

Induced pluripotent stem cells (iPSCs) represent a paradigm shift for central nervous system (CNS) disease modeling. Efforts in the field to develop more biologically relevant CNS disease models to provide screening assays useful for the pharmaceutical industry are described. Careful and strategic planning and shared resources will enable exponential advances in the field, leading to more sensitive and accurate screens for early diagnosis and allow the identification of patient-specific therapies.
Keywords: Central nervous system disease, iPSC, Cell-based screening, Genetic engineering
Abstract
Induced pluripotent stem cells (iPSCs) offer an opportunity to delve into the mechanisms underlying development while also affording the potential to take advantage of a number of naturally occurring mutations that contribute to either disease susceptibility or resistance. Just as with any new field, several models of screening are being explored, and innovators are working on the most efficient methods to overcome the inherent limitations of primary cell screens using iPSCs. In the present review, we provide a background regarding why iPSCs represent a paradigm shift for central nervous system (CNS) disease modeling. We describe the efforts in the field to develop more biologically relevant CNS disease models, which should provide screening assays useful for the pharmaceutical industry. We also provide some examples of successful uses for iPSC-based screens and suggest that additional development could revolutionize the field of drug discovery. The development and implementation of these advanced iPSC-based screens will create a more efficient disease-specific process underpinned by the biological mechanism in a patient- and disease-specific manner rather than by trial-and-error. Moreover, with careful and strategic planning, shared resources can be developed that will enable exponential advances in the field. This will undoubtedly lead to more sensitive and accurate screens for early diagnosis and allow the identification of patient-specific therapies, thus, paving the way to personalized medicine.
Introduction
The development of the central nervous system (CNS) occurs over a prolonged period. It starts from early embryonic development when the epiblast segregates to form the neuroectoderm. The neuroectoderm undergoes additional development to form the CNS and the peripheral nervous system (PNS). The neural tube, destined to form the CNS, undergoes classic morphogenetic movements. Rostrocaudal and dorsoventral specification gives rise to the forebrain (rostrally) and the spinal cord (caudally). The PNS is generated from the placodes in the skin and the neural crest stem cells. These cells migrate to give rise to sensory, enteric, and sympathetic neurons. They also support glia and a host of non-neural derivatives, including melanocytes and craniofascial mesenchyme [1–3].
Neural stem cells (NSCs) are the earliest born cells. They are often termed “neuroepithelial stem cells.” They first give rise to radial glial cells, which provide the scaffold on which subsequently born neurons migrate to reach appropriate positions in the developing brain. Oligodendrocyte precursors are born next, followed by astrocytes, which are born at or around the time of birth, followed by maturation of oligodendrocytes. During the first decade of life, oligodendrocytes myelinate the developing axons. During this period, the final number of neurons becomes specified, connections are pruned, and the adult structures are established. Although synaptic connections are dynamic, the total number of neurons remains relatively static throughout life. Astrocytes, in general, do not proliferate, unless they are responding to injury. Oligodendrocyte precursors persist in the adult brain and replenish the myelination of new connections that neurons might make. Overall, stem cells do not play a major role in repairing the adult brain. Estimates of NSC numbers suggest that they represent a very small fraction of the total number of cells present in the brain. Therefore, repair in the brain is believed to be induced by a glial response, which includes scarring and associated synaptic reorganization to achieve functional restoration, with oligodendrocytes, perhaps, myelinating the new connections [4].
Many of these processes have been modeled using animal models or in vitro culture systems. However, certain processes have clearly been difficult to model. For example, modeling the epithelial to mesenchymal transition that generates neural crest or modeling the neural tube folding defects that underlie several developmental abnormalities, such as spina bifida, are difficult to achieve with current in vitro technology. More success has been achieved by characterizing the properties of individual cells, modeling cell-to-cell interactions, and modeling some complex cellular interactions in a dish. Some of these successes include neuron-astrocyte interactions, in vitro models of the blood-brain barrier, synaptogenesis, and simplified models of learning and memory [5–8].
However, progress in modeling CNS diseases has been hampered by two important facts. First, acquiring human neural cells is difficult, because to accomplish this would require invasive surgery. Second, obtaining sufficient numbers of neurons, which do not proliferate from adult or cadaveric samples, has been limited for most applications. Given that neurons are postmitotic and rarely form tumors (with the possible exception of retinoblastomas and a subclass of medulloblastomas), this severely limits the sheer quantity of neurons available for investigation, even from tumor samples. Furthermore, glial tumors, which are relatively more abundant, appear to be irreversibly altered when propagated under standard culture conditions [9]. Much of our work on neuronal biology has used neuroblastomas from the PNS and pheochromocytomas of chromaffin cells, which are a non-neural endocrine derivative of the neural crest. In addition to the limitation of obtaining human neural cells in sufficient numbers, researchers have faced the uncomfortable realization that complex behavior simply cannot be modeled well in other species. Even more disconcerting is the species to species variability and the realization that even when the final output of a model seemed identical, different species can have variable responses to the exact same insult, leading to developing cures for one animal species (e.g., a mouse) that do not translate into cure for humans. Such has been the case with the knockout of SOD1 in mice to model familial amyotrophic lateral sclerosis, or the loss of c-ret signaling in modeling enteric disease. Thus, different species could respond to the same insult differently, and screening in animals could lead to a cure for the animal but not work for humans. Studies in mice have also led to the realization that the same gene knockouts can have a different phenotypic effect, depending on the genetic background of the rodent. Although this can be addressed by examining the phenotype in different rodent strains, it presents the challenge of how to assess the genetic background in human cells. For instance, how many different phenotypic cell lines should be examined to conclude that the results can be generalized to treat human patients with diverse genetic phenotypes? This second hurdle could be overcome with comparative data on patient cells to identify the patient characteristics that could be used to form subgroups and quantify the allelic variability and other attributes that will enhance the utility of the proposed therapy for a specified group of patients. However, this requires developing panels of lines that have been difficult to culture thus far.
Do iPSC-Derived Cells Resolve Some of These Issues?
iPSCs as a source of differentiated cells offer numerous possibilities and resolve some of the constraints with current non-iPSC based models. iPSCs are reprogrammed stem cells that are akin to embryonic stem cells (ESCs), with the advantage of being able to be generated from any individual at any point. This offers the advantage of modeling diseases across a lifetime in a patient-specific manner [10, 11]. Just as with their ESC counterparts, iPSCs can be maintained indefinitely, thus providing a limitless supply of well-characterized cells of a defined allelic phenotype. Equally important, these cells can be further differentiated into any cell type (or cocultures consisting of multiple cell types) and assessed to identify aberrant processes that can be targeted for the amelioration of diseases (Fig. 1). This offers the advantage of examining the effect of a perturbation on isogenic-differentiated cell populations. This was not feasible when we were limited to examining cell lines or even primary cells from rodents, other species, or even humans. Likewise, minimizing the confounding effect of allelic variability, as well as harnessing allelic variability, to understand the effect of modulators on primary gene disorders can be achieved using iPSCs from both diseased individuals and their closely related normal relatives [12, 13].
Figure 1.

Human iPSC-derived neural cells. Schematic summary of different types of neural cells that can be derived from human ESC/iPSC lines. The applications of iPSC-derived neural cells are listed in the square box on the right. Abbreviations: ESC, embryonic stem cell; iPSC, induced pluripotent stem cell; NSC, neural stem cell; OLIGO, oligodendrocyte; PNS, peripheral nervous system; Tox, toxicity.
Engineering iPSCs offers another unique advantage. iPSCs can be engineered using multiple methods to investigate how genetic alterations modulate physiological and disease processes. These engineered tools can be further applied in disease-pertinent cellular lineages and in developing isogenic and reporter cell lines [14]. Another advantage is the ability to derive iPSCs from different cellular sources. iPSCs can be reprogrammed from patient-derived cell sources, including blood [15–18], skin fibroblasts [19, 20], hair follicles [21], dental tissue [22], urine [23], and even postmortem tissue [24–26]. Another unique consideration is that patient cellular sources can be collected across a lifetime to examine the effects of the environment and age on the differentiation ability over time.
These advantages allow for investigations that are now no longer limited by tissue availability and access to isogenic controls. Additionally, iPSCs can be differentiated into various cell types of the CNS, including neurons, astrocytes, and oligodendrocytes [27–29]. Each of these neural cell types has been suggested or used for drug discovery and toxicity tests [29–31]. Given that many protocols that generate these CNS cell types have been reported [32–34], this also provides the opportunity for investigation of multiple cell types to elucidate differences between cell autonomous and cell intrinsic effects. It is also relatively straightforward to share samples and obtain independent validations, given the unlimited nature of the cell. Collectively, these advantages, illustrated in Figure 2, generate optimism that the drug discovery process can become more efficient and productive.
Figure 2.

iPSCs offer a paradigm shift in central nervous system (CNS) disease modeling. iPSCs can be reprogrammed from a variety of tissue sources and differentiated into many different cell types that would be useful for modeling disease aspects in the CNS. For instance, differentiation into midbrain dopaminergic neurons would provide a cell type applicable for modeling Parkinson’s disease. These disease models and neurodevelopmental processes can be developed into screening assays to interrogate further for drug screening and gene profiling studies. These screening applications can be aided further by the development of iPSC panels of controls, patient-specific lines, isogenic lines, engineered lines, and reporter lines. Abbreviations: iPSC, induced pluripotent stem cell; NSC, neural stem cell.
Although iPSCs offers unique advantages to screening in the CNS field, it is important to remember that iPSCs are certainly not a universal panacea and, certainly, iPSCs will not offer a solution many diseases (Table 1) [35]. For instance, iPSC lines might not be able to be derived from some disorders that affect DNA repair, aging disorders, and disease that affects protein pathology, such as prion protein disease. Genomic imprinting, in which epigenetic modifications in the DNA (e.g., DNA methylation or histone modification) can influence gene expression without altering the genomic sequence, could also prove difficult to model using an iPSC-based approach. In addition, disorders of the mitochondrial genome could be difficult to address. Likewise, even after the successful generation of iPSC lines, an unmet need still exists to construct biologically relevant three-dimensional (3D) models that can integrate multiple cell types. Similarly, achieving appropriate biological maturity to manifest late-stage disease phenotypes in a cell-based model to interrogate neurological diseases needs further development, although some early success has been reported [36]. Also, recapitulating in vivo interactions in a dish that have enabled us to model highly heterogeneous diseases could prove challenging. In addition, in vivo models in which human cells are placed in a relevant animal model could be difficult to generate, because we still have many limitations with developing humanized immune systems in rodents and viable immunosuppression models for xenotransplants. Finally, some common challenges in the reprogramming field need to be considered when using iPSC-based models. These include the heterogeneity of the reprogramming methods and efficiency, because they could lead to alterations in the iPSCs, which would make reproducibility difficult.
Table 1.
Limitations of iPSC-based models
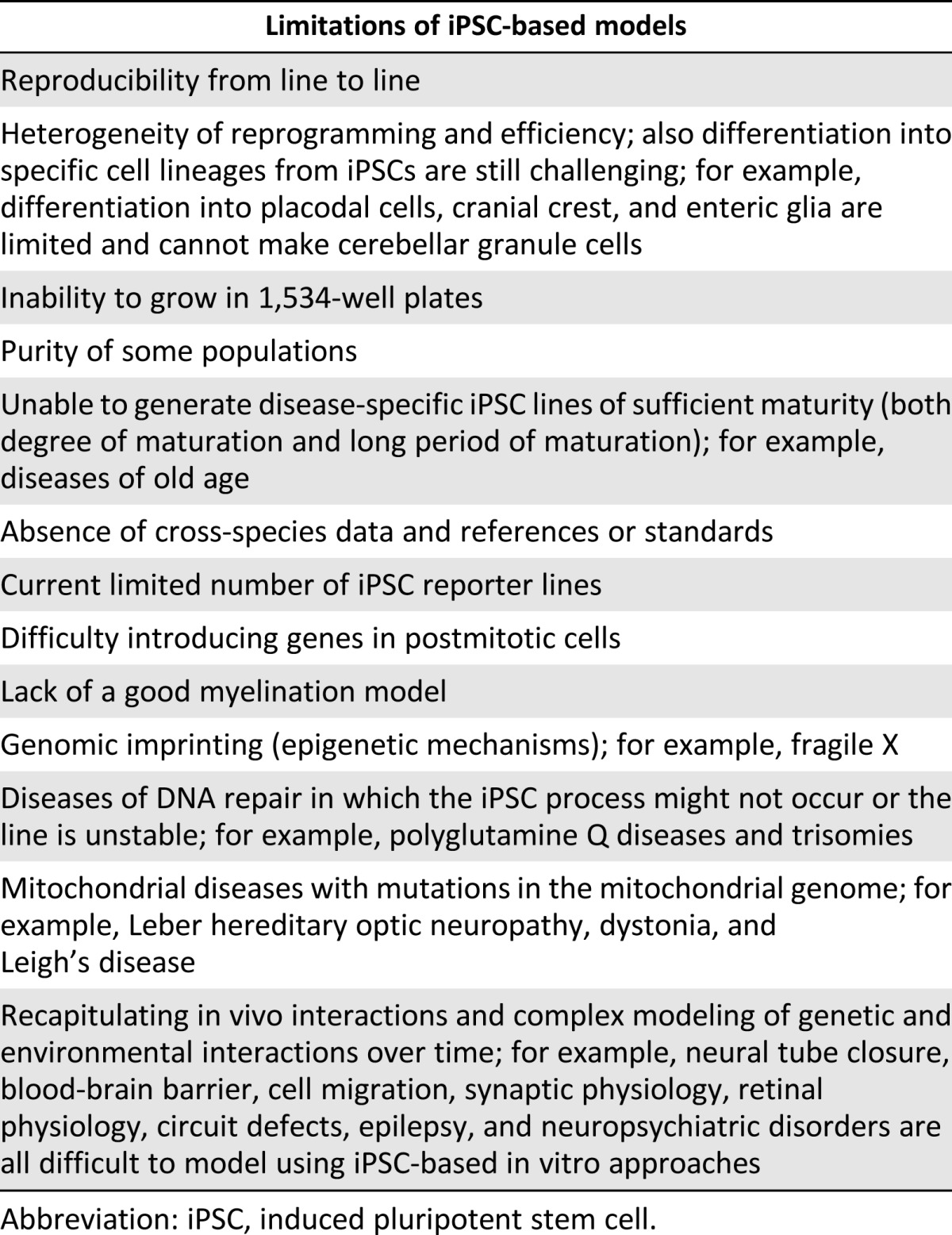
Overall, however, iPSCs clearly represent a radical advance in our capability of developing many simple models. These models are already proving useful for a field that has been severely limited by access to human cells. Nevertheless, significant challenges to expand the scope and utility of iPSCs remain, and these represent technical challenges for investigators to overcome.
Successes in Using iPSC-Based Models
Neurotoxic Developmental Assays and Survival Assays
Current toxicology studies do not effectively predict how a diverse population will react to drugs, pollutants, and other environmental chemicals. Moreover, these studies are expensive and time-consuming and often require a large number of animals for testing. Consequently, only a small number of chemicals have been evaluated using these methods. Current tests also provide little information on the modes and mechanisms of action, which are critical for understanding interspecies differences in toxicity. Furthermore, little to no information is available for assessing the variability in human susceptibility. In vitro mechanistic tests using human cells can provide rapid evaluations of a large number of chemicals, greatly reducing the need for live-animal use and might provide results potentially more relevant to human biology and human exposures. A fundamental understanding of the cellular responses to toxicants, combined with knowledge of tissue dosimetry in cell systems and in exposed human populations, will provide a suite of tools to permit more accurate predictions of the conditions under which humans can be expected to show pathway perturbations from toxicant exposure.
In collaboration with the National Institute of Environmental Health Sciences, we undertook a demonstration screen of 80 compounds (mostly developmental neurotoxicants) to demonstrate the feasibility of such an iPSC-based approach. We obtained cells in sufficient numbers and purity to rapidly evaluate the drug response using a 96-well format. Because the cells can be maintained in culture for prolonged periods, both viability assays and high content screening (HCS) can be performed. The effect of a single compound could be examined using cell viability assays (MTT- and/or ATP-based assays) on isogenic iPSC and iPSC-derived homogeneous neural populations (NSCs, neurons, and astrocytes). The 80 compounds used in our study produced specific cellular responses according to cell type and species, a critical step in validating this approach and developing it further to screen more compounds and for optimization for higher well formats to enable 384- and 1,536-based neurotoxicity assays.
Dissecting the contributions of enhancers and modulators on allelic diversity can now be ascertained. Well-characterized iPSC lines can be differentiated using identical differentiation protocols in human cell lines. Such efforts, thus far, have only been possible in rodents and have required a tremendous investment in developing inbred lines and diversity panels. We now have a model system in which rodent and human cell lines can be run in parallel.
The same strategy can be used to identify the mechanism of toxicity and the differential specificity of cellular responses to the same toxic agent. Our laboratories have optimized methods for feeder-free culture of pluripotent stem cells (PSCs) and PSC-derived NSCs to facilitate automated screening [37, 38]. With this optimized differentiation platform, we screened a collection of Food and Drug Administration (FDA)-approved drugs (∼1,000 compounds) to identify compounds that have differential toxicity to PSCs and their neural derivatives [37]. Using comparative screening of PSCs and PSC-derived homogenous NSCs, we were able to identify compounds that had differential toxicity to both cell populations. “Hits” obtained in the primary screen were then retested, and a small subset was assayed for dose-responsiveness. One confirmed dose-responsive compound, amiodarone, was further tested for toxicity in postmitotic neurons. We found amiodarone to be toxic to NSCs but not to postmitotic neurons. Using time-lapsed imaging, we were able to show that the cells died extremely rapidly within less than 4 hours and did so with little change in gene expression. Thus, the mechanism of death was likely osmotic changes resulting from blocking of a channel transporter [37].
Such rapid analysis with off-the-shelf human neural cells offers an unprecedented opportunity to understand neurotoxicity [39–41] in the developing nervous system. In addition, by combining this with HCS, one can rapidly dissect out the mechanism of action of the compound.
Modeling Monogenic CNS Disorders With iPSCs
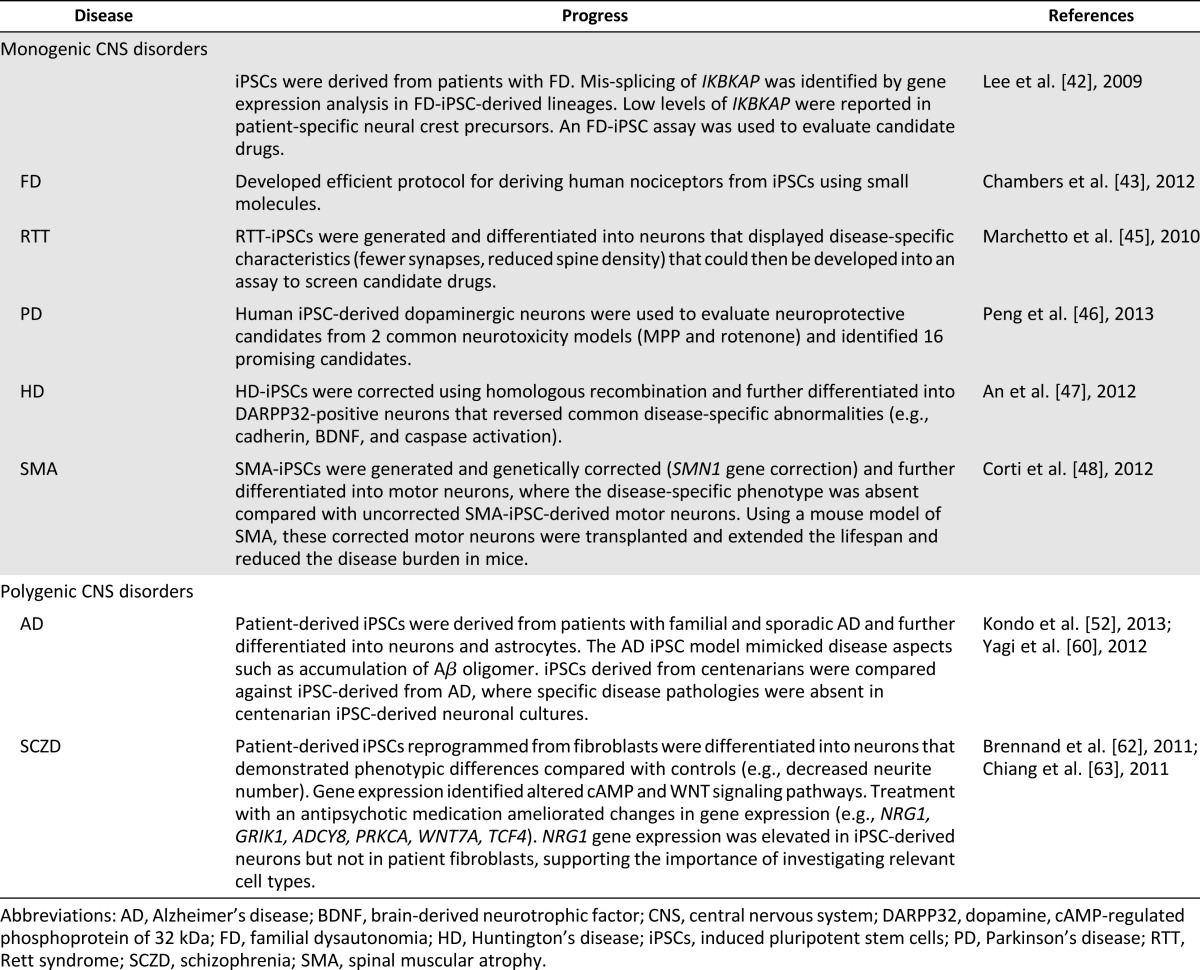
Modeling hereditary CNS disorders represents a unique advantage in which screens can be developed that takes advantage of the known genes that have been implicated in that particular disease and correlate it with a function that can be dissected out in vitro. By combining data from familial mutant lines with isogenic controls, in which the same gene is knocked out in a defined and well-characterized line, one can dissect out the role of the gene at high resolution. Testing the hypothesis in lines generated from patients who do not carry the mutation but display the same phenotype or introducing additional mutations will allow one to develop models of how genes interact. Such models were possible, although difficult, in mice but are now readily developed in human iPSCs because of the availability of lines and the advances in gene engineering technologies (Figs. 3, 4). We provide five examples (below) in which single genes can be corrected, knocked out, and engineered to develop high-throughput screens for drug discovery or to interrogate disease mechanisms (Table 2).
Figure 3.

Lineage-specific reporter line generation by zinc-finger nuclease (ZFN)-mediated gene targeting. The process for creating knock-in induced pluripotent stem cell (iPSC) lines by ZFN technology is illustrated. ZFN mRNAs and donor vector are inserted into the iPSC via nucleofection, and iPSCs that were successfully integrated were selected for via drug selection. An example of a donor vector with a reporter and a selection marker strategy is shown. The surviving clones (∼10–50 clones picked) are expanded for 2-4 weeks, with genomic DNA used to screen and confirm for mutations. The targeted iPSC lines are then assayed for pluripotency and genomic stability and other characterization desired. Abbreviations: 2A, 2A peptide; d, day; gDNA, genomic DNA; GFAP, Glial fibrillary acidic protein; LA, left homologous recombination arm; Neo, Neomycin resistant gene; PGK, phosphoglycerate kinase promoter; Puro, puromycin resistant gene; RA, right homologous recombination arm.
Figure 4.

Genetic engineering of induced pluripotent stem cells (iPSCs) by zinc-finger nuclease (ZFN)-mediated gene targeting. The process for creating knock-out iPSC lines by ZFN technology is illustrated. ZFN mRNAs are inserted into the iPSCs via nucleofection, and iPSCs that are successfully integrated are screened via polymerase chain reaction and sequencing. The selected clones are expanded for 2-4 weeks, and genomic DNA is used to confirm for heterozygote or homozygote. More than one round of nucleofection and screening can be required to generate homozygotes. The targeted iPSC lines are then assayed for pluripotency and genomic stability and other characterization desired. Abbreviations: d, day; gDNA, genomic DNA.
Table 2.
Successful modeling of CNS disorders with iPSC-based approaches
Modeling Familial Dysautonomia With iPSCs
Familial dysautonomia (FD) or hereditary sensory and autonomic neuropathy III (Riley-Day syndrome) affects the autonomic nervous system and is due to a point mutation in the gene for IκB kinase complex-associated protein (IKBKAP). This point mutation causes a tissue-specific splicing deficit in peripheral neurons, reduced levels of IKAP protein, and altered cell motility. Investigators derived iPSC lines from patients with FD, a fatal autosomal recessive disease, to model this disease in a tissue- and patient-specific manner that was further developed into assays to probe for candidate drugs [42].
The investigators were able to differentiate these FD-iPSC lines into all three germ layers and confirm the low levels of normal IKBKAP transcript in FD-iPSC derived neural crest precursors. In patients with FD, autonomic and sensory neurons have been lost; however, the exact mechanisms remain elusive, and, currently, no animal models are available to investigate FD disease pathology. These FD-iPSC models identified deficits in IKBKAP splicing and showed a reduced ability of FD-iPSC derived neural crest precursors to undergo neuronal differentiation and decreased migration in FD-iPSCs compared with control iPSC-derived neural crest precursors using the wound healing assay [42]. In turn, these models identified a candidate drug, kinetin, a plant hormone that promotes cell division. Acute treatment with this plant hormone was able to reduce the mutant IKBKAP splice form and increase normal IKBKAP levels. Chronic treatment increased the rate of neurogenesis and peripheral neuron markers but did not have significant effects on FD-iPSC neural crest precursor cell migration.
In addition to interrogating disease mechanisms and developing disease- and cell type-specific assays for novel drug discovery for the treatment of FD, progress has been made in differentiating neural crest stem cells into a specific type of sensory neuron, nociceptors. Chambers et al. have succeeded in directing differentiation from human PSCs to nociceptors using a cocktail of small molecules [43]. This has opened the door for investigating the transduction of pain mechanisms in a clinically relevant cell type.
Modeling Rett Syndrome With iPSCs
Rett syndrome (RTT) is a neurodevelopmental disorder due to a mutation in the X-linked gene encoding methyl-CpG-binding protein 2 [44]. Marchetto et al. recently developed a human model of RTT using an iPSC-based approach [45]. They generated iPSCs from fibroblasts taken from patients with RTT and controls. They then differentiated these iPSCs into neurons and found many disease characteristics. These included RTT-iPSC-derived neurons with fewer dendritic spines, fewer synapses, a decreased soma size, altered calcium signaling, and electrophysiological defects compared with control iPSC-derived neurons. These disease-specific characteristics were then used to test candidate drugs that would restore these deficits and altered responses toward the control levels. They found that insulin-like growth factor 1 increased the glutamatergic synapse number in treated RTT-derived neurons. Future studies should validate these disease specific deficits using high-throughput screens to identify the most robust models to be used for novel drug discovery.
Modeling Parkinson’s Disease With iPSCs
Parkinson’s disease (PD) is a neurodegenerative disorder primarily targeting dopaminergic neurons in which a specific brain region, the substantia nigra, is affected. Modeling this disease requires, first, generating iPSC-derived dopaminergic neurons and, second, challenging these cells with known toxins already developed in human and animal models to elucidate the mechanisms underlying the demise of these dopaminergic neurons. In a recent study, Peng et al. reported a screening platform with assays suitable for automated readout using primary dopaminergic neurons derived from PSCs and ran a small screen of compounds for neuroprotective effects in two toxic models (1-methyl-4-phenylpyridinium [MPP+]- and rotenone-induced toxicity) [46]. The investigators chose only known compounds that have been reported for neuroprotection in human immortalized cell lines, rodent primary cells, or in vivo in rodent cells. A positive feature of this strategy is that these compounds have been suggested to act via different protective mechanisms, making validating the mechanism of action easier. Of the 44 compounds screened, the investigators found only 18 were neuroprotective in human primary dopaminergic neurons, although all were reportedly protective in rodent or cell line models. Equally important, the compounds identified as effective were mostly those that have been used in human trials. These results clearly show that screens with primary cells derived from PSCs offer advantages compared with screens run in immortalized cell lines and those tested on rodent cells. Although it is difficult to run high-throughput screens with iPSCs, the investigators showed the assays can be used to study the mechanism of action and possible synergic effects of combinations of drugs. In addition, these assays allow investigators to evaluate the long-term effects of exposure to a particular treatment or drug, because the cells can be maintained in culture for prolonged periods.
Modeling Huntington’s Disease With iPSCs
Huntington’s disease (HD) is a neurodegenerative disease caused by CAG expansion that leads to a host of symptoms, including chorea, cognitive decline, and neuropsychiatric abnormalities. Researchers have generated HD-iPSCs and corrected HD-iPSCs using homologous recombination [47]. That report showed that the correction persisted in iPSC-derived DARPP-32 positive neurons both in vitro and in vivo. Additionally, the corrected HD-iPSCs reversed the abnormalities in the common HD signaling pathways (e.g., cadherin, BDNF, and caspase activation) and disease-specific phenotypic abnormalities. A relevant disease model with identical genetic backgrounds (isogenic controls) is a critical step to advancing new therapies and will pave the way for personalized medicine.
Modeling Spinal Muscular Atrophy Using iPSCs
Spinal muscular atrophy (SMA) is an autosomal recessive disease that has a defect in the SMN1 gene that leads to a loss of motor neurons. Because the SMN1 gene is mutated in SMA-affected individuals, correction of this deletion, occurring at exon 7, or other point mutations could provide a unique model system for investigating the SMA disease mechanisms using an iPSC-based model. A recent study generated iPSCs from skin fibroblasts from patients with SMA and genetically corrected these iPSCs [48]. The motor neurons differentiated from uncorrected SMA-iPSCs showed a disease-specific phenotype that was lost in the motor neurons derived from the corrected SMA-iPSCs. Moreover, in a mouse model of SMA, transplantation of these corrected motor neurons derived from SMA-iPSCs extended the life span and reduced the disease burden in the mice. If similar studies can be done in humans, this would suggest a clear therapeutic intervention for using corrected motor neurons derived from SMA-iPSCs.
Summary
As is clear from these examples, the general approach has been to (a) develop an assay using generic iPSC lines that are well characterized, (b) determine whether a phenotype can be obtained in vitro using patient-specific lines, and (c) assay compounds using standard viability, toxicity, survival, or functional assays as a readout and couple them with HCS to understand the mechanism of action. This process could provide a unique advantage for orphan and rare diseases for which tissue samples have often been rate limiting. With iPSC technology, this is no longer true. Standardized lines and the ability to engineer isogenic controls offer an opportunity for new investigators to enter this field. Several elegant results have been obtained by simply performing a drug-repurposing screen using already approved FDA compounds as a screening library. Such a screen at National Center for Advancing Translational Sciences led to the identification of δ-tocopherol and cyclodextrin as potential treatment of Niemann-Pick type C and identified six new classes of compounds for the treatment of Gaucher’s disease [49, 50].
Hasson et al. at the NIH were able to use siRNA libraries to identify key interacting proteins that might be important in the pathology of PD [51]. We believe iPSC-based screening of rare CNS diseases might be able to identify common mechanisms of action that contribute to multiple rare diseases that, collectively, are predicted to affect millions of Americans each year. Another benefit is that by identifying these mechanisms of action, it could facilitate the development of new therapeutic agents that might have wider uses and thus could attract more commercial attention.
Modeling Polygenic CNS Disorders With iPSCs
Although monogenic disorders represent the most obvious screening choice for iPSC-based research, the ability to obtain large panels of lines and develop complex culture assays suggest that iPSC technology could have some utility in our understanding of polygenic diseases and other complex disorders. This concept involves investigating disease-resistant and -susceptible mechanisms in disease-relevant cell types that will enable the discovery of novel therapies. Several investigators have shown promising results, and in the section below we provide some specific examples (Table 2).
Modeling Alzheimer’s Disease With iPSCs
In a recent study, iPSCs were generated from patients with familial and sporadic Alzheimer’s disease (AD) and were further differentiated into neurons and astrocytes [52]. One hypothesis of the contributing factors to the pathophysiology of AD is the accumulation of oligomeric forms of amyloid-β peptide (Aβ). In the AD iPSC-derived neurons and astrocytes, an accumulation of this Aβ oligomer was found, leading to endoplasmic reticulum (ER) and oxidative stress. Docosahexaenoic acid (DHA) treatment, an omega-3 fatty acid abundant in the CNS and a main constituent of the neuron’s plasma membrane, reduced the ER and oxidative stress response in AD-iPSC-derived neurons. Moreover, DHA is decreased in AD brains [53, 54], and decreased DHA serum content correlates with impaired cognitive ability [55–59]. Therefore, AD-iPSC-derived cells were able to model a particular aspect of the disease. This model can be further interrogated using drug screening to identify potential compounds to reduce ER and oxidative stress.
In addition to investigating the disease mechanisms of AD that go awry, another modeling approach is to investigate the mechanisms underlying healthy controls. An extreme example of this is the development of iPSC models from centenarians. This was done and compared with iPSC-derived neurons from those with familial Alzheimer’s disease and familial Parkinson’s disease [60]. Notably, disease-specific pathologic features were absent in the centenarian iPSC-derived neuronal cultures.
Modeling Schizophrenia With iPSCs
Schizophrenia (SCZD) is a neurodevelopmental disorder characterized by hallucinations, delusions, and cognitive deficits. It also has a high degree of heritability, with some estimates as high as 80% [61]. Given that SCZD is highly heritable, we anticipate that generating panels of iPSC lines according to subgrouping by gene expression or epigenetic patterns will lead to identifying interventions that are effective for the specific subgroups. This will enable clinicians to truly practice personalized medicine in a complex heterogeneous disease such as SCZD.
Modeling cellular characteristics of SCZD using iPSCs that were differentiated into neurons has been achieved [62]. Brennand et al. reprogrammed fibroblasts to generate iPSC lines that were then differentiated into neurons (mostly glutamatergic, ∼30% GABAergic and ∼10% dopaminergic) for both controls (n = 6) and SCZD (n = 4). Although this was only a small sample size, their study does present some interesting findings. First, SCZD neurons demonstrated phenotypic differences compared with controls (e.g., decreased neurite number, decreased neuronal connectivity, and decreased synaptic protein), and, at the same time, showed similar electrophysiological responses compared with the control neurons. Second, gene expression differences were also apparent where cAMP and WNT signaling pathway genes were altered in SCZD neurons. Third, treatment with an antipsychotic drug (loxapine) ameliorated the changes in gene expression (e.g., NRG1, GRIK1, ADCY8, PRKCA, WNT7A, TCF4). Finally, NGR1 gene expression was elevated in SCZD neurons compared with the controls and was specific for the relevant cell type, because no differences were found between the SCZD and control fibroblasts.
It is also feasible now to generate integration-free SCZD iPSC lines from skin biopsies [63]. This advent permits the generation of more consistent iPSC lines that are not plagued with the potential of foreign DNA integration, which can disrupt the host’s genome and lead to unintended consequences (e.g., reactivation of oncogenes). Moreover, iPSCs can also be generated using human postmortem tissue (e.g., dura mater and scalp) to make use of postmortem human samples from large cohorts of well-characterized subjects stored in brain banks.
Other Innovative Strategies Using iPSC-Based Screens
This section provides additional examples for other innovative strategies using iPSC-based screens. These innovative strategies include modeling neurotrauma, modeling CNS infections, screening prospectively to personalize therapy, complementing adverse event screening during clinical trials, developing diversity panels, conducting genome-wide association study (GWAS) screens, and developing shared resources. Collectively, these innovative strategies can be used to create more advanced iPSC-based screens that are more sensitive and effective predictors for disease susceptibility and treatment.
Modeling Neurotrauma With iPSCs
To improve the treatment of patients and thereby decrease the associated mortality, morbidity, and cost, several in vivo models of CNS injury have been developed and characterized during the past two decades. To complement the ability of these in vivo models to reproduce the sequelae of human CNS injury, in vitro models of neuronal injury have also been developed [64]. Despite the inherent simplifications of these in vitro systems, many aspects of the post-traumatic sequelae, including primary, secondary, and tertiary responses to blast injuries, can be faithfully reproduced in cultured cells. The changes that have been monitored include ultrastructural changes, ionic derangements, alterations in electrophysiology, free radical generation, and alterations in gene expression.
Additional development of these models has been limited owing to the lack of availability of human cells and the ability to grow cells for prolonged periods. However, iPSCs and other pluripotent cells allow one to overcome this limitation, and many investigators have now begun to transfer their rodent in vitro models to in vivo models. Advances in 3D culture and the ability to develop myelination models has further improved the fidelity of the models. These models have also been extended to investigate radiation damage.
Modeling Infections in the CNS
Lafaille et al. engineered iPSCs from children with inborn errors of toll-like receptor 3 (TLR3) immunity, who are prone to herpes simplex virus 1 (HSV-1) encephalitis (HSE), and used these cells to pinpoint the underlying causes of the disease [65]. They illustrated that HSE involves nonhematopoietic CNS resident cells and that iPSC-derived neurons were far more susceptible than UNC93B-deficient patients and controls. These iPSC lines were differentiated into purified populations of NSCs, neurons, astrocytes, and oligodendrocytes. More importantly, the team showed that the induction of interferon-β (IFN-β) and/or IFN-λ1 in response to stimulation by the double-stranded RNA analog polyinosinic-polycytidylic acid [poly(I:C)] was dependent on TLR3 and UNC93B in all cells tested. However, the induction of IFN-β and IFN-λ1 in response to HSV-1 infection was impaired selectively in UNC93B-deficient neurons and oligodendrocytes. These cells were also much more susceptible to HSV-1 infection than were the control cells, although the UNC93B-deficient NSCs and astrocytes were not. TLR3-deficient neurons were also susceptible to HSV-1 infection. The rescue of UNC93B- and TLR3-deficient cells with the corresponding wild-type allele showed that the genetic defect was the cause of the poly(I:C) and HSV-1 phenotypes. The viral infection phenotype was rescued further by treatment with exogenous IFN-α or IFN-β (IFN-α/β), but not IFN-λ1. Thus, impaired TLR3- and UNC93B-dependent IFN-α/β intrinsic immunity to HSV-1 in the CNS, in the neurons and oligodendrocytes in particular, might underlie the pathogenesis of HSE in children with TLR3-pathway deficiencies.
Screening Prospectively to Personalize Therapy
Cancer biologists have suggested that tumor recurrence likely occurs because a rare tumor population escapes standard therapy. If the population was isolated and approved drugs were tested, perhaps a tailored optimized cocktail of drugs could be rapidly developed to administer to a patient in whom the therapy has failed. This strategy has shown some success and is a novel approach to screening with immediate benefits.
iPSC-based therapy to treat gliomas is gaining speed through the use of engineering iPSC-derived NSCs as vehicles to deliver anticancer gene therapy [66]. Other investigators have argued that the process of iPSC generation might mimic the changes that occur in cancer and that studying the process and drugs that affect the process could provide clues to cancer therapy.
Adverse Event Screening During Drug Trials
An important issue for the pharmaceutical industry has been the risk of an unexpected rare adverse event that will be missed as the drug proceeds through the standard clinical evaluation phase, necessitating a late recall. Drugs have been withdrawn despite their efficacy in a large population because of this risk to a rare population. iPSCs offer a solution to this problem. Patients with adverse events can be identified, and their iPSCs can be generated and used to extensively map the drug response and compare it to that of patients who had a beneficial effect. This sort of testing could allow one to salvage drugs as biomarkers and could be developed further to identify patients who should not be administered such a drug. Likewise, in most clinical trials, some patients will have a treatment response and others will not. Thus, comparing responders to nonresponders could provide novel insights and help stratify patients for therapy. No fundamental technical issue exists in performing such screens and the size of the screens is not large; thus, it is simply a matter of time and a decision to implement.
Developing Diversity Panels
One can imagine developing panels of lines that are genetically diverse and testing them routinely with any new approved drug to reduce the risk of unexpected events when a drug is widely used. Such a panel does not exist and would require a public-private partnership. However, it is certainly technically feasible, and we believe its benefits will far outweigh the cost associated with such an endeavor. Efforts to develop these lines are being initiated (Fig. 4), and, with careful attention to ensuring adequate diversity, we might soon have a panel of iPSC lines for such a purpose.
GWAS Type Screens
iPSCs have also offered a solution to the allelic variability issue illustrated in pharmacogenomics [67]. The problem is simply that humans are outbred, which means the allelic variability reflected in gene expression and variation in gene levels is in the same range as across species. This has been the basis of the reluctance in using primary cells, because we do not know how normal the response of any given line will be. With iPSCs, we can generate a large number of lines and return to the same allelic phenotype because of the intrinsic lack of senescence in the cells. Thus, we can achieve many of the advantages of an immortalized cell line without its disadvantages. Efforts along these lines have already begun. Disease-specific panels in which sporadic and familial cases from patients with the same disease are obtained using a standardized method will allow one to test hypotheses and drugs in a panel to identify unique and uniform responses and dissect stochastic differences from biologically relevant ones. Panels for PD, HD, and cardiac hypertrophy have been developed. More ambitious proposals have suggested that a similar panel-based approach can be successfully implemented for polygenic disorders. These proposals would include even larger polygenic disease panels in which screening is combined with whole genome, exome, and epigenome sequencing. This is an ambitious idea but, if successful, could completely change how we perform screens.
Developing Shared Resources
Too often resources are not developed to allow the field to advance rapidly. We suggest that the development of several shared resources will enable exponential advances in the field. These shared resources include (a) access to disease panels of iPSCs and their differentiated/engineered products, (b) access to common controls, (c) access to common patient data for selecting appropriate samples, and (d) sophisticated bioinformatic tools that can integrate patient data and cell assay data in a cloud-based format for global use. These resources, if developed and implemented strategically, will allow preventative medicine to be fully exploited such that disease-specific, cell-based screens can be personalized for each patient to, not only predict susceptibility to disease, but also to develop innovative therapies to cure that disease.
Conclusion
iPSC-based CNS disease modeling holds tremendous potential. The cells can be used in multiple ways. No doubt exists that additional screens such as those related to myelination, novel screens related to modeling the blood-brain barrier or neural folding, or entirely new modes of therapy will be developed. Our sense is that the current successes are only the tip of the iceberg. As investigators become more familiar with the system and the costs of such assays continue to decline, one will see even more innovation in this area of toxicology research.
Author Contributions
X.Z. and M.R.: conception and design, financial support, administrative support, collection and/or assembly of data, manuscript writing, final approval of manuscript; J.G.H.: conception and design, manuscript writing; A.S., N.M., and Y.P.: manuscript writing.
Disclosure of Potential Conflicts of Interest
X.Z. has uncompensated ownership interest in XCell Science Inc.
References
- 1.Galvin JE, Ginsberg SD. Expression profiling and pharmacotherapeutic development in the central nervous system. Alzheimer Dis Assoc Disord. 2004;18:264–269. [PubMed] [Google Scholar]
- 2.Hua JY, Smith SJ. Neural activity and the dynamics of central nervous system development. Nat Neurosci. 2004;7:327–332. doi: 10.1038/nn1218. [DOI] [PubMed] [Google Scholar]
- 3.Lewis MH. Environmental complexity and central nervous system development and function. Ment Retard Dev Disabil Res Rev. 2004;10:91–95. doi: 10.1002/mrdd.20017. [DOI] [PubMed] [Google Scholar]
- 4.Maier IC, Schwab ME. Sprouting, regeneration and circuit formation in the injured spinal cord: Factors and activity. Philos Trans R Soc Lond B Biol Sci. 2006;361:1611–1634. doi: 10.1098/rstb.2006.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ricci G, Volpi L, Pasquali L, et al. Astrocyte-neuron interactions in neurological disorders. J Biol Phys. 2009;35:317–336. doi: 10.1007/s10867-009-9157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mundy WR, Radio NM, Freudenrich TM. Neuronal models for evaluation of proliferation in vitro using high content screening. Toxicology. 2010;270:121–130. doi: 10.1016/j.tox.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Vernon H, Clark K, Bressler JP. In vitro models to study the blood brain barrier. Methods Mol Biol. 2011;758:153–168. doi: 10.1007/978-1-61779-170-3_10. [DOI] [PubMed] [Google Scholar]
- 8.Naik P, Cucullo L. In vitro blood-brain barrier models: Current and perspective technologies. J Pharm Sci. 2012;101:1337–1354. doi: 10.1002/jps.23022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noble M, Mayer-Pröschel M. Growth factors, glia and gliomas. J Neurooncol. 1997;35:193–209. doi: 10.1023/a:1005898228116. [DOI] [PubMed] [Google Scholar]
- 10.Laustriat D, Gide J, Peschanski M. Human pluripotent stem cells in drug discovery and predictive toxicology. Biochem Soc Trans. 2010;38:1051–1057. doi: 10.1042/BST0381051. [DOI] [PubMed] [Google Scholar]
- 11.Mohamet L, Miazga NJ, Ward CM. Familial Alzheimer’s disease modelling using induced pluripotent stem cell technology. World J Stem Cells. 2014;6:239–247. doi: 10.4252/wjsc.v6.i2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar KK, Aboud AA, Bowman AB. The potential of induced pluripotent stem cells as a translational model for neurotoxicological risk. Neurotoxicology. 2012;33:518–529. doi: 10.1016/j.neuro.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shtrichman R, Germanguz I, Itskovitz-Eldor J. Induced pluripotent stem cells (iPSCs) derived from different cell sources and their potential for regenerative and personalized medicine. Curr Mol Med. 2013;13:792–805. doi: 10.2174/1566524011313050010. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Rao M. Gene targeting in human pluripotent stem cells. Methods Mol Biol. 2011;767:355–367. doi: 10.1007/978-1-61779-201-4_26. [DOI] [PubMed] [Google Scholar]
- 15.Giorgetti A, Montserrat N, Rodriguez-Piza I, et al. Generation of induced pluripotent stem cells from human cord blood cells with only two factors: Oct4 and Sox2. Nat Protoc. 2010;5:811–820. doi: 10.1038/nprot.2010.16. [DOI] [PubMed] [Google Scholar]
- 16.Dowey SN, Huang X, Chou BK, et al. Generation of integration-free human induced pluripotent stem cells from postnatal blood mononuclear cells by plasmid vector expression. Nat Protoc. 2012;7:2013–2021. doi: 10.1038/nprot.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Li X, Zhao H, et al. Efficient induction of pluripotent stem cells from menstrual blood. Stem Cells Dev. 2013;22:1147–1158. doi: 10.1089/scd.2012.0428. [DOI] [PubMed] [Google Scholar]
- 18.Merling RK, Sweeney CL, Choi U, et al. Transgene-free iPSCs generated from small volume peripheral blood non-mobilized CD34+ cells. Blood. 2013;121:e98–e107. doi: 10.1182/blood-2012-03-420273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Somers A, Jean JC, Sommer CA, et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells. 2010;28:1728–1740. doi: 10.1002/stem.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petit I, Kesner NS, Karry R, et al. Induced pluripotent stem cells from hair follicles as a cellular. Stem Cell Res (Amst) 2012;8:134–140. doi: 10.1016/j.scr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Yan X, Qin H, Qu C, et al. iPS cells reprogrammed from human mesenchymal-like stem/progenitor cells of dental tissue origin. Stem Cells Dev. 2010;19:469–480. doi: 10.1089/scd.2009.0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou T, Benda C, Dunzinger S, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7:2080–2089. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]
- 24.Meske V, Albert F, Wehser R, et al. Culture of autopsy-derived fibroblasts as a tool to study systemic alterations in human neurodegenerative disorders such as Alzheimer’s disease—methodological investigations. J Neural Transm. 1999;106:537–548. doi: 10.1007/s007020050177. [DOI] [PubMed] [Google Scholar]
- 25.Hjelm BE, Rosenberg JB, Szelinger S, et al. Induction of pluripotent stem cells from autopsy donor-derived somatic cells. Neurosci Lett. 2011;502:219–224. doi: 10.1016/j.neulet.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bliss LA, Sams MR, Deep-Soboslay A, et al. Use of postmortem human dura mater and scalp for deriving human fibroblast cultures. PLoS One. 2012;7:e45282. doi: 10.1371/journal.pone.0045282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chamberlain SJ, Li XJ, Lalande M. Induced pluripotent stem (iPS) cells as in vitro models of human neurogenetic disorders. Neurogenetics. 2008;9:227–235. doi: 10.1007/s10048-008-0147-z. [DOI] [PubMed] [Google Scholar]
- 28.Paşca SP, Portmann T, Voineagu I, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song B, Sun G, Herszfeld D, et al. Neural differentiation of patient specific iPS cells as a novel approach to study the pathophysiology of multiple sclerosis. Stem Cell Res (Amst) 2012;8:259–273. doi: 10.1016/j.scr.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Yahata N, Asai M, Kitaoka S, et al. Anti-Aβ drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS One. 2011;6:e25788. doi: 10.1371/journal.pone.0025788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Efthymiou A, Shaltouki A, Steiner JP, et al. Functional screening assays with neurons generated from pluripotent stem cell-derived neural stem cells. J Biomol Screen. 2014;19:32–43. doi: 10.1177/1087057113501869. [DOI] [PubMed] [Google Scholar]
- 32.Chambers SM, Fasano CA, Papapetrou EP, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emdad L, D’Souza SL, Kothari HP, et al. Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev. 2012;21:404–410. doi: 10.1089/scd.2010.0560. [DOI] [PubMed] [Google Scholar]
- 34.Liu Q, Pedersen OZ, Peng J, et al. Optimizing dopaminergic differentiation of pluripotent stem cells for the manufacture of dopaminergic neurons for transplantation. Cytotherapy. 2013;15:999–1010. doi: 10.1016/j.jcyt.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 35.Vojnits K, Bremer S. Challenges of using pluripotent stem cells for safety assessments of substances. Toxicology. 2010;270:10–17. doi: 10.1016/j.tox.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Miller JD, Ganat YM, Kishinevsky S, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han Y, Miller A, Mangada J, et al. Identification by automated screening of a small molecule that selectively eliminates neural stem cells derived from hESCs but not dopamine neurons. PLoS One. 2009;4:e7155. doi: 10.1371/journal.pone.0007155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swistowski A, Peng J, Han Y, et al. Xeno-free defined conditions for culture of human embryonic stem cells, neural stem cells and dopaminergic neurons derived from them. PLoS One. 2009;4:e6233. doi: 10.1371/journal.pone.0006233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egawa N, Kitaoka S, Tsukita K, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells Sci Transl Med 2012;4:145ra104. [DOI] [PubMed] [Google Scholar]
- 40.Mackay-Sim A. Patient-derived stem cells: Pathways to drug discovery for brain diseases. Front Cell Neurosci. 2013;7:29. doi: 10.3389/fncel.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haggarty SJ, Perlis RH. Translation: Screening for novel therapeutics with disease-relevant cell types derived from human stem cell models. Biol Psychiatry. 2014;75:952–960. doi: 10.1016/j.biopsych.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee G, Papapetrou EP, Kim H, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chambers SM, Qi Y, Mica Y, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. 2012;30:715–720. doi: 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 45.Marchetto MC, Carromeu C, Acab A, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng J, Liu Q, Rao MS, et al. Using human pluripotent stem cell-derived dopaminergic neurons to evaluate candidate Parkinson’s disease therapeutic agents in MPP+ and rotenone models. J Biomol Screen. 2013;18:522–533. doi: 10.1177/1087057112474468. [DOI] [PubMed] [Google Scholar]
- 47.An MC, Zhang N, Scott G, et al. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11:253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corti S, Nizzardo M, Simone C, et al. Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy Sci Transl Med 2012;4:165ra162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu M, Liu K, Swaroop M, et al. δ-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J Biol Chem. 2012;287:39349–39360. doi: 10.1074/jbc.M112.357707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pineda M, Perez-Poyato MS. Current and future therapies for Niemann-Pick C disease. Exp Opin Orphan Drugs. 2013;1:915–923. [Google Scholar]
- 51.Hasson SA, Kane LA, Yamano K, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013;504:291–295. doi: 10.1038/nature12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kondo T, Asai M, Tsukita K, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 53.Söderberg M, Edlund C, Kristensson K, et al. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids. 1991;26:421–425. doi: 10.1007/BF02536067. [DOI] [PubMed] [Google Scholar]
- 54.Prasad MR, Lovell MA, Yatin M, et al. Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res. 1998;23:81–88. doi: 10.1023/a:1022457605436. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki H, Park SJ, Tamura M, et al. Effect of the long-term feeding of dietary lipids on the learning ability, fatty acid composition of brain stem phospholipids and synaptic membrane fluidity in adult mice: A comparison of sardine oil diet with palm oil diet. Mech Ageing Dev. 1998;101:119–128. doi: 10.1016/s0047-6374(97)00169-3. [DOI] [PubMed] [Google Scholar]
- 56.Kyle DJ, Schaefer E, Patton G, et al. Low serum docosahexaenoic acid is a significant risk factor for Alzheimer’s dementia. Lipids. 1999;34(suppl):S245. doi: 10.1007/BF02562306. [DOI] [PubMed] [Google Scholar]
- 57.Gamoh S, Hashimoto M, Hossain S, et al. Chronic administration of docosahexaenoic acid improves the performance of radial arm maze task in aged rats. Clin Exp Pharmacol Physiol. 2001;28:266–270. doi: 10.1046/j.1440-1681.2001.03437.x. [DOI] [PubMed] [Google Scholar]
- 58.Ikemoto A, Ohishi M, Sato Y, et al. Reversibility of n-3 fatty acid deficiency-induced alterations of learning behavior in the rat: Level of n-6 fatty acids as another critical factor. J Lipid Res. 2001;42:1655–1663. [PubMed] [Google Scholar]
- 59.Catalan J, Moriguchi T, Slotnick B, et al. Cognitive deficits in docosahexaenoic acid-deficient rats. Behav Neurosci. 2002;116:1022–1031. doi: 10.1037//0735-7044.116.6.1022. [DOI] [PubMed] [Google Scholar]
- 60.Yagi T, Kosakai A, Ito D, et al. Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS One. 2012;7:e41572. doi: 10.1371/journal.pone.0041572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cardno AG, Marshall EJ, Coid B, et al. Heritability estimates for psychotic disorders: The Maudsley twin psychosis series. Arch Gen Psychiatry. 1999;56:162–168. doi: 10.1001/archpsyc.56.2.162. [DOI] [PubMed] [Google Scholar]
- 62.Brennand KJ, Simone A, Jou J, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chiang CH, Su Y, Wen Z, et al. Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol Psychiatry. 2011;16:358–360. doi: 10.1038/mp.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morrison B, III, Saatman KE, Meaney DF, et al. In vitro central nervous system models of mechanically induced trauma: A review. J Neurotrauma. 1998;15:911–928. doi: 10.1089/neu.1998.15.911. [DOI] [PubMed] [Google Scholar]
- 65.Lafaille FG, Pessach IM, Zhang SY, et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature. 2012;491:769–773. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee EX, Lam DH, Wu C, et al. Glioma gene therapy using induced pluripotent stem cell derived neural stem cells. Mol Pharm. 2011;8:1515–1524. doi: 10.1021/mp200127u. [DOI] [PubMed] [Google Scholar]
- 67.Zhu H, Lensch MW, Cahan P, et al. Investigating monogenic and complex diseases with pluripotent stem cells. Nat Rev Genet. 2011;12:266–275. doi: 10.1038/nrg2951. [DOI] [PubMed] [Google Scholar]