) Oxygen-independent alkane formation by non-heme iron-dependent cyanobacterial aldehyde decarbonylase: Investigation of kinetics and requirement for an external electron donor. Biochemistry 50, 10743–10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandelia M. E.; Li N.; Norgaard H.; Warui D. M.; Rajakovich L. J.; Chang W. C.; Booker S. J.; Krebs C.; Bollinger J. M. (2013) Substrate-triggered addition of dioxygen to the diferrous cofactor of aldehyde-deformylating oxygenase to form a diferric-peroxide intermediate. J. Am. Chem. Soc. 135, 15801–15812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul B.; Das D.; Ellington B.; Marsh E. N. G. (2013) Probing the mechanism of cyanobacterial aldehyde decarbonylase using a cyclopropyl aldehyde. J. Am. Chem. Soc. 135, 5234–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D.; Ellington B.; Paul B.; Marsh E. N. G. (2014) Mechanistic insights from reaction of α-oxiranyl-aldehydes with cyanobacterial aldehyde deformylating oxygenase. ACS Chem. Biol. 9, 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unpublished. Structure solved by Joint Center of Structural Genomics (protein database entry PDB|2OC5|A).
- Baik M. H.; Newcomb M.; Friesner R. A.; Lippard S. J. (2003) Mechanistic studies on the hydroxylation of methane by methane monooxygenase. Chem. Rev. 103, 2385–2419. [DOI] [PubMed] [Google Scholar]
- Sjoberg B. M. (1997) Ribonucleotide reductases—A group of enzymes with different metallosites and a similar reaction mechanism. In Metal Sites in Proteins and Models, pp 139–173, Springer, New York. [Google Scholar]
- Stubbe J.; Nocera D. G.; Yee C. S.; Chang M. C. Y. (2003) Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer?. Chem. Rev. 103, 2167–2201. [DOI] [PubMed] [Google Scholar]
- Theil E. C.; Goss D. J. (2009) Living with iron (and oxygen): Questions and answers about iron homeostasis. Chem. Rev. 109, 4568–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallar B. J.; Lipscomb J. D. (1996) Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem. Rev. 96, 2625–2657. [DOI] [PubMed] [Google Scholar]
- Feig A. L.; Lippard S. J. (1994) Reactions of non-heme iron(II) centers with dioxygen in biology and chemistry. Chem. Rev. 94, 759–805. [Google Scholar]
- Lange S. J.; Que L. (1998) Oxygen activating nonheme iron enzymes. Curr. Opin. Chem. Biol. 2, 159–172. [DOI] [PubMed] [Google Scholar]
- Khara B.; Menon N.; Levy C.; Mansell D.; Das D.; Marsh E. N. G.; Leys D.; Scrutton N. S. (2013) Production of propane and other short-chain alkanes by structure-based engineering of ligand specificity in aldehyde-deformylating oxygenase. ChemBioChem 14, 1204–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberg A.; Nordlund P.; Eklund H. (1993) Unusual clustering of carboxyl side chains in the core of iron-free ribonucleotide reductase. Nature 361, 276–278. [DOI] [PubMed] [Google Scholar]
- Sazinsky M. H.; Merkx M.; Cadieux E.; Tang S.; Lippard S. J. (2004) Preparation and X-ray structures of metal-free dicobalt and dimanganese forms of soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath). Biochemistry 43, 16263–16276. [DOI] [PubMed] [Google Scholar]
- Nordlund P.; Eklund H. (1993) Structure and function of the Escherichia coli ribonucleotide reductase protein R2. J. Mol. Biol. 232, 123–164. [DOI] [PubMed] [Google Scholar]
- Waugh M. W.; Marsh E. N. G. (2014) Solvent isotope effects on alkane formation by cyanobacterial aldehyde deformylating oxygenase and their mechanistic implications. Biochemistry 53, 5537–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology, Macromolecular Crystallography, Part A (Carter C. W. Jr., and Sweet R. M., Eds.), pp 307–326, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potterton E.; Briggs P.; Turkenburg M.; Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; van Aalten D. M. F. (2004) PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O., Vonrhein C., and Womack T. (2011) BUSTER 2.10.0 ed., Global Phasing Ltd., Cambridge, U.K. [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; et al. (2010) MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.