

Abstract
We have identified a novel 7.7 Mb del(8)(q23.2q24.11) in a patient progressing to acute myeloid leukemia (AML) following a 12-year stable phase of chronic myelomonocytic leukemia (CMML). A surprisingly high JAK2+ allelic burden of 92% at the time of AML led us to delineate the molecular aberrations relevant for leukemogenesis. While a frameshift mutation in the TET2 gene was stably present throughout the course of disease the JAK2 mutation was acquired after initial diagnosis of CMML. At progression aCGH revealed del(8q)(q23.2q24.11) encompassing various cancer relevant genes of which RAD21 and CSMD3 are of particular interest.
Keywords: del(8q), CMML, TET2, JAK2, Transformation
Highlights
-
•
Unraveling the molecular events in a CMML-patient progressing to AML.
-
•
Stable TET2 mutation throughout the course of disease; JAK2 mutation was acquired.
-
•
Array CGH at progression revealed a novel del(8q).
-
•
del(8q) encompasses genes of reported relevance in carcinogenesis.
-
•
RAD21 and CSMD3 are of particular interest.
1. Introduction
Chronic myelomonocytic leukemia (CMML) is a clonal hematopoietic stem cell neoplasm with overlapping myelodysplastic and myeloproliferative features. While progression to acute myeloid leukemia (AML) is common, the driving molecular aberrations involved in transformation remain obscure [1]. Clonal cytogenetic abnormalities predict adverse prognosis and are evident in up to 40% of cases [2]. Next generation sequencing has identified molecular aberrations in approximately 90% of CMML patients with TET2 gene mutations (TET2+) being particularly frequent. On the contrary, the JAK2 V617F mutation (JAK2+) is uncommon [3]. Concomitant TET2+ and JAK2+ have been described in chronic myeloproliferative disorders, but none of these are sufficient to drive blastic transformation. Moreover, transformation of myeloproliferative disorders most often results in loss of JAK2+ indicating the transformation to arise in a JAK2-negative subclone [4]. We here report a case of TET2+ CMML who acquired JAK2+ and progressed into overt AML 12 years after initial diagnosis. Array comparative genomic hybridization (aCGH) revealed a novel del(8q)(q23.2q24.11) (del(8q)) comprising genes with recently reported relevance in cancer, including AML.
2. Case presentation
In 1998, a 64-year old male presented with pleural effusion, pericarditis, and leukocytosis of 38×109/L with 30% monocytes. A complete overview of the clinical course of disease is given in Table 1. A bone marrow (BM) biopsy revealed CMML-1. There was no splenomegaly or cytopenias. In 2004, pleural effusion reemerged and microscopy of pleural fluid revealed numerous CD68 positive monocytes. Lymph node enlargement emerged in 2007 and a biopsy showed infiltration of 50% fully differentiated monocytes. Despite these various CMML-related manifestations, no cytopenias were evident. However, in 2010 BM failure developed with decreasing hemoglobin and thrombocyte counts and increasing leukocyte count. Morphology revealed persistent CMML-1. Nine months later, in June 2011 at the age of 76 the patient was admitted with abdominal pain and severely deranged hematopoiesis and was diagnosed with AML. Flow cytometry of peripheral blood showing 40% myeloblasts staining positive for CD34, CD117, HLA-DR, and CD13. A CT scan showed splenomegaly, and signs of malignant infiltration in the renal parenchyma and the soft tissue along the lower thoracic vertebras, suspicious of myelosarcomas. The patient received palliative care, but succumbed from progressive renal failure in July 2011.
Table 1.
Longitudinal registration of disease stage, laboratory values, cytogenetic and molecular analyses.
| December 1998 | June 2001 | September 2010 | June 2011 | |
|---|---|---|---|---|
| Clinical features | Pericarditis and pleuritis | Routine follow-up | Progressive BM failure | BM failure, splenomegaly, and possible myelosarcoma |
| Lab results | ||||
| Hemoglobin (mmol/L) | 7.9 | 8.5 | 5.1 | 5.3 |
| Platelets (109/L) | 206 | 241 | 218 | 25 |
| Leukocytes (109/L) | 23.7 | 33.9 | 55.5 | 84.2 |
| Neutrophils (109/L) | 12.6 | 8.8 | 46.6 | 61.5 |
| Monocytes (109/L) | 5.45 | 18.6 | 3.03 | 4.21 |
| Morphology | CMML-1 | CMML-1 | CMML-1 | Dry tap, inconclusive |
| Blasts incl. promonocytes in BM | <5% | <5% | <5% | 20%a |
| Flow cytometry | ND | ND | CMML (BM) | AML (PB) |
| 3% CD34+CD117+myeloblasts | 40% CD34+CD117+myeloblasts | |||
| 84% CD64+myelomonocytoid cells | 10% co-expression of CD64 | |||
| G-banding | ND | 46,XY[20] | 46,XY,dup(9)(p21p21)[6]/46,XY[19] | 45,X–Y[5]/46,XY[20] |
| TET2mutation | Positive | ND | Positive | Positive |
| JAK2mutation | Negative | ND | 93% of BM MNCs | 92% of PB MNCs |
| Array CGH | ND | ND | del(8)(q23.2q24.11) | ND |
| Interphase nuclei FISH | ND | BM | BM | PB |
| Neg. for del(8q) | 88% cells with del(8q) | 15% cells with del(8q) | ||
BM: bone marrow; ND: not done; PB: peripheral blood; MNCs: mononuclear cells.
Blast count in PB, due to dry tap.
Routine molecular analysis at AML diagnosis showed a JAK2+ allelic burden of 92% of mononuclear cells (MNCs) from peripheral blood (PB). Since JAK2+ is usually lost upon transformation this led us to retrospectively delineate the longitudinal evolution of molecular aberrations at critical time points, summarized in Table 1.
In short, DNA was extracted from diagnostic paraffin embedded BM biopsies (1998), from thawed BM MNCs from the time of disease progression (2010), and from PB MNCs at the time of overt AML (2011). JAK2+ was analyzed for by standard quantitative polymerase chain reaction (qPCR). While the diagnostic CMML sample was negative for JAK2+, the allelic burden was above the 90% level both in the transformation- and AML phase of disease. The entire coding sequences and all splice sites of the TET2 gene (exons 3–11) were scanned for mutations by PCR combined with denaturing gradient gel electrophoresis (DGGE) as described in Ref. [5]. In all samples, a stable frameshift mutation in exon 6 was found (c.3658_3658del A, Thr1220fs. RefSeq NM_001127208.2). DNA extracted from a benign skin biopsy from 1990 proved negative for TET2 mutations, hence excluding germline mutation (Fig. 1A). By multiplex fluorescent-labeled PCR followed by capillary gel electrophoresis we retrospectively analyzed MNCs from the time of AML-diagnosis for recurrent AML related mutations, namely FLT3-ITD, FLT3-D835, cKIT D816V, IDH1 R132, CEBPA, NPM1 exon 12, and WT1 exon 7 of which all came out negative. Standard G-band karyotyping was not performed at initial diagnosis in 1998, but the karyotype was 46,XY[20] two and a half years from diagnosis in stable phase of disease. However, in September 2010 the karyotype had changed to 46,XY,dup(9)(p21p21)[6]/46,XY[19].
Fig. 1.
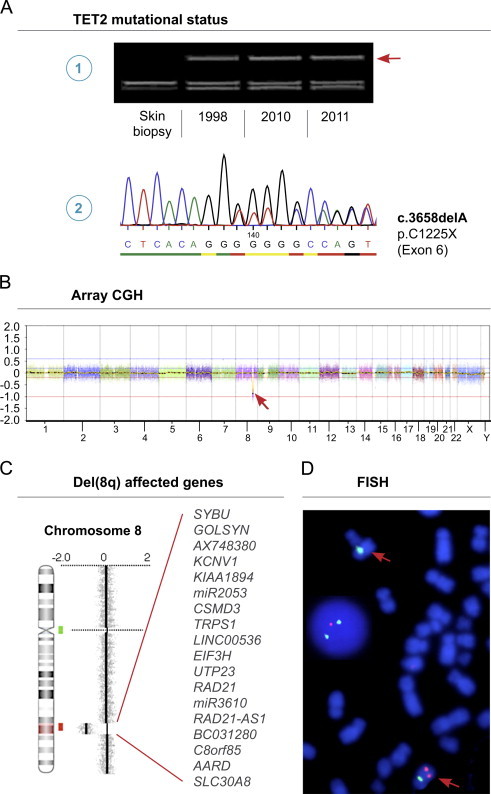
Output data of applied analyses. (A) DGGE analysis showing mutant TET2 being present in all samples as indicated by the heteroduplex band (arrow) (1). Sanger sequencing of the heteroduplex band showing presence of both mutant (del A) and normal alleles (reverse strand shown) (2). (B) High-resolution aCGH analysis at the time of progressive BM failure in 2010 revealing a submicroscopic deletion at chromosome 8 (arrow). The X-axis at the bottom denotes the chromosomal position. (C) Zoom view of genomic profile at chromosome 8 showing the deleted region 8q23.2–24.11 as indicated by red shade on chromosome 8 ideogram. The genes located in the deleted region are listed. Locations of BAC probes for subsequent FISH are indicated (red and green bars). (D) Confirmatory FISH using BAC probe RP11-11A18 at 8q23.3 in the CSMD3 gene (red) and centromeric SE 8 probe (green) showing the deletion on one chromosome 8 (red arrows) in metaphases and interphase nuclei (1R2G signal pattern). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Since progressive BM failure was not evident until September 2010, we considered this time point pivotal in AML transformation. Consequently, we focused on identifying candidate aberrations of potential impact by applying aCGH using human Cancer CytoChip at this time point. Regions of gain or loss contained within copy number variable regions (CNVs) were discarded. Reference genome was NCBI build 36.1 (hg18). High-resolution aCGH analysis revealed an interstitial deletion in chromosome 8 at bands q23.2-q24.11 of maximal size 7,7 Mb (position 110.679.916–118.334.826) (Fig. 1B and C). To confirm this finding and to longitudinally analyze for the del(8q), we performed fluorescence in situ hybridization (FISH) with the following directly labeled probes: RP11-11A18 (SpectrumOrange-labeled, BlueGnome, Cambridge, UK) located at 8q23.3 (position 113.491.837–113.659.918) and centromere SE 8 probe (FITC-labeled, Kreatech, Amsterdam, The Netherlands). Chromosomes and nuclei were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride. The FISH analysis confirmed the presence of del(8q) in the progression- and AML phase and absence of del(8q) in the stable phase of CMML in 2001 (Fig. 1D). Array CGH did not detect the dup(9)(p21p21) found with standard G-banding karyotyping in September 2010, since the fraction of dup(9) positive cells is below the sensitivity of aCGH.
The region 8q23.2-q24.11 encompasses genes as depicted in Fig. 1C. We performed an NCBI gene database and PubMed search on each of these genes for their reported relevance in carcinogenesis. Importantly, several of these are of notified relevance; TRPS1 and EIF3H are reported as candidate oncogenes with increased expression in breast cancer [6,7]. In a study of non-small cell lung carcinomas the CUB and sushi multiple domains 3 gene (CSMD3) was identified as the second most frequently mutated gene and loss of function cause increased proliferation of airway epithelial cells in vitro [8]. While the CSMD gene family has recently been suggested tumor suppressor genes in colorectal cancer [9], the role of CSMD3 in hematological cancers remains to be elucidated. Strikingly, RAD21 has been associated with AML as it encodes a protein in the cohesin complex, which is involved in chromosome segregation during cell division, regulation of transcription, and DNA double strand break repair. Recently, genes belonging to the cohesin complex were described as novel recurrent and mutually exclusive mutations in myeloid neoplasms [10].
3. Conclusion
We have longitudinally described the molecular aberrations present at critical time points from initial diagnosis of CMML to overt AML applying a comprehensive panel of molecular analyses. Firstly, the consistent and non-germ line TET2+ in all stages of disease verify that the AML stage indeed was transformed from CMML and not a de novo event. Secondly, in this case, TET2+ and JAK2+ represent founder mutations. It cannot be ruled out that the massive JAK2+ allelic burden have contributed to genomic instability and, speculatively, also influenced the proliferative potential of the leukemic blasts. Thirdly, aCGH revealed a novel del(8q). Our results indicate that the leukemic transformation of an otherwise long-standing and stable CMML could have been driven by loss of gene function resulting from this exact deletion. Of the deleted genes, RAD21 and CSMD3 are of particular interest, since their loss of function have recently been reported to play a role in carcinogenesis. However, their role in hematological cancers remains to be studied. Finally, this case report emphasizes the relevance of aCGH in the search for candidate genes involved in leukemogenesis, not least in heterogeneous neoplasms such as CMML.
Conflict of interest
We wish to confirm that there are no known conflicts of interest associated with this publication.
There has been no significant financial support for this work that could have influenced its outcome.
Contributions
All authors contributed to the writing, experimentation, and preparation of the manuscript.
We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship.
We further confirm that the order of authors listed in the manuscript has been approved by all of us.
We confirm that we have given due consideration to the protection of intellectual property associated with this work and that there are no impediments to publication, including the timing of publication.
Acknowledgments
The authors would like to thank Mariann Guldmann-Christensen M.Sc., Ph.D., from the Department of Pathology, Aarhus University Hospital for helping with DNA extraction from paraffin-embedded tissue and Marcus Celik Hansen, M.Sc., for graphical support. The Karen Elise Jensen Foundation and the Danish Cancer Society (Grant no. R40-A2094-11-S2) supported the study.
References
- 1.Patnaik M.M., Parikh S.A., Hanson C.A., Tefferi A. Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol. 2014;165:273–286. doi: 10.1111/bjh.12756. [DOI] [PubMed] [Google Scholar]
- 2.Such E., Cervera J., Costa D., Sole F., Vallespi T., Luno E. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96:375–383. doi: 10.3324/haematol.2010.030957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itzykson R., Kosmider O., Renneville A., Gelsi-Boyer V., Meggendorfer M., Morabito M. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31:2428–2436. doi: 10.1200/JCO.2012.47.3314. [DOI] [PubMed] [Google Scholar]
- 4.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24:1128–1138. doi: 10.1038/leu.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asmar F., Punj V., Christensen J., Pedersen M.T., Pedersen A., Nielsen A.B. Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica. 2013;98:1912–1920. doi: 10.3324/haematol.2013.088740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu J., Su P., Jia M., Wu X., Zhang H., Li W. TRPS1 expression promotes angiogenesis and affects VEGFA expression in breast cancer. Exp Biol Med. 2014;239:423–429. doi: 10.1177/1535370214523904. [DOI] [PubMed] [Google Scholar]
- 7.Mahmood S.F., Gruel N., Chapeaublanc E., Lescure A., Jones T., Reyal F. A siRNA screen identifies RAD21, EIF3H, CHRAC1 and TANC2 as driver genes within the 8q23, 8q24.3 and 17q23 amplicons in breast cancer with effects on cell growth, survival and transformation. Carcinogenesis. 2014;35:670–682. doi: 10.1093/carcin/bgt351. [DOI] [PubMed] [Google Scholar]
- 8.Liu P., Morrison C., Wang L., Xiong D., Vedell P., Cui P. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis. 2012;33:1270–1276. doi: 10.1093/carcin/bgs148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang R., Song C. Loss of CSMD1 or 2 may contribute to the poor prognosis of colorectal cancer patients. Tumour Biol. 2014;35:4419–4423. doi: 10.1007/s13277-013-1581-6. [DOI] [PubMed] [Google Scholar]
- 10.Kon A., Shih L.Y., Minamino M., Sanada M., Shiraishi Y., Nagata Y. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45:1232–1237. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]