

Background: The enzymatic activities of mammalian P4-ATPases are incompletely characterized.
Results: ATP11A and ATP11C catalyze flipping of NBD-PS and NBD-PE, whereas ATP8B1 preferentially catalyzes flipping of NBD-PC. Furthermore, some PFIC1 mutants of ATP8B1 failed to flip PC.
Conclusion: ATP11A/ATP11C and ATP8B1/ATP8B2 preferentially translocate aminophospholipids and PC, respectively.
Significance: This is the first evidence showing that the PC-flipping activity of ATP8B1 is associated with the episode of PFIC1.
Keywords: ATPase, Lipid Bilayer, Lipid Transport, Phospholipid, Plasma Membrane
Abstract
Type IV P-type ATPases (P4-ATPases) are believed to translocate aminophospholipids from the exoplasmic to the cytoplasmic leaflets of cellular membranes. The yeast P4-ATPases, Drs2p and Dnf1p/Dnf2p, flip nitrobenzoxadiazole-labeled phosphatidylserine at the Golgi complex and nitrobenzoxadiazole-labeled phosphatidylcholine (PC) at the plasma membrane, respectively. However, the flippase activities and substrate specificities of mammalian P4-ATPases remain incompletely characterized. In this study, we established an assay for phospholipid flippase activities of plasma membrane-localized P4-ATPases using human cell lines stably expressing ATP8B1, ATP8B2, ATP11A, and ATP11C. We found that ATP11A and ATP11C have flippase activities toward phosphatidylserine and phosphatidylethanolamine but not PC or sphingomyelin. By contrast, ATPase-deficient mutants of ATP11A and ATP11C did not exhibit any flippase activity, indicating that these enzymes catalyze flipping in an ATPase-dependent manner. Furthermore, ATP8B1 and ATP8B2 exhibited preferential flippase activities toward PC. Some ATP8B1 mutants found in patients of progressive familial intrahepatic cholestasis type 1 (PFIC1), a severe liver disease caused by impaired bile flow, failed to translocate PC despite their delivery to the plasma membrane. Moreover, incorporation of PC mediated by ATP8B1 can be reversed by simultaneous expression of ABCB4, a PC floppase mutated in PFIC3 patients. Our findings elucidate the flippase activities and substrate specificities of plasma membrane-localized human P4-ATPases and suggest that phenotypes of some PFIC1 patients result from impairment of the PC flippase activity of ATP8B1.
Introduction
The lipid bilayers of cellular membranes exhibit asymmetric lipid distributions. In mammalian cells, the aminophospholipids phosphatidylserine (PS)2 and phosphatidylethanolamine (PE) are abundant in the cytoplasmic leaflet, whereas phosphatidylcholine (PC) and sphingomyelin (SM) are enriched in the exoplasmic leaflet. Type IV P-type ATPases (P4-ATPases), also called aminophospholipid translocases, are essential for generation and maintenance of phospholipid asymmetry in lipid bilayers (1, 2). Regulated exposure of PS in the exoplasmic leaflet is critical for some biological processes such as apoptotic cell death, platelet coagulation, and fusion of muscle cells (3–5), indirectly indicating the importance of lipid asymmetry at steady state. In yeasts, P4-ATPases form heteromeric complexes with members of the CDC50 protein family (6, 7). We and others recently showed that most of the 14 mammalian P4-ATPases associate with CDC50 to exit the endoplasmic reticulum (ER) and reach their appropriate subcellular destinations (8–12). The yeast P4-ATPases, Drs2p and Dnf1p/Dnf2p, flip NBD-labeled PS (NBD-PS) at the Golgi complex and NBD-PC at the plasma membrane, respectively (13, 14). Several key residues have been proposed to determine the phospholipid specificities of Drs2p and Dnf1p (15, 16). By contrast, although an understanding of the enzymatic properties of mammalian P4-ATPases is essential to determining their physiological functions, the flippase activities and substrate specificities of the mammalian P4-ATPases have not been extensively characterized.
Patients with progressive familial intrahepatic cholestasis (PFIC) exhibit hepatic and systemic accumulation of bile salts, jaundice, and pruritus and usually develop end stage liver disease. PFIC type 1 (PFIC1) and a less severe condition, benign recurrent intrahepatic cholestasis 1 (BRIC1), result from defects in the P4-ATPase ATP8B1 (17, 18), whereas PFIC2 and PFIC3 are attributed, respectively, to defects in ABCB11 (also referred to as bile salt-exporting pump protein) and ABCB4, which is involved in biliary PC excretion (19–21). A previous study reported that exogenous expression of ATP8B1 in the CHO-K1 mutant cell line UPS-1, which is defective in PS translocation, promoted PS translocation from the exoplasmic to the cytoplasmic leaflet (8). In contrast, depletion of ATP8B1 by RNAi had no effect on PS translocation in Caco-2 cells (22), and another study proposed that ATP8B1 acts as a cardiolipin importer (23). Therefore, it remains uncertain whether PS is a bona fide substrate of ATP8B1.
In this study, we established an assay for plasma membrane-localized phospholipid flippases by modifying previously described methods (8, 24). Using this method, we showed that ATP11A and ATP11C catalyze flipping of NBD-PS and NBD-PE but not NBD-PC or NBD-SM, whereas ATP8B1 preferentially catalyzes flipping of NBD-PC. Furthermore, we found that some PFIC1-type mutants of ATP8B1 failed to flip PC and that exogenous expression of ABCB4 diminished PC translocation mediated by ATP8B1.
EXPERIMENTAL PROCEDURES
Plasmids
P4-ATPase cDNAs were cloned separately into the pENTR3C vector (Invitrogen) as described previously (12). The ABCB4 cDNA was a kind gift from Kazumitsu Ueda (Kyoto University). Point mutations of ATP8B1, ATP8B2, ATP11A, ATP11C, and ABCB4 were introduced into the cDNAs using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). The DEST region from pcDNA6.2/V5-DEST (Invitrogen) encompassing the attR1 site, ccdB gene, chloramphenicol resistance gene, and attR2 site was cloned into the pMXs-neo and pMXs-puro expression vectors (25) with a C-terminal HA tag (pMXs-neo-DEST-HA) and a C-terminal FLAG tag (pMXs-puro-DEST-FLAG), respectively. The pMXs-neo and pMXs-puro vectors and the pEF-gag-pol plasmid were kind gifts from Toshio Kitamura (The University of Tokyo). To construct pMXs-puro, the neomycin resistance gene was replaced with the puromycin resistance gene. Transfer of the genes to pMXs-neo-DEST-HA or pMXs-puro-DEST-FLAG was performed using the Gateway system (Invitrogen). The pCMV-VSVG-RSV-Rev plasmid was a kind gift from Hiroyuki Miyoshi (RIKEN BioResource Center). The pCAG-based vector for expression of P4-ATPase with a C-terminal HA tag and the pcDNA3-based vector for expression of CDC50A with an N-terminal FLAG tag were described previously (12).
Antibodies, Reagents, and Immunofluorescence Analysis
Sources of antibodies used in the present study were as follows: polyclonal rabbit anti-giantin, Covance; monoclonal mouse anti-protein-disulfide isomerase (1D3), Enzo Life Sciences; monoclonal mouse anti-transferrin receptor (TfnR) (H68. 4), Zymed Laboratories Inc.; monoclonal rat anti-HA (3F10), Roche Applied Science; monoclonal mouse anti-β-tubulin, Millipore; monoclonal mouse anti-FLAG (M2), Sigma; polyclonal rabbit anti-FLAG, Sigma; monoclonal mouse anti-DYKDDDDK (1E6), Wako; Alexa Fluor 488-conjugated monoclonal mouse anti-CD147 (HIM6), BioLegend; Alexa Fluor-conjugated secondary antibodies, Molecular Probes; Cy3-, DyLight649-, and horseradish peroxidase-conjugated secondary antibodies, Jackson ImmunoResearch Laboratories. Alexa Fluor 555-conjugated wheat germ agglutinin was purchased from Invitrogen. The NBD-labeled phospholipids (Avanti Polar Lipids) used were NBD-PS (1-oleoyl-2-[6-[(7-nitro-2-,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphoserine), NBD-PE (1-oleoyl-2-[6-[(7-nitro-2-,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphoethanolamine), NBD-PC (1-oleoyl-2-[6-[(7-nitro-2-,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphocholine), and NBD-SM(N-[6-[(7-nitro-2-,3-benzoxadiazol-4-yl)amino]hexanoyl]sphingosine 1-phosphocholine). Immunostaining was performed as described previously (26) and visualized using an Axiovert 200MAT microscope (Carl Zeiss, Thornwood, NY).
Cell Culture and Establishment of Stable Cell Lines
Mouse interleukin-3 (IL-3)-dependent Ba/F3 cells were maintained in RPMI 1640 medium containing 10% fetal calf serum and recombinant mouse IL-3 as described (27). HeLa cells were cultured as described previously (12). CHO-K1 cells were grown in a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 medium supplemented with 10% fetal calf serum, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin.
To transiently express P4-ATPases, HeLa cells were transfected with a pCAG-HA-based vector carrying P4-ATPase cDNA and a pcDNA3-FLAG-based vector carrying CDC50A cDNA (12) using X-tremeGENE9 (Roche Applied Science). Two days later, the transfected cells were fixed for immunofluorescence analysis. To establish cell lines stably expressing each P4-ATPase, HeLa cells were transfected by lipofection with a pCAG-HA-based P4-ATPase vector. Colonies derived from single cells were isolated and selected in the presence of G418.
For retroviral production, pMXs-neo-derived vectors for expression of HA-tagged P4-ATPases were introduced into Plat-E cells (provided by Toshio Kitamura) (28) or co-transfected with pEF-gag-pol and pCMV-VSVG-RSV-Rev into HEK293T cells. pMXs-puro-derived vector for expression of FLAG-tagged ABCB4 was co-transfected with pEF-gag-pol and pCMV-VSVG-RSV-Rev into HEK293T cells. The resultant retroviruses were concentrated as described previously (24) and then used to infect Ba/F3, CHO-K1, or HeLa cells to establish stable cell lines. The infected cells were selected in medium containing G418 (1 mg/ml) or G418 (1 mg/ml) and puromycin (1 μg/ml). A mixed population of drug-resistant cells was used for the flippase assay and immunoblot analysis.
Surface Biotinylation
Cells were washed three times with chilled PBS containing 0.1 mm CaCl2 and 0.1 mm MgCl2 (PBS++) and then incubated with 2 mm sulfo-NHS-LC-biotin (Thermo Scientific) in PBS++ at 4 °C for 30 min. To stop the biotinylation reaction, the cells were washed three times with chilled PBS++ containing 100 mm glycine and 0.3% BSA and then washed twice more with chilled PBS. The cells were then lysed in lysis buffer (20 mm HEPES-KOH (pH 7.4) containing 1% Nonidet P-40, 150 mm NaCl, and protease inhibitor mixture (Complete Mini, Roche Applied Science)) for 30 min at 4 °C. To remove cellular debris and insoluble materials, the lysates were centrifuged at maximum speed at 4 °C for 20 min in a microcentrifuge. To precipitate the biotinylated proteins, the supernatant was incubated at 4 °C for 4 h with streptavidin-agarose beads (Thermo Scientific) pre-equilibrated with lysis buffer. The streptavidin beads were washed three times with lysis buffer, twice with high salt buffer (20 mm HEPES-KOH (pH 7.4), 500 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40), and once with 20 mm HEPES-KOH (pH 7.4). Proteins were eluted from the beads with SDS-PAGE sample buffer, denatured at 37 °C for 2 h, and subjected to immunoblot analysis. Blots were developed using a Chemi-Lumi One L kit (Nacalai Tesque), recorded on an LAS-3000 bioimaging system (Fujifilm), and quantified using Image Gauge software (Version 4.0; Fujifilm).
Flippase Assay
Incorporation of NBD-phospholipids was analyzed by flow cytometry as described (24). In brief, HeLa or CHO-K1 cells were detached from dishes in PBS containing 5 mm EDTA and then harvested by centrifugation. Ba/F3 cells were harvested from suspension culture by centrifugation. Cells (1 × 106 cells/sample) were washed and equilibrated at 15 °C (or 4 or 37 °C as indicated) for 15 min in 500 μl of Hanks' balanced salt solution (pH 7.4) containing 1 g/liter glucose (HBSS-glucose). An equal volume of 2 μm NBD-phospholipid in HBSS-glucose was added to the cell suspension and incubated at 15 °C (or 4 or 37 °C as indicated). At each time point, 200 μl of cell suspension was collected and mixed with 200 μl of ice-cold HBSS-glucose containing 5% fatty acid-free BSA (PAA Laboratories GmbH) to extract NBD-lipids incorporated into the exoplasmic leaflet of the plasma membrane as well as unincorporated NBD-lipids. Next, 10,000 cells were analyzed with a FACSCalibur (BD Biosciences) to measure fluorescence of NBD-lipids translocated into the cytoplasmic leaflet of the plasma membrane. The mean of fluorescence intensities per cells was calculated. Propidium iodide-positive cells (i.e. dead cells) were excluded from the analysis. To compare the flippase activities between WT and mutants of P4-ATPases, the mean of fluorescence intensities was normalized with the surface expression level of P4-ATPases and their mutants. Briefly, basal activity of parental cells (A) was subtracted from the activity of each stable cell line (B), and then the value was divided by the surface expression level of P4-ATPases, which was quantified using Image Gauge software and normalized with the level of the internal control (TfnR).
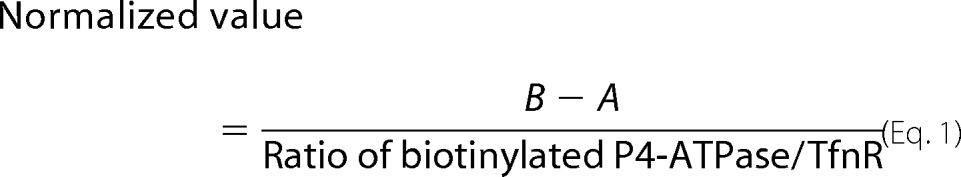 |
RESULTS
ATP11C Flips PS and PE
To establish an assay system for flippase activities of P4-ATPases in mammalian cells, we first used a Ba/F3 pro-B cell line stably expressing ATP11C. We chose this system for two reasons. 1) ATP11C localizes to the plasma membrane (12) (Fig. 1), and 2) pro-B cells derived from ATP11C mutant mice exhibit a lower PS-flipping activity than wild-type cells (29), indicating that ATP11C flips PS in pro-B cells. Ba/F3 cells stably expressing ATP11C(WT) or ATP11C(E184Q), an ATPase-deficient mutant, were established by infection with recombinant retrovirus and subsequent selection in the presence of antibiotics. The glutamate residue at position 184 is conserved in all P-type ATPases; the mutation of this residue abolishes dephosphorylation of a catalytically critical aspartate residue (30), resulting in a defect in ATPase activity. Comparable expression levels and plasma membrane localization of ATP11C(WT) and ATP11C(E184Q) were confirmed by immunoblot and immunofluorescence analyses, respectively (Fig. 1A and B). We assayed the flippase activities of ATP11C by incubating the cells in the presence of NBD-labeled PS, PE, PC, or SM at 15 °C (to minimize endocytic incorporation of the fluorescent phospholipids (24)) followed by extraction with fatty acid-free BSA of fluorescent phospholipids that were unincorporated or still in the exoplasmic leaflet of the plasma membrane. When added to parental Ba/F3 cells, levels of non-extractable NBD-labeled phospholipids increased in a time-dependent manner as a result of their translocation from the exoplasmic to the cytoplasmic leaflet by endogenous flippases and/or scramblases (Fig. 1C, −, black open squares). In Ba/F3 cells stably expressing ATP11C(WT), the levels of non-extractable NBD-PS and NBD-PE, but not NBD-PC or NBD-SM, increased faster than those in control cells (parental cells or cells infected with an empty vector; Fig. 1, C and D). By contrast, expression of ATP11C(E184Q) did not significantly increase the BSA-non-extractable level of any tested NBD-phospholipid relative to the corresponding level in control cells (Fig. 1, C and D), indicating that the increase in the levels of incorporated NBD-PS and NBD-PE in the ATP11C-expressing cells is dependent on the ATPase cycle. Thus, our assay confirmed the PS flippase activity of ATP11C reported previously (29, 31) and revealed that ATP11C can also translocate PE from the exoplasmic to the cytoplasmic leaflet of the plasma membrane. NBD-PS was incorporated much faster than other NBD-phospholipids (Fig. 1C); therefore, the level of non-extractable NBD-PS essentially reached a plateau after 20 min of incubation probably due to depletion of the fluorescent substrate.
FIGURE 1.

Flippase activities of ATP11C across the leaflets of the plasma membrane in Ba/F3 cells. Parental Ba/F3 cells (−) or Ba/F3 cells infected with empty retrovirus vector (Vector) or recombinant virus vector encoding HA-tagged ATP11C(WT) or ATP11C(E184Q) were either processed for immunoblot analysis with antibodies against HA and TfnR (as an internal control) (A) or stained with Alexa Fluor 555-conjugated wheat germ agglutinin (WGA) to visualize the plasma membrane followed by immunostaining for HA and giantin (a Golgi marker) (B). Scale bar, 10 μm. C, Ba/F3 cells were incubated with the indicated NBD-lipids at 15 °C for the indicated times (x axis). After extraction with fatty acid-free BSA, the residual fluorescence intensity associated with the cells was determined by flow cytometry. Graphs are representative of two independent experiments, and results display averages from triplicates ±S.D. D, -fold increase of NBD-lipid uptake compared with parental cells (−) is shown at the 10- (NBD-PS) or 15-min (NBD-PE, -PC, and -SM) time point from C. Graphs are representatives of three independent experiments, and results display averages from triplicates ±S.D. (*, p < 0.0001; **, p < 0.0005). Error bars represent S.D.
ATP11A and ATP11C Specifically Flip Aminophospholipids
Mutant P4-ATPase proteins with alterations in the catalytically critical aspartate residue, which undergoes phosphorylation and dephosphorylation in the ATPase cycle, are commonly used as ATPase-deficient mutants in yeast (32). We found that such aspartate mutants of ATP8B1, ATP11A, and ATP11C failed to localize to the plasma membrane and were instead retained in the ER even when coexpressed with CDC50A (Fig. 2, A–D) in HeLa cells. Therefore, the aspartate mutants could not exit from the ER, whereas the glutamate mutants could be transported normally to the plasma membrane but would not have enzymatic activities. Given that the aspartate mutant, but not the glutamate mutant, of yeast Drs2p fails to interact with CDC50 (33), these observations indicate that aspartate mutants of P4-ATPases cannot exit the ER due to a defect in their CDC50 binding ability. Therefore, in this study, we used the glutamate mutants as ATPase-deficient mutants.
FIGURE 2.

Plasma membrane localization of P4-ATPases and their ATPase-deficient mutants in HeLa cells. HeLa cells were transiently co-transfected with expression vectors for FLAG-tagged CDC50A and HA-tagged ATP8B1(WT), ATP8B1(E234Q), or ATP8B1(D454G) (a BRIC mutant, see Fig. 7) (A); ATP8B2(WT) or ATP8B2(E204Q) (B); ATP11A(WT), ATP11A(E186Q), or ATP11A(D414N) (C); or ATP11C(WT), ATP11C(E184Q), or ATP11C(D412N) (D). Before fixation, cells were incubated with Alexa Fluor 488-conjugated anti-CD147 antibody for 5 min at room temperature to label the plasma membrane. The fixed cells were then incubated with anti-HA and anti-FLAG (M2) antibodies followed by Cy3-conjugated anti-rat and DyLight649-conjugated anti-mouse secondary antibodies. For protein-disulfide isomerase (PDI; an endoplasmic reticulum marker) staining, fixed cells were incubated with anti-HA, anti-FLAG, and anti-protein-disulfide isomerase antibodies followed by Cy3-conjugated anti-rat, Alexa Fluor 647-conjugated anti-rabbit, and Alexa Fluor 488-conjugated anti-mouse secondary antibodies. Insets indicate FLAG-CDC50A-expressing cells. Scale bars, 10 μm.
To systematically evaluate the phospholipid flippase activities and substrate specificities of plasma membrane-localizing human P4-ATPases, we established several clones of HeLa cells stably expressing wild-type or glutamate mutant ATP8B1, ATP8B2, ATP11A, or ATP11C. To exclude potential clonal effects, we established several cell lines stably expressing C-terminally HA-tagged ATP8B1, ATP8B2, ATP11A, or ATP11C and then examined their expression levels by immunoblot analysis (Fig. 3, A and C). We also analyzed their delivery to the plasma membrane by surface biotinylation analysis (Fig. 3, B and D) or co-immunostaining with a plasma membrane marker (data not shown). Despite the fact that transiently expressed P4-ATPases generally require exogenous coexpression of CDC50 to exit the ER and reach their final cellular destinations (9, 10, 12) (Fig. 2), in our stably expressing cell lines, these P4-ATPase proteins were detected at the plasma membrane without coexpression of exogenous CDC50 (Fig. 3, B and D), indicating that the endogenous CDC50 level was sufficient for the localization of these proteins in the stable cell lines probably due to their moderate expression levels.
FIGURE 3.

Cell surface expression of P4-ATPases in HeLa stable cell lines. HeLa cell lines stably expressing HA-tagged P4-ATPase were established by plasmid transfection followed by selection in the presence of antibiotics. Two or three independent clones of cells expressing ATP8B1(WT), ATP8B2(WT), ATP11A(WT), or ATP11C(WT) or one clone expressing ATP8B1(E234Q), ATP11A(E186Q), or ATP11C(E184Q) was examined. A and C, the total expression level of the P4-ATPase in each cell line was analyzed by immunoblotting with anti-HA and anti-TfnR antibodies (as an internal control). 4% of the input of the biotinylation reaction was loaded in each lane. Arrow, ATP8B2; open arrow, ATP8B1. B and D, the cell surface level of the P4-ATPase in each cell line was analyzed after surface biotinylation. Proteins precipitated with streptavidin-agarose beads were subjected to immunoblot analysis for HA and TfnR (as an internal control). The numbers show the relative expression level of proteins, which were normalized with the level of the internal control, TfnR, and were used for normalizing the enzymatic activities shown in Fig. 4 (J and K). In A and B, open and solid arrows indicate positions of ATP8B1 and ATP8B2, respectively. In A–D, middle panels show overexposures. WT(vi), cell lines established by infection of recombinant retrovirus for each P4-ATPase.
To determine the substrate specificities and flippase activities of these P4-ATPases, we made use of an assay system established using Ba/F3 cells (Fig. 4). When added to parental HeLa cells, the levels of NBD-labeled phospholipids (PS, PE, PC, or SM) that were non-extractable with fatty acid-free BSA increased in a time-dependent manner as a result of translocation of these lipids from the exoplasmic to the cytoplasmic leaflet of the plasma membrane (Fig. 4, A–D, −, black open squares) catalyzed by endogenous flippases and/or scramblases. Like Ba/F3 cells (Fig. 1C), parental HeLa cells incorporated NBD-PS faster than other NBD-lipids (Fig. 4A). HeLa cells expressing ATP11A or ATP11C exhibited increased levels of BSA-non-extractable NBD-PS and NBD-PE compared with parental cells (Fig. 4, A, B, F, G, and I). By contrast, the level of uptake of NBD-PC or NBD-SM did not change in these stable cell lines relative to the corresponding level in parental cells (Fig. 4, C, D, G, H, and I), indicating that ATP11A and ATP11C are aminophospholipid-specific flippases. Moreover, the level of BSA-non-extractable NBD-PS or NBD-PE did not increase in cells expressing ATP11A(E186Q) or ATP11C(E184Q) (Fig. 4, E, F, J, and K), supporting the idea that the observed flipping activity is dependent on the ATPase cycle of ATP11A or ATP11C. To exclude the possibility that the lack of significant flippase activity toward PS and PE of ATP11A(E186Q) in the clonal cell line was due to an expression level lower than that of ATP11A(WT) (Fig. 3, C and D), we established HeLa cells stably expressing ATP11A(WT) or ATP11A(E186Q) by retroviral infection (Fig. 5, A and B) and subjected the cells to the flippase assay. Even if the expression level of ATP11A(E186Q) was slightly higher than that of ATP11A(WT), cells expressing ATP11A(E186Q) did not show flipping activities toward any NBD-lipids examined (Fig. 5, C–F). We also compared the activities between WT and glutamate mutants by normalizing fluorescence intensities with the surface expression level of P4-ATPases (Figs. 4, J and K, and 5, G and H). To minimize the effect of endocytic incorporation of fluorescent phospholipids, in subsequent flippase assays, we incubated cells at 15 °C for 10 or 15 min (a previous study reported ATP-dependent flipping of phospholipids in erythrocytes at temperatures as low as 5 °C (34)). We also examined translocation of NBD-lipids at 4 °C, a temperature at which endocytosis is completely blocked. ATP11A-expressing cells exhibited higher rates of translocation of NBD-PS and NBD-PE than parental cells even at 4 °C (Fig. 6, A and B, respectively), indicating that the substrate-specific incorporation is not due to endocytosis. From these results, we draw two main conclusions. 1) Flippase activities and substrate specificities can be examined using the cell lines stably expressing human P4-ATPases, which localize to the plasma membrane, and 2) ATP11A and ATP11C exhibit specific flippase activities toward PS and PE in an ATPase cycle-dependent manner.
FIGURE 4.

Flippase activities of P4-ATPases across the leaflets of the plasma membrane in HeLa cells. A–D, parental HeLa cells (−; open squares) and a cell line stably expressing ATP8B1 (clone 12; blue), ATP11A (clone 14; green), or ATP11C (clone 38; orange) were incubated with the indicated NBD-lipids at 15 °C for the indicated times (x axis). After extraction with fatty acid-free BSA, the residual fluorescence intensity associated with the cells was determined by flow cytometry. Graphs are representatives of two independent experiments, and results display averages from triplicates ±S.D. E–H, each of the clonal cell lines shown in Fig. 2 was incubated with the indicated NBD-lipids at 15 °C for 15 min (or 10 min for NBD-PS). After extraction with fatty acid-free BSA, the residual fluorescence intensity associated with the cells was determined by flow cytometry. -Fold increase of NBD-lipid uptake compared with parental HeLa cells (−) is shown. Graphs are representative of at least two independent experiments, and results display averages from triplicates ±S.D. (*, p < 0.001; **, p < 0.005). Error bars represent S.D. I, a histogram of a representative experiment displaying the differences in the fluorescence intensity among parental HeLa cells in the absence (w/o NBD-lipids; dashed line) or presence (gray area) of NBD-lipids or a cell line stably expressing ATP8B1 (clone 12; blue), ATP11A (clone 14; green), or ATP11C (clone 38; orange) in the presence of indicated NBD-lipids at 15 °C for the indicated times. J and K, the enzymatic activities of WTs and glutamate mutants were normalized with the level of biotinylated P4-ATPases shown in Fig. 3D.
FIGURE 5.

Cell surface expression and flippase activities of P4-ATPases in retrovirus-infected HeLa stable cells. HeLa cells stably expressing the indicated HA-tagged P4-ATPases were established by infection with recombinant retrovirus without clonal selection and processed for analysis of the total (A) or cell surface (B) expression level of the P4-ATPase proteins as described in the legend for Fig. 3. The middle panels of A and B show overexposures of the blots shown in the top panels. Cells expressing ATP8B1, ATP8B2, and ATP11A were incubated with NBD-PS (C), NBD-PE (D), NBD-PC (E), or NBD-SM (F) at 15 °C for 15 min (or 10 min for NBD-PS). After extraction with fatty acid-free BSA, the residual fluorescence intensity associated with the cells was determined by flow cytometry. -Fold increase of NBD-lipid uptake relative to parental HeLa cells (−) is shown. The graphs are representative of two independent experiments, and results display averages from triplicates ±S.D. (*, p < 0.001). Error bars represent S.D. G–I, the enzymatic activities were normalized with the level of biotinylated P4-ATPases shown in B.
FIGURE 6.
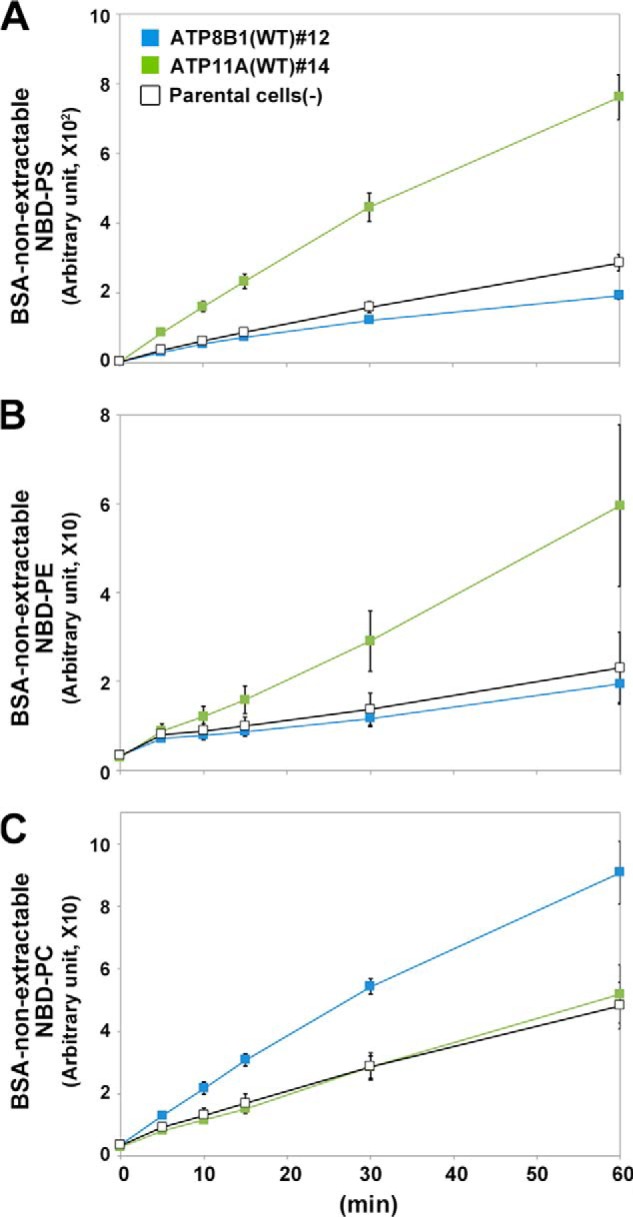
Flippase activities of P4-ATPases at 4 °C. HeLa cell lines stably expressing the HA-tagged ATP11A(clone 14) and ATP8B1(clone 12) were incubated with NBD-PS (A), NBD-PE (B), or NBD-PC (C) at 4 °C for the indicated times. The graph displays averages from two independent experiments ±S.D. Error bars represent S.D.
ATP8B1 Preferentially Flips PC
ATP8B1 has been hypothesized to translocate PS for the following reasons. 1) Previous studies showed that ATP8B1 translocated PS in CHO-K1 cells, although the flippase activities toward other phospholipids were not carefully examined (8, 35). 2) Chenodeoxycholate treatment caused canalicular accumulation of NBD-PS in cultured ATP8B1 knockdown hepatocytes (36). 3) Infusion of taurocholate resulted in higher recovery of PS in bile from Atp8b1G308V/G308V mutant mice relative to wild-type mice (37). However, several controversies remain. 1) In Caco-2 cells, depletion of ATP8B1 by RNAi was reported to have no effect on PS translocation (22). 2) Another study proposed that ATP8B1 acts as a cardiolipin importer (23). Therefore, it remains uncertain whether PS is a bona fide substrate for ATP8B1.
Unexpectedly, stable expression of ATP8B1 in HeLa cells increased the level of BSA-non-extractable NBD-PC at 15 °C in a time-dependent manner relative to parental cells (Fig. 4, C, G, and I). In contrast, stable ATP8B1 expression failed to increase the levels of BSA-non-extractable NBD-PS (Fig. 4, A, E, and I), NBD-PE (Fig. 4, B, F, and I), and NBD-SM (Fig. 4, D, H, and I). Moreover, even when endocytosis was blocked by incubation at 4 °C, ATP8B1-expressing cells exhibited increased translocation of NBD-PC, but not NBD-PS or NBD-PE, relative to the parental cells (Fig. 6C). ATP8B2, whose primary sequence is 54% identical to that of ATP8B1, also exhibited a significant flippase activity toward NBD-PC (Fig. 4, E–H). To exclude potential clonal effects, we also confirmed the flippase activities of ATP8B1 and ATP8B2 toward NBD-PC using stably expressing HeLa cells established by retroviral infection (Fig. 5, A and B). We observed that ATP8B1(WT) and ATP8B2(WT) exhibited PC flippase activities (Fig. 5, C–F). To exclude a possibility that the lack of significant PC flippase activity of ATP8B1(E234Q) in the clonal cell line was due to an expression level lower than that of ATP8B1(WT) (Fig. 3A and B), we established HeLa cells stably expressing the mutant of ATP8B1 by retroviral infection (Fig. 5, A and B). However, the total and surface expression levels of ATP8B1(E234Q) were improved but still lower than those of ATP8B1(WT) (Figs. 5, A and B, and 7, A and B), suggesting that the ATP8B1 mutant might be unstable in cells for an unknown reason. Although we could not exclude the possibility that the lack of PC-flipping activity of ATP8B1(E234Q) might be due to its lower expression level in the cells, ATP8B1(E234Q) did not show a significant PC flippase activity compared with ATP8B1(WT) after normalization with the expression level of the proteins (Fig. 5I). Conversely, although the expression level of ATP8B2(E204Q) was comparable with that of ATP8B2(WT) (Fig. 5, A and B), ATP8B2(E204Q) did not flip any NBD-lipids examined (Fig. 5, C–F and I), indicating that the flippase activity of ATP8B2 toward PC is ATPase cycle-dependent.
FIGURE 7.

Flippase activity of ATP8B1 across the leaflets of the plasma membrane in CHO-K1 cells. CHO-K1 cell lines stably expressing HA-tagged ATP8B1(WT) or ATP8B1(E234Q) were established by infection with recombinant retrovirus followed by selection in the presence of antibiotics. A, the total expression level of ATP8B1 was analyzed by immunoblotting with antibodies against HA and TfnR (as an internal control). 4% of the input of the biotinylation reaction was loaded in each lane. B, the cell surface expression level of ATP8B1 was determined by surface biotinylation analysis. The middle panels show overexposures. C, cells were incubated with NBD-lipids and processed for flippase assays as described in the legend for Fig. 4. -Fold increase of NBD-lipid uptake relative to parental CHO-K1 cells (−) is shown. The results display averages from triplicates ±S.D. (*, p < 0.001). Error bars represent S.D.
Because a previous study reported that ATP8B1 exhibited PS flippase activity in CHO-K1-derived cells at 15 °C (8), we next established CHO-K1 cells stably expressing ATP8B1(WT) or ATP8B1(E234Q) (Fig. 7). Plasma membrane localization of these P4-ATPases was confirmed by surface biotinylation (Fig. 7, A and B). Consistent with the results obtained using HeLa cells, CHO-K1 cells expressing ATP8B1(WT) exhibited PC flippase activity but no significant activity toward PS or PE (Fig. 7C).
Analyses of ATP8B1 Missense Mutations Found in PFIC1 and BRIC1 Patients
Genetic analyses of PFIC1 and BRIC1 patients have identified a wide range of ATP8B1 mutations that give rise to a variety of symptoms (17, 18). However, the relationships between types of missense mutations and flippase activity have not been explored in part because the substrate specificity of ATP8B1 was uncertain. Many missense mutant ATP8B1 proteins found in PFIC1 are degraded before delivery to the plasma membrane (38, 39) (Fig. 8A). Indeed, Atp8b1G308V/G308V mutant mice express no detectable ATP8B1 protein (40). Therefore, to date, there has been no evidence that PFIC1 is caused by a deficiency of ATP8B1 enzymatic activity rather than a lack of the ATP8B1 protein itself. We therefore set out to investigate whether certain missense mutations of ATP8B1 found in PFIC1 (L127P, L288S, G308V, and G1040R) and BRIC1 (D70N, I344F, D454G, and I661T) affect the flippase activity of this protein (18). When expressed in HeLa cells by recombinant retrovirus infection, total protein levels of the L288S, G308V, G1040R, D454G, and I661T mutants were greatly reduced compared with the level of ATP8B1(WT) (Fig. 8, A and B), although ATP8B1(I661T) and a low level of ATP8B1(G1040R) were detected on the plasma membrane (Fig. 8B) as reported previously (38, 39). Incubation of the stable cell lines with MG-132, a proteasome inhibitor, resulted in modest recovery of the protein levels of these mutants (Fig. 8C) as reported previously (38, 39), suggesting that the mutated proteins are degraded by the proteasome. However, MG-132 treatment did not significantly increase the cell surface levels of these mutants (Fig. 8, D and E, right panels), suggesting that these mutant proteins are misfolded and undergo degradation before their delivery to the plasma membrane.
FIGURE 8.

Localization and flippase activity of ATP8B1 mutants associated with cholestasis. HeLa cell lines stably expressing the HA-tagged ATP8B1 mutants indicated were established by infection of recombinant retroviral vectors. A and C, the total expression level of each ATP8B1 protein was analyzed by immunoblotting with anti-HA and anti-TfnR antibodies. 4% of the input of the biotinylation reaction was loaded in each lane. B and D, cell surface expression level of each ATP8B1 protein was analyzed after surface biotinylation. The numbers in B show the relative expression level of proteins, which were normalized with the level of the internal control, TfnR. C and D, the cells were treated with 10 μm MG-132 for 12 h. E, cells expressing the indicated ATP8B1 protein were processed for immunostaining. Before fixation, cells were incubated with Alexa Fluor 488-conjugated anti-CD147 antibody for 5 min at room temperature to label the plasma membrane. The fixed cells were then incubated with anti-HA antibody followed by Cy3-conjugated anti-rat secondary antibody. Cells treated with MG-132 are indicated. Scale bar, 10 μm. Each cell line was incubated with NBD-PC (F) or NBD-PS (H) and then processed for the flippase assay as described in the legend for Fig. 4. -Fold increase of NBD-lipid uptake compared with parental HeLa cells (−) is shown. The graph is representative of two independent experiments, and results display averages from triplicates ±S.D. (*, p < 0.001). Error bars represent S.D. G, the PC flippase activities were normalized with the level of biotinylated P4-ATPases shown in B.
L127P and I344F Mutations Abolish the PC Flippase Activity of ATP8B1
In striking contrast to these mutants, the levels of the D70N, L127P, and I344F mutants on the plasma membrane were comparable with that of ATP8B1(WT) (Fig. 8, A and B). The D70N and L127P mutants are properly localized to the plasma membrane and are able to interact with CDC50 protein (38, 39). Therefore, we next explored the flippase activities of these mutants toward PC and PS. Importantly, the L127P and I344F mutations abolished the PC flippase activity of ATP8B1 (Fig. 8, F and G). In contrast, the D70N mutant retained the flippase activity (Fig. 8, F and G); in light of this observation and the fact that the D70N mutation is present on 0.5% of normal human chromosomes (18), it is possible that this mutation is not directly related to the etiology of the disease. None of the mutants examined exhibited significant PS flippase activity (Fig. 8H), indicating that the missense mutations in ATP8B1 did not scramble the substrate preferences of the enzyme. Together, these results suggest that the phenotypes of PFIC1 patients harboring at least some of these mutations result from a defect in ATP8B1 flippase activity toward PC as well as from a reduction in the level of ATP8B1 protein.
ABCB4 Diminished PC Flipping Mediated by ATP8B1
PFIC3 is caused by mutations in the ABCB4 gene and is characterized by the absence of PC in bile. ABCB4 has been identified as the PC translocator from the cytoplasmic to the exoplasmic leaflet of the canalicular membrane (20). Therefore, we hypothesized that coexpression of ABCB4 would reverse the increased PC translocation in ATP8B1-expressing cell lines. To address this hypothesis, we established cell lines stably expressing FLAG-tagged ABCB4(WT) or ABCB4(K435M), which is nonfunctional due to a mutation in the Walker A motif in the nucleotide-binding domain (41), with or without ATP8B1-HA. In both the singly and doubly expressing cell lines, comparable expression levels of ABCB4(WT) and ABCB4(K435M) were confirmed by immunoblotting, and the plasma membrane localization of all proteins was visualized by immunostaining with anti-FLAG antibody (Fig. 9, A and B). Comparable expression levels of ATP8B1 were also confirmed, although in cells coexpressing ABCB4(WT) and ATP8B1, the expression levels of both proteins were slightly elevated relative to those in cells expressing either protein alone (Fig. 9A); this result is consistent with a previous report (42). ABCB4(WT) and ABCB4(K435M) colocalized with ATP8B1 at the plasma membrane (Fig. 8B). Importantly, coexpression of ATP8B1 with ABCB4(WT), but not ABCB4(K435M), significantly suppressed PC incorporation at all time points examined relative to that in cells expressing only ATP8B1 (Fig. 9, C and D). Because we could not detect the enzymatic activity of ABCB4 at 15 °C in this experiment (data not shown), we performed the NBD-PC incorporation assay at 37 °C. To minimize potential NBD-PC incorporation by endocytosis, we compared the incorporation at the 5-min time point (Fig. 9D). Although we did not detect a decrease in PC incorporation in cells expressing ABCB4 alone at 5 min, we did detect a small but significant decrease at 30 min relative to parental cells or cells expressing ABCB4(K435M) (Fig. 9C). Taken together, these data indicate that ATP8B1-mediated PC translocation can be reversed by coexpression of ABCB4, a PC floppase, although the PC incorporation was not reduced to the control level.
FIGURE 9.

Exogenous expression of ABCB4 antagonizes PC flipping mediated by ATP8B1. HeLa cell lines stably expressing HA-tagged ATP8B1, FLAG-tagged ABCB4(WT), FLAG-tagged ABCB4(K435M), ATP8B1-HA + FLAG-ABCB4(WT), or ATP8B1-HA + FLAG-ABCB4(K435M) were established by infection with recombinant retroviral vectors. A, the total expression level of each protein was analyzed by immunoblotting with anti-HA, anti-DYKDDDDK (which recognizes the FLAG epitope), and anti-β-tubulin antibodies. B, cells expressing the indicated proteins singly or doubly were processed for immunostaining with anti-HA and anti-DYKDDDDK antibodies followed by Cy3-conjugated anti-rat and DyLight649-conjugated anti-mouse secondary antibodies. Scale bar, 20 μm. C, cells were incubated with NBD-PC at 37 °C for the indicated times (x axis). After extraction with fatty acid-free BSA, the residual fluorescence intensity associated with the cells was determined by flow cytometry. Graphs display averages from three independent experiments ±S.D. (*, p < 0.005). D, -fold increase of NBD-PC uptake compared with parental HeLa cells (−) at the 5-min time point of C is shown. Graphs display averages from three independent experiments ±S.D. (*, p < 0.001 versus −; #, p < 0.001 versus ATP8B1). Error bars represent S.D.
DISCUSSION
In this study, we elucidated the flippase activities and substrate specificities of plasma membrane-localized human P4-ATPases using stably expressing cell lines. We found that ATP11A and ATP11C flip aminophospholipids PS and PE, whereas ATP8B1 and ATP8B2 flip PC. In parental HeLa cells, NBD-PS was incorporated much faster than other NBD-phospholipids probably due to high basal PS-flipping activities. Despite the high endogenous PS flippase activity, mild expression of ATP11A or ATP11C further increased the translocation of PS and PE but not PC, indicating that ATP11A and ATP11C are aminophospholipid-specific flippases and that the enzymatic activities of P4-ATPases in stable cell lines can be examined even in the presence of endogenous flippases. We have found that ATP8B1 and ATP8B2 translocate PC rather than PS and that the former exhibits PC-specific flippase activity even at 4 °C (Fig. 6), although the activity is much lower than that at 15 °C. However, because the high endogenous PS-flipping activities may have concealed a hypothetical low PS flippase activity of ATP8B1 and ATP8B2, we cannot exclude the possibility that ATP8B1 also has flippase activity toward PS.
The major constituents of bile are bile salts, PC, cholesterol, and bilirubin. PC, a crucial constituent of bile, associates with bile salts to prevent their detergent activities from causing biliary toxicity, and it is also necessary for maintaining the solubility of cholesterol in bile (20, 21). ABCB4 is a phospholipid translocator that is highly expressed in the canalicular membrane and involved in biliary PC excretion (43). The liver damage in PFIC3 (mutations in ABCB4) patients is likely caused by the absence of biliary PC (20, 21). Overexpression of ABCB4 is toxic to HEK293T cells, but this toxicity can be reversed by coexpression of ATP8B1 and CDC50A (42). Given that exogenously expressed ABCB4 can antagonize the PC flippase activity of ATP8B1 (Fig. 9), the impaired balance between flip and flop of PC is implicated in the toxicity of ABCB4 overexpression.
Analyses of PFIC1 and BRIC1 patients have identified a wide range of ATP8B1 mutations (17, 18). However, the relationships between types of missense mutations and the flippase activity have not been explored, and many missense mutants of ATP8B1 found in PFIC1 are degraded before delivery to the plasma membrane (38, 39). Stone et al. (44) introduced PFIC1-type mutations into the yeast plasma membrane P4-ATPase Dnf2p and examined whether the mutations affected its PC-flipping activity. In that study, Dnf2p mutant corresponding to ATP8B1(L127P) flipped NBD-PC as efficiently as wild-type Dnf2p. By contrast, the I344F mutation reduced the PC-flipping activity, although the expression level or stability of the mutant Dnf2p was not affected. In this study, both the L127P and I344F mutants of ATP8B1 were delivered normally to the plasma membrane but had no PC flippase activity (Fig. 8), suggesting that phenotypes of PFIC1 patients with these mutations may be caused by the lack of enzymatic activity of ATP8B1. Thus, the L127P mutation, unlike its corresponding Dnf2p mutation L264P, impaired the enzymatic activity of ATP8B1. Although we do not know the exact reason for the difference between the yeast and human P4-ATPases, the simplest explanation is that they have different enzymatic properties; for example, aspartate mutants of human P4-ATPases could not exit the ER (Fig. 2), whereas the aspartate mutant of Drs2p can properly localize to the Golgi apparatus (32, 45).
How is the impaired flippase activity of ATP8B1 responsible for the symptoms of PFIC1? One possibility is that a reduction in the PC-flipping activity in PFIC1 patients changes the lipid composition in the canalicular membrane and thereby changes membrane fluidity, resulting in impaired functions of canalicular membrane proteins such as ABCB11. Because PFIC1 patients exhibit decreased excretion of bile salts and Atp8b1G308V/G308V mutant mice exhibit normal ABCB11 localization to the canalicular membrane (37), it is likely that impaired ATP8B1 activity may affect ABCB11 function. Furthermore, in Atp8b1G308V/G308V mice, infusion of taurocholate increases recovery of PS in bile relative to that in wild-type mice and promotes the accumulation of vesicular structures in the canalicular lumen (37). The defect in the flip-flop balance of PC (flip < flop) in Atp8b1G308V/G308V mutant mice might increase the volume of the outer leaflet of the canalicular membrane, leading to a change in membrane curvature (34, 46) and thus allowing membranes to protrude; shedding of the membrane protrusions may result in formation of vesicular structures in the canalicular lumen. Because of the presence of vesicular structures generated from the canalicular membrane, PS, which usually localizes to the cytoplasmic leaflet, could be recovered in bile in Atp8b1G308V/G308V mutant mice. Although it remains unknown whether the enzymatic activity of ATP8B1 is necessary for bile excretion in vivo, our findings provide important insights into the pathophysiological mechanisms underlying PFIC.
In this study, we systematically evaluated the enzymatic activities of human P4-ATPases for the first time. Knowledge of these activities is essential for achieving an understanding of the physiological roles of P4-ATPases in mammalian cells.
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Special Coordination Fund for Promoting Science and Technology; the Takeda Science Foundation; and the Inamori Foundation.
- PS
- phosphatidylserine
- P4-ATPase
- type IV P-type ATPase
- PE
- phosphatidylethanolamine
- PC
- phosphatidylcholine
- SM
- sphingomyelin
- NBD
- nitrobenzoxadiazole
- PFIC
- progressive familial intrahepatic cholestasis
- BRIC
- benign recurrent intrahepatic cholestasis
- ER
- endoplasmic reticulum
- TfnR
- transferrin receptor
- NHS
- N-hydroxysuccinimide
- HBSS
- Hanks' balanced salt solution.
REFERENCES
- 1. Graham T. R. (2004) Flippases and vesicle-mediated protein transport. Trends Cell Biol. 14, 670–677 [DOI] [PubMed] [Google Scholar]
- 2. Pomorski T., Holthuis J. C., Herrmann A., van Meer G. (2004) Tracking down lipid flippases and their biological functions. J. Cell Sci. 117, 805–813 [DOI] [PubMed] [Google Scholar]
- 3. Zwaal R. F., Schroit A. J. (1997) Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 89, 1121–1132 [PubMed] [Google Scholar]
- 4. van den Eijnde S. M., van den Hoff M. J., Reutelingsperger C. P., van Heerde W. L., Henfling M. E., Vermeij-Keers C., Schutte B., Borgers M., Ramaekers F. C. (2001) Transient expression of phosphatidylserine at cell-cell contact areas is required for myotube formation. J. Cell Sci. 114, 3631–3642 [DOI] [PubMed] [Google Scholar]
- 5. Williamson P., Schlegel R. A. (2002) Transbilayer phospholipid movement and the clearance of apoptotic cells. Biochim. Biophys. Acta 1585, 53–63 [DOI] [PubMed] [Google Scholar]
- 6. Saito K., Fujimura-Kamada K., Furuta N., Kato U., Umeda M., Tanaka K. (2004) Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 3418–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furuta N., Fujimura-Kamada K., Saito K., Yamamoto T., Tanaka K. (2007) Endocytic recycling in yeast is regulated by putative phospholipid translocases and the Ypt31p/32p-Rcy1p pathway. Mol. Biol. Cell 18, 295–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paulusma C. C., Folmer D. E., Ho-Mok K. S., de Waart D. R., Hilarius P. M., Verhoeven A. J., Oude Elferink R. P. (2008) ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology 47, 268–278 [DOI] [PubMed] [Google Scholar]
- 9. Bryde S., Hennrich H., Verhulst P. M., Devaux P. F., Lenoir G., Holthuis J. C. (2010) CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J. Biol. Chem. 285, 40562–40572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Velden L. M., Wichers C. G., van Breevoort A. E., Coleman J. A., Molday R. S., Berger R., Klomp L. W., van de Graaf S. F. (2010) Heteromeric interactions required for abundance and subcellular localization of human CDC50 proteins and class 1 P4-ATPases. J. Biol. Chem. 285, 40088–40096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coleman J. A., Molday R. S. (2011) Critical role of the β-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J. Biol. Chem. 286, 17205–17216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takatsu H., Baba K., Shima T., Umino H., Kato U., Umeda M., Nakayama K., Shin H.-W. (2011) ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J. Biol. Chem. 286, 38159–38167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pomorski T., Lombardi R., Riezman H., Devaux P. F., van Meer G., Holthuis J. C. (2003) Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol. Biol. Cell 14, 1240–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Natarajan P., Wang J., Hua Z., Graham T. R. (2004) Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc. Natl. Acad. Sci. 101, 10614–10619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baldridge R. D., Xu P., Graham T. R. (2013) Type IV P-type ATPases distinguish mono- versus diacyl phosphatidylserine using a cytofacial exit gate in the membrane domain. J. Biol. Chem. 288, 19516–19527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baldridge R. D., Graham T. R. (2012) Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc. Natl. Acad. Sci. 109, E290–E298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bull L. N., van Eijk M. J., Pawlikowska L., DeYoung J. A., Juijn J. A., Liao M., Klomp L. W., Lomri N., Berger R., Scharschmidt B. F., Knisely A. S., Houwen R. H., Freimer N. B. (1998) A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 18, 219–224 [DOI] [PubMed] [Google Scholar]
- 18. Klomp L. W., Vargas J. C., van Mil S. W., Pawlikowska L., Strautnieks S. S., van Eijk M. J., Juijn J. A., Pabón-Peña C., Smith L. B., DeYoung J. A., Byrne J. A., Gombert J., van der Brugge G., Berger R., Jankowska I., Pawlowska J., Villa E., Knisely A. S., Thompson R. J., Freimer N. B., Houwen R. H., Bull L. N. (2004) Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 40, 27–38 [DOI] [PubMed] [Google Scholar]
- 19. Davit-Spraul A., Gonzales E., Baussan C., Jacquemin E. (2009) Progressive familial intrahepatic cholestasis. Orphanet J. Rare Dis. 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nicolaou M., Andress E. J., Zolnerciks J. K., Dixon P. H., Williamson C., Linton K. J. (2012) Canalicular ABC transporters and liver disease. J. Pathol. 226, 300–315 [DOI] [PubMed] [Google Scholar]
- 21. Jacquemin E. (2012) Progressive familial intrahepatic cholestasis. Clin. Res. Hepatol. Gastroenterol. 36, Suppl. 1, S26–S35 [DOI] [PubMed] [Google Scholar]
- 22. Verhulst P. M., van der Velden L. M., Oorschot V., van Faassen E. E., Klumperman J., Houwen R. H., Pomorski T. G., Holthuis J. C., Klomp L. W. (2010) A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology 51, 2049–2060 [DOI] [PubMed] [Google Scholar]
- 23. Ray N. B., Durairaj L., Chen B. B., McVerry B. J., Ryan A. J., Donahoe M., Waltenbaugh A. K., O'Donnell C. P., Henderson F. C., Etscheidt C. A., McCoy D. M., Agassandian M., Hayes-Rowan E. C., Coon T. A., Butler P. L., Gakhar L., Mathur S. N., Sieren J. C., Tyurina Y. Y., Kagan V. E., McLennan G., Mallampalli R. K. (2010) Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat. Med. 16, 1120–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suzuki J., Umeda M., Sims P. J., Nagata S. (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 [DOI] [PubMed] [Google Scholar]
- 25. Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. (2003) Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 31, 1007–1014 [PubMed] [Google Scholar]
- 26. Shin H.-W., Morinaga N., Noda M., Nakayama K. (2004) BIG2, a guanine nucleotide exchange factor for ADP-ribosylation factors: its localization to recycling endosomes and implication in the endosome integrity. Mol. Biol. Cell 15, 5283–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fukunaga R., Ishizaka-Ikeda E., Nagata S. (1990) Purification and characterization of the receptor for murine granulocyte colony-stimulating factor. J. Biol. Chem. 265, 14008–14015 [PubMed] [Google Scholar]
- 28. Morita S., Kojima T., Kitamura T. (2000) Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 29. Yabas M., Teh C. E., Frankenreiter S., Lal D., Roots C. M., Whittle B., Andrews D. T., Zhang Y., Teoh N. C., Sprent J., Tze L. E., Kucharska E. M., Kofler J., Farell G. C., Bröer S., Goodnow C. C., Enders A. (2011) ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat. Immunol. 12, 441–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anthonisen A. N., Clausen J. D., Andersen J. P. (2006) Mutational analysis of the conserved TGES loop of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 281, 31572–31582 [DOI] [PubMed] [Google Scholar]
- 31. Segawa K., Kurata S., Yanagihashi Y., Brummelkamp T. R., Matsuda F., Nagata S. (2014) Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168 [DOI] [PubMed] [Google Scholar]
- 32. Chen C. Y., Ingram M. F., Rosal P. H., Graham T. R. (1999) Role for Drs2p, a P-type ATPase and potential aminophospholipid translocase, in yeast late Golgi function. J. Cell Biol. 147, 1223–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lenoir G., Williamson P., Puts C. F., Holthuis J. C. (2009) Cdc50p plays a vital role in the ATPase reaction cycle of the putative aminophospholipid transporter Drs2p. J. Biol. Chem. 284, 17956–17967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seigneuret M., Devaux P. F. (1984) ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proc. Natl. Acad. Sci. 81, 3751–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ujhazy P., Ortiz D., Misra S., Li S., Moseley J., Jones H., Arias I. M. (2001) Familial intrahepatic cholestasis 1: studies of localization and function. Hepatology 34, 768–775 [DOI] [PubMed] [Google Scholar]
- 36. Cai S. Y., Gautam S., Nguyen T., Soroka C. J., Rahner C., Boyer J. L. (2009) ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology 136, 1060–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paulusma C. C., Groen A., Kunne C., Ho-Mok K. S., Spijkerboer A. L., Rudi de Waart D., Hoek F. J., Vreeling H., Hoeben K. A., van Marle J., Pawlikowska L., Bull L. N., Hofmann A. F., Knisely A. S., Oude Elferink R. P. (2006) Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology 44, 195–204 [DOI] [PubMed] [Google Scholar]
- 38. Folmer D. E., van der Mark V. A., Ho-Mok K. S., Oude Elferink R. P., Paulusma C. C. (2009) Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology 50, 1597–1605 [DOI] [PubMed] [Google Scholar]
- 39. van der Velden L. M., Stapelbroek J. M., Krieger E., van den Berghe P. V., Berger R., Verhulst P. M., Holthuis J. C., Houwen R. H., Klomp L. W., van de Graaf S. F. (2010) Folding defects in P-type ATP 8B1 associated with hereditary cholestasis are ameliorated by 4-phenylbutyrate. Hepatology 51, 286–296 [DOI] [PubMed] [Google Scholar]
- 40. Pawlikowska L., Groen A., Eppens E. F., Kunne C., Ottenhoff R., Looije N., Knisely A. S., Killeen N. P., Bull L. N., Elferink R. P., Freimer N. B. (2004) A mouse genetic model for familial cholestasis caused by ATP8B1 mutations reveals perturbed bile salt homeostasis but no impairment in bile secretion. Hum. Mol. Genet. 13, 881–892 [DOI] [PubMed] [Google Scholar]
- 41. Morita S. Y., Kobayashi A., Takanezawa Y., Kioka N., Handa T., Arai H., Matsuo M., Ueda K. (2007) Bile salt-dependent efflux of cellular phospholipids mediated by ATP binding cassette protein B4. Hepatology 46, 188–199 [DOI] [PubMed] [Google Scholar]
- 42. Groen A., Romero M. R., Kunne C., Hoosdally S. J., Dixon P. H., Wooding C., Williamson C., Seppen J., Van den Oever K., Mok K. S., Paulusma C. C., Linton K. J., Oude Elferink R. P. (2011) Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology 141, 1927–1937.e1-4 [DOI] [PubMed] [Google Scholar]
- 43. Buschman E., Arceci R. J., Croop J. M., Che M., Arias I. M., Housman D. E., Gros P. (1992) mdr2 encodes P-glycoprotein expressed in the bile canalicular membrane as determined by isoform-specific antibodies. J. Biol. Chem. 267, 18093–18099 [PubMed] [Google Scholar]
- 44. Stone A., Chau C., Eaton C., Foran E., Kapur M., Prevatt E., Belkin N., Kerr D., Kohlin T., Williamson P. (2012) Biochemical characterization of P4-ATPase mutations identified in patients with progressive familial intrahepatic cholestasis. J. Biol. Chem. 287, 41139–41151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gall W. E., Geething N. C., Hua Z., Ingram M. F., Liu K., Chen S. I., Graham T. R. (2002) Drs2p-dependent formation of exocytic clathrin-coated vesicles in vivo. Curr. Biol. 12, 1623–1627 [DOI] [PubMed] [Google Scholar]
- 46. Devaux P. F. (1991) Static and dynamic lipid asymmetry in cell membranes. Biochemistry 30, 1163–1173 [DOI] [PubMed] [Google Scholar]