

Abstract
We report here a series of five chemically diverse scaffolds that have in vitro activities on replicating and hypoxic nonreplicating bacilli by targeting the respiratory bc1 complex in Mycobacterium tuberculosis in a strain-dependent manner. Deletion of the cytochrome bd oxidase generated a hypersusceptible mutant in which resistance was acquired by a mutation in qcrB. These results highlight the promiscuity of the bc1 complex and the risk of targeting energy metabolism with new drugs.
TEXT
Tuberculosis remains a leading cause of death despite more than half a century of chemotherapeutic intervention. Bedaquiline (BDQ), the first FDA-approved antitubercular in more than 4 decades, was recently accepted by the U.S. FDA for the treatment of multidrug- and extensively drug-resistant disease (1). This diarylquinoline drug is thought to target the Mycobacterium tuberculosis ATP synthase, a critical component of mycobacterial energy metabolism (2). Recently, a new clinical candidate based on an imidazo[1,2-α]pyridine scaffold was disclosed (3). We previously described this scaffold from a screening effort specifically designed to identify inhibitors of respiratory enzymes, and it subsequently appeared from several other screening efforts (4–6). Mutants resistant to this series were mapped to an amino acid mutation in the QcrB subunit of the menaquinol cytochrome c oxidoreductase (bc1 complex), which is part of the bc1-aa3-type cytochrome c oxidase complex (3, 5).
We previously established that the majority of clinical strains of M. tuberculosis tested were susceptible to full growth inhibition by the imidazo[1,2-α]pyridines (4, 7, 8) but found that the laboratory-adapted strains H37Rv, CDC1551, and Erdman overcame this growth inhibition, despite the ability of these respiratory inhibitors to block the transfer of electrons to resazurin (Tables 1 and 2, compound 2; Fig. 1). The outgrowth observed with the laboratory-adapted strains was not due to the acquisition of resistance, since the cells showed the same phenomenon of outgrowth upon subculturing in drug-free medium followed by repeated MIC testing. We hypothesized that the ability of M. tuberculosis H37Rv to overcome bc1 complex inhibition was due to the upregulation of the cytochrome bd oxidase and cydDC genes (4), an alternative respiratory complex. Indeed, standing cultures that mimic the less oxygenated physiology of cells in MIC assays of the laboratory-adapted strains had higher basal expression levels of the cydA gene than did sensitive clinical strains (see Table S1 in the supplemental material). Although the laboratory and clinical K14b0DS strains upregulated cydA in response to QcrB inhibition, the final levels of cytochrome bd oxidase in the clinical strain did not match those evident in the laboratory-adapted strain (see Fig. S1 in the supplemental material). (Strain K14b0DS was collected with NIAID institutional review board approval under protocol [ClinicalTrials.gov identification no. NCT00341601].)
TABLE 1.
In vitro evaluation of compounds that target the bc1 complex
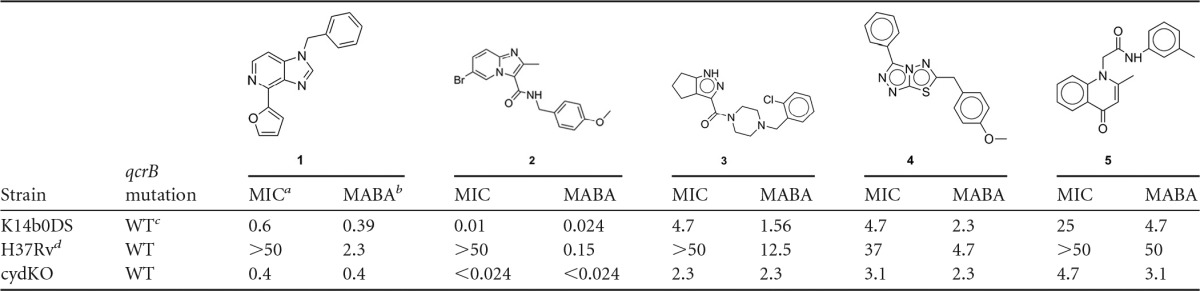
MIC values are in μM.
Microplate alamarBlue assay (MABA) results (the concentration of compound that inhibits resazurin reduction by >90%) are in μM.
WT, wild type.
Similar results were seen with the laboratory strains CDC1551 and Erdman.
TABLE 2.
Resistance to compounds that target the bc1 complex
| Protein | qcrB mutation | Fold resistance to compound: |
||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| Qcr1 | A317V | >128 | 940.0 | >22 | 16.0 | >11 |
| Qcr2 | M342T | 24.1 | 78.0 | 8.3 | 8.0 | 1.3 |
| Qcr3 | W312G | 94.9 | 313.0 | >22 | 8.0 | >11 |
| Qcr4 | A396T | 5.9 | 7.0 | 16.1 | 6.1 | 5.3 |
| Qcr5 | M342I | 32.1 | 30.0 | >22 | 3.0 | 0.3 |
| Qcr6 | A317T | 8.0 | 60.0 | 21.7 | 11.8 | >11 |
| Qcr7 | S182P | 48.7 | 20.0 | >22 | 8.0 | >11 |
FIG 1.

Treatment of M. tuberculosis isolates with inhibitors of the bc1 complex results in inhibition of resazurin reduction. M. tuberculosis isolates were treated with 2-fold serial dilutions of compound 2 (lanes i, iii, and v) or isoniazid (lanes ii, iv, and vi) for 2 weeks followed by addition of alamarBlue. Lanes i and ii, H37Rv; lanes iii and iv, K14b0DS; lanes v and vi, cydKO.
The ability of laboratory-adapted strains to overcome bc1 complex inhibition by upregulation of the cytochrome bd oxidase led us to speculate that a knockout mutant of this complex in the laboratory strain H37Rv would remain susceptible to such inhibitors. We deleted this oxidase in H37Rv by replacing a 221-bp MluI fragment in the cydABDC operon with the aph gene encoding an aminoglycoside phosphotransferase. The mutant lacks the 3' end of the cydB gene (encoding subunit II of the cytochrome bd oxidase), the entire cydD gene, and the 5' end of cydC (where cydDC encodes a transporter involved in cytochrome biogenesis). The cytochrome bd oxidase mutant (cydKO) was found to be highly susceptible to the imidazo[1,2-α]pyridines (Table 1). To map the binding site of the inhibitors in the bc1 complex, we generated mutants resistant to compound 1 in the cydKO strain. Mutants were raised at a frequency of 10−8, and sequencing of the qcrB gene revealed single-nucleotide polymorphisms (SNPs) leading to 7 different amino acid mutations that all mapped to the stigmatellin binding site of the bc1 complex (9) (Table 2).
We identified four other diverse chemotypes that exhibited the same time-dependent inability to maintain growth inhibition of M. tuberculosis H37Rv while maintaining the ability to inhibit resazurin reduction (Table 1) and rapidly inhibit ATP production with kinetics similar to those of BDQ and the protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) (see Fig. S2 in the supplemental material). The cydKO mutant was hypersusceptible to these compounds, whereas the qcrB mutants in the genetic cydKO background were resistant to them; mutations at Met342 did not confer resistance to compound 5, suggesting that this compound does not interact with this residue. Compound 1 is an imidazo[4,5-c]pyridine that was previously reported to have submicromolar potency against M. tuberculosis (10). The ability of all these scaffolds to inhibit the mycobacterial bc1 complex raised the concern that they might similarly inhibit mitochondrial respiration, which can lead to deleterious side effects during chemotherapy in vivo. Therefore, we conducted cytotoxicity testing against HepG2 cells using an assay which forces cells to use mitochondrial respiration by growth in galactose (11). It was found that compounds 3, 4, and 5 inhibited mitochondrial respiration (see Table S2 in the supplemental material), further reinforcing the notion that these compounds affect respiratory cytochrome function. A homology model of QcrB was constructed with multiple cytochrome b homologues (PDB no. 3CX5 [yeast], 1NTM [bovine], 1ZRT [Rhodobacter capsulatus], 2QJP [Rhodobacter sphaeroides], and 3H1H [chicken]) using MP-T (12) and MODELLER (13). The mutated residues were found to lie in close proximity in the pocket that binds stigmatellin, a known inhibitor in other species, suggesting a mechanism analogous to stigmatellin involving disruption of electron transport to the iron-sulfur protein. Although part of the pocket lies in a region of significant divergence in M. tuberculosis, it was nevertheless possible to dock the compounds in a plausible binding mode consistent with the mutational results using the software GOLD (14), allowing an elastic distance potential to the mutant residues (Fig. 2).
FIG 2.

Modeling of the imidazopyridine binding pocket of QcrB showing docking poses for the two scaffolds. (A) Docking of imidazo[1,2-α]pyridine (compound 2) in the QcrB homology model; (B) docking of imidazo[4,5-c]pyridine (compound 1) in QcrB; (C) docking of compound 3; (D) docking of compound 4. Compounds are shown in magenta, iron-sulfur protein in gray, and QcrB in rainbow spectrum; resistance-conferring residues are labeled and shown in stick representation.
The discovery of five distinct scaffolds that all target the M. tuberculosis bc1 complex suggests that this membrane target is promiscuous, as reported for several other membrane-associated targets (15). The hypersusceptibility of the cydKO mutant, in combination with the discordance between growth inhibition and resazurin reduction, offers a means of identifying such inhibitors early in the drug discovery process.
Since the discovery of BDQ, there has been significant enthusiasm for new targets that might disrupt energy metabolism, which is now perceived as a metabolic vulnerability in nonreplicating M. tuberculosis (16). Our results suggest that the pathways involved in respiration and energy metabolism show significant metabolic plasticity and can be altered during long-term laboratory adaption of clinical isolates. Laboratory passage of M. tuberculosis strains has likely resulted in many adaptations to in vitro growth over time (17). In this instance, the tight control of the cydAB genes in strain H37Rv seems to have been lost, allowing a buffered response to the reduced functioning of the bc1 complex. Significantly, our results show that M. tuberculosis has the respiratory flexibility to not only survive but also actually grow using the alternative cytochrome bd oxidase complex during chemical inhibition of the bc1 complex, consistent with observations in Mycobacterium smegmatis (18). Our understanding of the regulation of the respiratory network in M. tuberculosis is minimal, although it is known that the cytochrome bd oxidase complex is upregulated under hypoxic conditions (19). These results argue for caution when assuming that an effect observed on aerobic organisms (such as those found in sputum) will translate into an effect on microaerophilic organisms (such as those found in lesions) and further highlight the risk of targeting respiration in drug development without understanding the relative contribution of the respiratory complexes in human granuloma.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded, in part, by the Intramural Research Program of the NIAID and by grants from the Foundation for the National Institutes of Health with support from the Bill & Melinda Gates Foundation (to C.E.B.) and the South African Medical Research Council (to V.M.).
We thank Gail Louw for assistance with reverse transcriptase quantitative PCR.
Footnotes
Published ahead of print 25 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03486-14.
REFERENCES
- 1.Mahajan R. 2013. Bedaquiline: first FDA-approved tuberculosis drug in 40 years. Int. J. Appl. Basic Med. Res. 3:1–2. 10.4103/2229-516X.112228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 3.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim SA, Nam J, Kang H, Kwon H, Oh CT, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SP, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han SJ, No Z, Lee J, Brodin P, Cho SN, Nam K, Kim J. 2013. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19:1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 4.Moraski GC, Markley LD, Hipskind PA, Boshoff H, Cho S, Franzblau SG, Miller MJ. 2011. Advent of imidazo[1,2-a]pyridine-3-carboxamides with potent multi- and extended drug resistant antituberculosis activity. ACS Med. Chem. Lett. 2:466–470. 10.1021/ml200036r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abrahams KA, Cox JA, Spivey VL, Loman NJ, Pallen MJ, Constantinidou C, Fernandez R, Alemparte C, Remuinan MJ, Barros D, Ballell L, Besra GS. 2012. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS One 7:e52951. 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mak PA, Rao SP, Ping Tan M, Lin X, Chyba J, Tay J, Ng SH, Tan BH, Cherian J, Duraiswamy J, Bifani P, Lim V, Lee BH, Ling Ma N, Beer D, Thayalan P, Kuhen K, Chatterjee A, Supek F, Glynne R, Zheng J, Boshoff HI, Barry CE, III, Dick T, Pethe K, Camacho LR. 2012. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 7:1190–1197. 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moraski GC, Markley LD, Chang M, Cho S, Franzblau SG, Hwang CH, Boshoff H, Miller MJ. 2012. Generation and exploration of new classes of antitubercular agents: the optimization of oxazolines, oxazoles, thiazolines, thiazoles to imidazo[1,2-a]pyridines and isomeric 5,6-fused scaffolds. Bioorg. Med. Chem. 20:2214–2220. 10.1016/j.bmc.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moraski GC, Markley LD, Cramer J, Hipskind PA, Boshoff H, Bailey M, Alling T, Ollinger J, Parish T, Miller MJ. 2013. Advancement of imidazo[1,2-a]pyridines with improved pharmacokinetics and nanomolar activity against Mycobacterium tuberculosis. ACS Med. Chem. Lett. 4:675–679. 10.1021/ml400088y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esser L, Quinn B, Li YF, Zhang M, Elberry M, Yu L, Yu CA, Xia D. 2004. Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for the cytochrome bc(1) complex. J. Mol. Biol. 341:281–302. 10.1016/j.jmb.2004.05.065. [DOI] [PubMed] [Google Scholar]
- 10.Khoje AD, Charnock C, Wan B, Franzblau S, Gundersen LL. 2011. Synthesis and antimycobacterial activities of non-purine analogs of 6-aryl-9-benzylpurines: imidazopyridines, pyrrolopyridines, benzimidazoles, and indoles. Bioorg. Med. Chem. 19:3483–3491. 10.1016/j.bmc.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 11.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. 2007. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 97:539–547. 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 12.Hill JR, Deane CM. 2013. MP-T: improving membrane protein alignment for structure prediction. Bioinformatics 29:54–61. 10.1093/bioinformatics/bts640. [DOI] [PubMed] [Google Scholar]
- 13.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 14.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. 2003. Improved protein-ligand docking using GOLD. Proteins 52:609–623. 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 15.Goldman RC. 2013. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis? Tuberculosis 93:569–588. 10.1016/j.tube.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Rao SP, Alonso S, Rand L, Dick T, Pethe K. 2008. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 105:11945–11950. 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domenech P, Rog A, Moolji JU, Radomski N, Fallow A, Leon-Solis L, Bowes J, Behr MA, Reed MB. 2014. Origins of a 350-kilobase genomic duplication in Mycobacterium tuberculosis and its impact on virulence. Infect. Immun. 82:2902–2912. 10.1128/IAI.01791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, Dawes SS, Rubin H, Coppel RL, Mizrahi V. 2005. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 187:6300–6308. 10.1128/JB.187.18.6300-6308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE., III 2004. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 279:40174–40184. 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.