

Abstract
The route of O2 to and from the high-spin heme in heme–copper oxidases has generally been believed to emulate that of carbon monoxide (CO). Time-resolved and stationary infrared experiments in our laboratories of the fully reduced CO-bound enzymes, as well as transient optical absorption saturation kinetics studies as a function of CO pressure, have provided strong support for CO binding to on the pathway to and from the high-spin heme. The presence of CO on suggests that O2 binding may be compromised in CO flow-flash experiments. Time-resolved optical absorption studies show that the rate of O2 and NO binding in the bovine enzyme (1 × 108 M−1 s−1) is unaffected by the presence of CO, which is consistent with the rapid dissociation (t1/2 = 1.5 μs) of CO from . In contrast, in Thermus thermophilus (Tt) cytochrome ba3 the O2 and NO binding to heme a3 slows by an order of magnitude in the presence of CO (from 1 × 109 to 1 × 108 M−1 s−1), but is still considerably faster (~10 μs at 1 atm O2) than the CO off-rate from CuB in the absence of O2 (milliseconds). These results show that traditional CO flow-flash experiments do not give accurate results for the physiological binding of O2 and NO in Tt ba3, namely, in the absence of CO. They also raise the question whether in CO flow-flash experiments on Tt ba3 the presence of CO on impedes the binding of O2 to or, if O2 does not bind to prior to heme a3, whether the complex sterically restricts access of O2 to the heme. Both possibilities are discussed, and we argue that O2 binds directly to heme a3 in Tt ba3, causing CO to dissociate from in a concerted manner through steric and/or electronic effects. This would allow to function as an electron donor during the fast (5 μs) breaking of the O–O bond. These results suggest that the binding of CO to on the path to and from heme a3 may not be applicable to O2 and NO in all heme-copper oxidases. This article is part of a Special Issue entitled: Vibrational Spectroscopies in Molecular Bioenergetics.
Keywords: Time-resolved infrared spectroscopy, Time-resolved infrared linear dichroism, Thermus thermophilus ba3, Photolabile O2 and NO complex, CO photodissociation and recombination, dynamics
1. Introduction
Heme–copper oxidases, which include the cytochrome and ubiquinol oxidases, play a crucial role in energy production by aerobic organisms [1–3]. Their primary function is to catalyze the reduction of dioxygen to water using electrons from respiratory electron transport. The energy made available by the reaction generates a transmembrane electrochemical proton gradient that drives ATP synthesis [4]. These enzymes are responsible for over 90% of biological dioxygen reduction and for nearly half of the redox energy of cellular respiration [5,6]. Significantly, cytochrome c oxidase is inhibited by nitric oxide (NO) [7], a signaling molecule involved in diverse biochemical and physiological processes [8]. This inhibition of cytochrome oxidase may play an important role in regulating cellular respiration [7,9]. Several bacterial heme–copper oxidases are also able to catalyze the reduction of nitric oxide (NO) to nitrous oxide (N2O) [10–13]; however, there are confiicting reports whether the bovine cytochrome oxidase has NO reductase activity [14,15].
The heme–copper oxidases are subdivided into three families, denoted A, B, and C [16,17], and all three families contain a high-spin heme (a3, o3 or b3) which together with a copper center, CuB, forms the binuclear heme–copper site of O2 binding and reduction. While sequence homology of the catalytic subunit containing the binuclear center is high between the bovine enzyme and the bacterial Rhodobacter sphaeroides (Rs) and Paracoccus denitrificans (Pd) aa3 oxidases (54 and 55%, respectively), it is much lower in Thermus thermophilus (Tt) ba3 and Pseudomonas stutzeri cbb3 oxidases (23 and 15%, respectively). Despite this diversity, CuB in its oxidized form in all three oxidase families is trigonally ligated to the imidazole side chains of three conserved histidines, and the high-spin iron is ligated to an invariant histidine on the “proximal” side (opposite to the O2 binding site) of the heme; in the cbb3 oxidases, the proximal histidine is hydrogen bonded to the carboxylate of a glutamate residue [18]. All heme–copper oxidases also contain a post-translational modification, a cross-link between C6 of a tyrosine residue (Tyr 244 in the bovine enzyme) and the ε-nitrogen of one of the histidine ligands to CuB [19–21]. In the cbb3 oxidases, the tyrosine originates from a different helix than in the A-and B-type oxidases [18].
Knowledge of the structural and dynamic features of the binuclear active site and the intraprotein channel(s) for ligation and proton transfer is critical for understanding the mechanisms of O2 reduction, inhibition by NO, and NO reduction. The first step in O2 reduction and NO inhibition and reduction relies on access of O2 and NO, respectively, to the active site and how the protein environment modulates this access. Based on crystal structures, ligand pathways have been postulated for many heme–copper oxidases [18,20,22–25]. While there are significant sequence and structural similarities among these ligand pathways, there are variations in the global structure of the catalytic subunit, which may reflect the different functional environments of these enzymes. For instance, recent crystallographic studies of xenon (Xe) binding in Tt ba3 [26] indicated that a constriction point in the oxygen channel of the aa3 oxidases [24,27,28] is not present in Tt ba3. This observation suggests easier access of ligands to the binuclear site in ba3, which may be related to the different physiological requirements of the aa3 and ba3 oxidases [29].
Migration of O2 through a ligand channel to the active site is followed by O2 binding to the binuclear center. The role of CuB in transporting ligands such as O2 and NO to and from the high-spin heme is of particular interest. Carbon monoxide (CO), a competitive inhibitor of O2 reduction in cytochrome oxidase, has frequently been used as a model for O2 binding [30–34], and the reactions following photodissociation of CO have been thought to exemplify the pathways of O2 to and from the active site. This knowledge, however, is only relevant to the physiological O2 reduction as long as the coordination chemistry of CO mimics that of O2. The photodissociation of CO from the high-spin heme in the presence of O2 has also been used extensively to initiate the O2 reduction reaction [2,3,35–39]. However, the fate of the photodissociated CO may compromise the O2 (or NO) binding and electron transfer dynamics [40]. This issue can only be addressed by exploring the photodissociation and recombination dynamics of the CO ligand, and by comparing the binding of O2 and NO in the heme–copper oxidases in the presence of CO to the binding of these ligands under more realistic physiological conditions, namely, in the absence of CO.
In this review, we summarize vibrational and UV–vis spectroscopic studies of 1) the flash-induced photodissociation and rebinding of CO in the heme–copper oxidases, and 2) the reaction of O2 with these enzymes in the presence and absence of CO. These results provide insight into the ligand binding dynamics of the heme-copper oxidases and how the protein environment modulates the ligand pathways and metal centers for different physiological environments.
2. CO photolysis and recombination dynamics
2.1. Fourier transform infrared (FTIR) experiments: CO binding to
Characterization of the O2 binding site is critical for understanding the mechanism of O2 reduction to water and for elucidation of the protein structures that facilitate this reaction. Infrared spectroscopy provides a direct approach for studying the binding of ligands to heme proteins, including the heme–copper oxidases [30–34,41]. Carbon monoxide (CO) is commonly used as an infrared probe for O2 binding because it generally binds to the same sites as O2 and because of its strong infrared absorption and non-reducible nature. The frequencies and bandwidths of the CO infrared stretching bands can give valuable information not only about the identity of the metal center to which CO binds but also about the environment surrounding the CO ligand [31,34,41]. For example, CO binding to the high-spin heme in the bovine enzyme gives rise to a major peak at 1963 cm−1 (the Fe-bound C–O stretching frequency) in the infrared spectrum [31,34]. Infrared spectroscopy has also allowed us to follow the binding of CO to , a metal center that is inaccessible to other spectroscopic techniques. Pioneering FTIR studies by Alben and coworkers showed that the photodissociated CO binds to in mitochondrial preparations under cryogenic conditions based on the infrared frequency at 2062 cm−1 [30,42].
Our laboratories have carried out extensive infrared studies of the photodissociation and recombination of the fully reduced CO-bound heme–copper oxidases [31,32,33,43–48]. FTIR difference spectra (dark minus light) were recorded over a wide temperature range (21–298 K) for the bovine enzyme, and Tt caa3 and ba3 [33]. Fig. 1 shows the spectra recorded between ~22 and 300 K for the bovine enzyme (left panels) and Tt ba3 (right panels). In addition to the major heme a3-CO band at 1963 cm−1, the bovine enzyme has two minor bands at 1949 and 1944 cm−1. At 21 K, Tt caa3 shows two positive peaks at 1953 and 1947 cm−1 representing the heme a3-CO [33], while CO binding to heme a3 in Tt ba3 gives rise to two major positive bands at 1974 and 1983 cm−1 (Fig. 1, right panels). The FTIR difference spectra show negative peaks at 2066, 2054 and 2039 cm−1 for the bovine enzyme, at 2060 and 2036 cm−1 for Tt caa3 [33] and at 2054 cm−1 for Tt ba3, which are attributed to CO binding to .
Fig. 1.
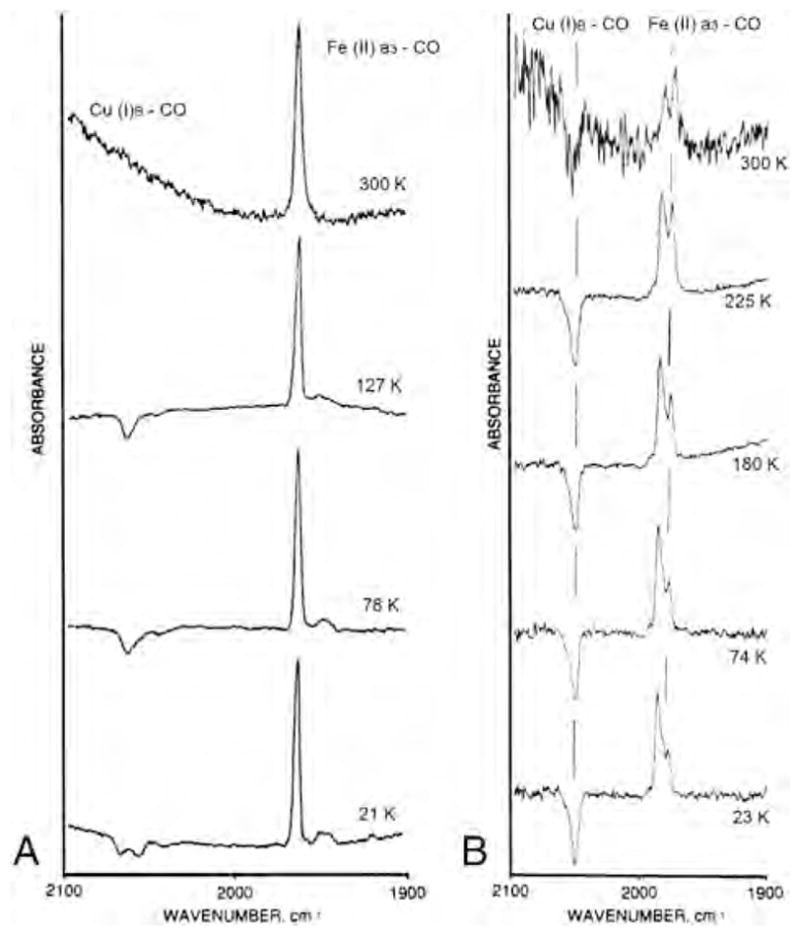
FTIR difference spectra (dark minus light) of carbonmonoxy fully reduced bovine cytochrome aa3 (left) and Thermus thermophilus cytochrome ba3 (right) at various temperatures. The 300 K light spectrum of the ba3 enzyme was recorded under continuous photolysis. Conditions are those described in [32] and [33].
The complex in the bovine and Tt caa3 enzymes is kinetically stable below 140 K and 170 K, respectively, but dissociates at higher temperature. However, for Tt ba3 we were able to observe binding of CO to upon continuous photolysis at room temperature in the FTIR dark-minus-light difference spectrum (Fig. 1, right panel, top spectrum). The differences in the CO stretching frequency for the Fe–CO (and ) infrared absorption among these three oxidases indicate significant variations in the details of the CO binding and the stability of the intermediate. Nonetheless, the bandwidths of the IR peaks remain narrow over a large temperature range for all the oxidases, indicating a very homogeneous environment around the CO ligand. A recent combined crystallographic and infrared spectral study supports CO binding to in Tt ba3 following photolysis of CO from heme a3 [49].
We were also able to follow CO transfer from to between 158 and 179 K in the bovine enzyme, 175–195 K in Tt caa3 and 205–230 K in Tt ba3 [33]. Taking into account the relative absorptivities of heme and copper CO-complexes, the relative integrated areas of the Fe–CO and infrared peaks represent quantitative transfer of CO from the heme to CuB following CO photodissociation, supporting a closed pocket isolated from the surrounding medium [32]. The activation parameters derived from an Eyring plot of the CO recombination in the three enzymes (Fig. 2) are listed in Table 1.
Fig. 2.
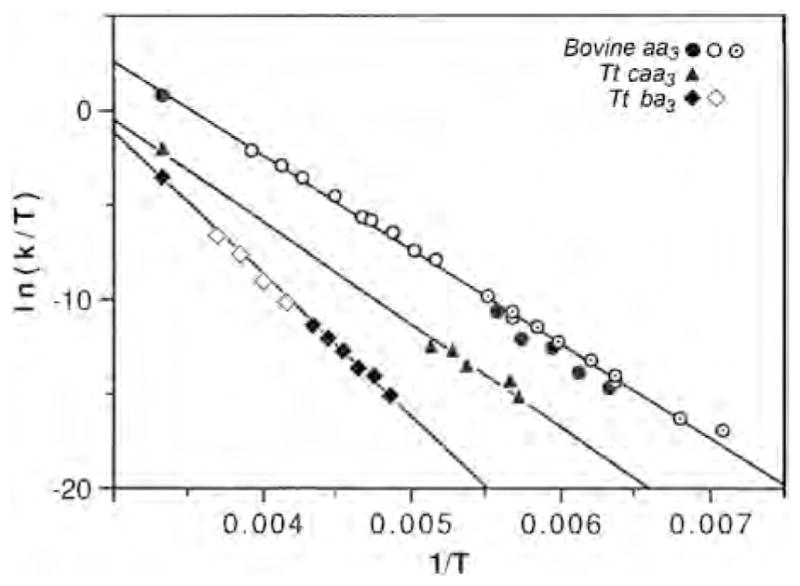
The Eyring plot for T. thermophilus cytochrome ba3–CO, T. thermophilus caa3–CO and bovine aa3–CO recombination, measured from the Fe–CO infrared peaks (low temperature) and by kinetic UV–vis spectrophotometry at room temperature. Data are from current work and [32,33]. For the bovine enzyme, open circles are from Sharrock and Yonetani [77] and the circles with concentric dots are from Fiamingo et al. [42].
Table 1.
Activation parameters for Tt ba3, Tt caa3 and bovine aa3.
| ΔH‡ (kcal/mol) | ΔS‡ (cal/mol-K) | |
|---|---|---|
| T. thermophilus ba3 | 14.9 | −5 |
| T. thermophilus caa3 | 10.8 | −16 |
| Bovine aa3 | 10.0 | −12 |
The multiple infrared heme–CO stretching bands represent discrete CO conformers of different structures that interconvert rapidly (on the FTIR time scale, i.e. minutes) [31,33]. For Tt ba3, the Fe–CO conformers interconvert down to ~150 K, and they are energetically close as reflected by their relative populations between 180 K and room temperature. The thermodynamic parameters, ΔH0 = 0.84 +/− 0.17 kcal/mol and ΔS0 of 3.5 +/− 0.9 cal/mol-K, were obtained for the interconversion of the 1983 and 1974 cm−1 conformers based on the relative areas of the two conformers as a function of temperature [33]. These conformers were also observed in the CO-FTIR spectra of intact plasma membranes.
2.2. Resonance Raman experiments: multiple Fe–Im(N) conformers in Tt ba3
We explored the different heme–copper oxidases by resonance Raman spectroscopy [48,50,51]. The resonance Raman spectra of the unliganded reduced Tt ba3 in the low-frequency region, which contains the out-of-plane iron-imidazole nitrogen, Fe–N(Im), stretching vibration, show two bands around 192 and 208 cm−1 (Fig. 3). The use of isotopically enriched 57Fe (95%) confirmed the assignments of these bands as the Fe–N(Im) stretching frequencies [51]. In contrast, the bovine enzyme shows a single Fe–N(Im) frequency at 214 cm−1. The relative intensities of the two conformers in ba3 are temperature dependent over a large range and track those of the Fe-CO peaks in the infrared spectra. This is reflected in almost identical thermodynamic parameters based on the Fe–CO (see above) and Fe–Im(N) conformer (ΔH0 = 0.75 +/−1.2 kcal/mol and ΔS0 of 2.1 +/− 1.2 cal/mol-K) thermodynamics plot (Fig. 4). This suggests that the Fe–CO infrared conformers and the Fe–N(Im) conformers arise from the same conformational changes, although the small thermodynamics values do not indicate major global structural changes. The different conformers may represent rotamers of the imidazole plane about the Fe–N axis, giving rise to different steric interactions between the imidazole and the heme of possible relevance to the coordination at the heme.
Fig. 3.

Resonance Raman spectra showing the Fe–N(Im) stretching peaks of T. thermophilus cytochrome ba3 as a function of temperature.
Fig. 4.
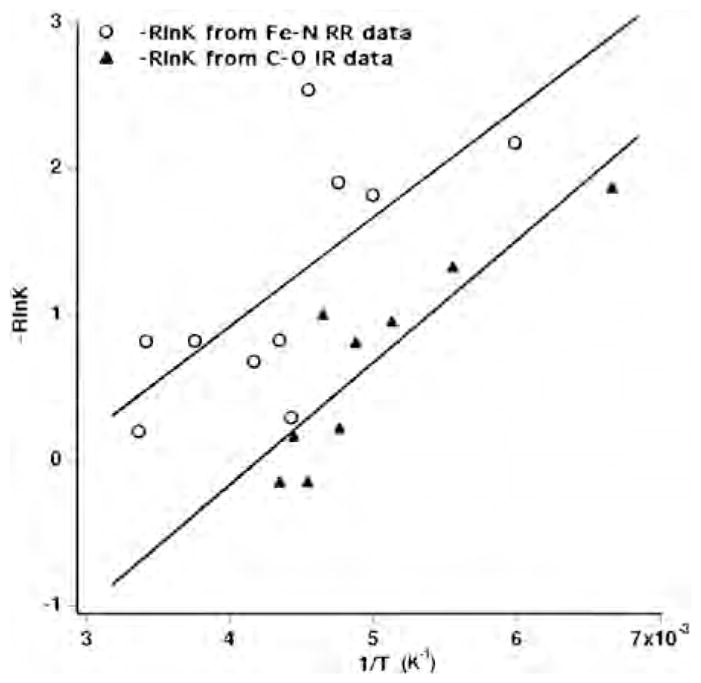
Conformer thermodynamics. The temperature dependent intensities of the 192 and 208 cm−1 Fe-N(Im) bands in the resonance Raman spectra of Tt ba3 and those of the two major Fe-CO stretching peaks at 1973 and 1984 cm−1.
2.3. Dynamic time-resolved infrared (TRIR) experiments: Transient binding of CO to at ambient temperature
While the low-temperature FTIR measurements provided important information about CO binding to in the various heme–copper oxidases, it was imperative to demonstrate whether the photodissociated CO could also bind to at room temperature, particularly with respect to CO flow-flash experiments that rely on the photolability of the CO complex to initiate the reaction with O2. Time-resolved infrared (TRIR) spectroscopy of the photodissociated CO-bound bovine enzyme in our laboratories provided the first evidence for CO binding to at room temperature following photodissociation of CO from heme a3 [43]. Fig. 5 (top) displays the infrared transient at 2061 cm−1 due to the complex, and a single exponential fit shows that the transient decays with a half-life of 1.5 μs (Fig. 5, bottom). The time resolution of these early experiments was 200 ns but later experiments showed that CO binds to within 3 ps [45]. More recent studies by others have shown photoinitiated CO ligand transfer to CuB of 60 fs [52]. The subsequent recombination of CO with Fea3 occurs with an observed rate constant of ~90 s−1 at 1 atm of CO [32].
Fig. 5.

(Top) The room temperature transients of bovine heart cytochrome aa3–CO following photolysis of CO from the heme. (Bottom) A single exponential fit to the transient decay (see ref. [43]) for details.
The photodissociated CO also binds to in Tt caa3 and ba3 at room temperature following photodissociation of CO from heme a3 as reflected by the infrared transients at 2036 cm−1 and 2054 cm−1, respectively. In Tt caa3, CO equilibrates with the surroundings on a microsecond time scale, 2 × 104 s−1, and rebinds to heme a3 with an apparent rate constant of 40 s−1 [47]. The room temperature infrared transients for Tt ba3-CO before photolysis (1974 cm−1; heme a3-CO bound) and after photolysis (2053 cm−1; ) are shown in Fig. 6. The transient is much more stable in Tt ba3 than in the bovine enzyme, and based on a single exponential fit decays with an apparent lifetime of ~46 ms, which is the same within experimental error as the value of 50 ms obtained for CO rebinding to heme a3 (Fig. 6). These values are similar to those obtained previously (~30 s−1) by time-resolved step-scan FTIR difference spectroscopy [53]. However, our UV–vis time-resolved optical absorption measurements show that the photodissociated CO rebinds to heme a3 in ba3 with an apparent lifetime of 260 ms, which is significantly slower than observed in our TRIR experiment and previous time-resolved step-scan FTIR measurements [53]. The cause of the discrepancy between the two approaches is unknown and is currently under investigation. Regardless, the slow CO dissociation from in Tt ba3 raises the question whether the photodissociated CO interferes with the reaction of Tt ba3 with O2. TRIR spectroscopy has also demonstrated the binding of the photodissociated CO to in other heme–copper oxidases. For example, in Escherichia coli bo3, CO dissociation from was reported with a rate constant of ~500 s−1 [47], although later studies reported a multiphasic dissociation of CO from on both microsecond and millisecond time scales [54]. The CO recombines with heme o3 with an apparent rate constant of 40 s−1 (25 ms) based on UV–vis time-resolved optical absorption measurements in our laboratory.
Fig. 6.
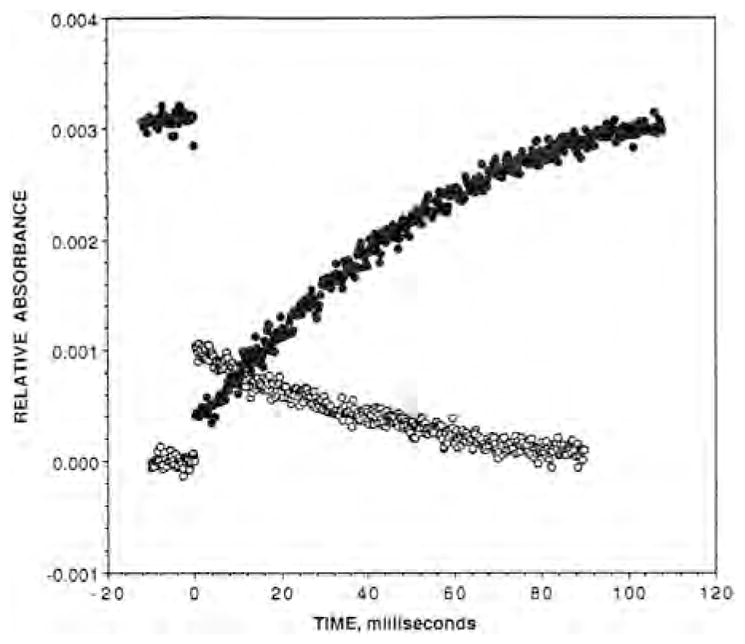
The post-photodissociation TRIR transient absorbance trace recorded at the maximum of the Fea3–CO absorbance peak at 1974 cm−1 (filled circles) and the absorbance peak at 2053 cm−1 (open circles).
2.4. Time-resolved infrared linear dichroism: the orientation of CO in heme a3–CO and CuB–CO complexes of bovine aa3
TRIR linear dichroism (TRIRLID) measurements allowed us to determine the orientation of the C–O bond axis with respect to the heme normal in the complex of the bovine enzyme as well as the photoproduct [44]. The TRIRLID is based on the differential absorption (photoselection) of parallel versus perpendicular polarized infrared light with respect to that of the photodissociation pulse and is measured as a function of the transmittance of the infrared probe beam. The results of such an experiment for both the Fea3–CO complex and the CuB–CO photoproduct of the bovine enzyme are shown in Fig. 7. TRIRLID experiments gave an angle between the heme normal and the C–O bond vector of 21° (+/−2°) for the in the bovine enzyme, which was supported by subsequent picosecond TRIR measurements [46]. For the , a discrete angle (α) rather than a distribution of angles was assumed because the IR peak is very narrow, indicating the absence of inhomogeneous broadening [46]. The angle of 21° is in good agreement with an analogous angle of 16.6° derived based on the heme–CO bound crystal structure [55].
Fig. 7.

(A) The TRIR linear dichroism signals of CO-bound bovine heart cytochrome oxidase between −5 and 30 μs with respect to the time of the photodissociation pulse. The upper traces are for the Fe–CO complex and the lower traces for the CuB–CO photoproduct. The polarization of the infrared probe beam relative to the photo-dissociation pulse is indicated. The solid traces represent perpendicular polarization and the dashed traces represent the parallel polarization. (B) The time-dependence of the polarization ratio, R(t) =(ΔAperpendicular /ΔAparallel) (see ref. [44] for further details).
Modified from [44].
The TRIR linear dichroism technique also allowed us to determine the orientation of the C–O bond axis with respect to the heme normal in the transient photoproduct in the bovine enzyme. The TRIRLID signal for the transient photoproduct was practically the same for the parallel and perpendicular polarization of the infrared probe beam with respect to the photodissociation pulse (Fig. 7A, lower traces). The near absence of linear dichroism was interpreted in terms of angle, α, of 51 +/−3° between the heme normal and the C–O bond vector of the complex. Subsequent picosecond infrared measurements gave α of 55 +/− 3° [46]. It should be noted that the α values for the heme iron and CuB are cone half-angles because the TRIRLID does not provide angular orientation of the Fea3C–O and CuB–CO vectors. Recent x-ray structure of the CO-bound bovine enzyme at 100 K indicates that CO is bound to CuB in a side-on fashion, with metal-to-carbon and metal-to-oxygen atom distances of 2.4 and 2.7 Å, respectively, indicating a weak CuB–CO bond [55]. Based on the crystal structure of the proposed complex (the crystal structure of the CO derivative determined at 100 K), the angle between the heme normal and the C–O bond vector for the complex is 65.5°, somewhat higher than observed in solution in the TRIRLID measurements. Based on a recent x-ray crystal structure of the CO-bound Tt ba3 and the photodissociated product [49], the angle between the heme normal and the C–O bond vector is 66° for and 64° for . Although the C–O bond vector for the complex makes a similar angle with respect to the heme normal in both the Tt ba3 (64°) and bovine enzymes (65.5°), the CO is oriented within the binuclear center quite differently in these two enzymes as illustrated in Fig. 8. In the Tt ba3 complex, the carbon atom is bonded to at a distance of 1.9 Å (top panel), while the oxygen atom is bonded to Fea3 at a distance of 2.3 Å [49]. In addition, in the Tt ba3 photoproduct, the ligand is directly above the Fea3 atom along the heme normal vector. By contrast, the CO ligand is bound quite weakly to CuB in the bovine CuB–CO photoproduct, and the CO ligand is not above the Fea3 atom, but is displaced away from CuB and the K-proton channel and toward the ligand entrance channel (Fig. 8, lower panel).
Fig. 8.

Top panel: The Tt ba3 CuB–CO transient photoproduct (PDB 3QJR, 49). Lower panel: The bovine CuB–CO transient photoproduct (PDB 3AG2, 55). In both panels, the left figure (Top) is viewed from the positive side of membrane, while the right figure (Side) is viewed from the ligand entrance channel. The “Side” view is generated from the “Top” by a left-handed 90° rotation about the horizontal axis.
2.5. Transient UV–vis spectroscopy: the photodissociation and recombination dynamics of CO-cytochrome oxidase
While infrared spectroscopy has structural specificity and allows us to follow species that are inaccessible by other spectroscopic approaches, such as in cytochrome oxidase, transient UV–visible kinetic studies of the CO photodissociation and rebinding in a variety of heme proteins have provided important information about the heme environment and the dynamics of CO in the active site cavity and its migration pathways through the proteins [56,57]. In bovine cytochrome oxidase, an increase in the intensity of the α-band (615 nm) on picosecond time scale was observed following the initial femtosecond events accompanying the photodissociation of CO from heme a3 [32]. A subsequent decrease in the α-band was observed on ~1 μs times scale, simultaneously with the loss of CO from . These picosecond and microsecond changes were associated with structural effects at heme a3 following the formation and dissociation of the complex [32]. The observed pseudo-first-order rate of rebinding of the flash-dissociated CO to cytochrome a3 of the bovine heart enzyme showed the onset of saturation at [CO] >1 mM (PCO ~1–22 atm) when measured at room temperature on micro- and millisecond time scales. This was interpreted in terms of CO, and by extension other ligands such as O2, first binding to a non-heme site, i.e. , as it migrated to heme a3. Saturation kinetics was also reported for the rebinding of the photodissociated CO to E. coli bo3 ubiquinol oxidase as a function of CO concentration [58]. Interestingly, two mutant oxidases (His333Leu and His334Leu), in which the CuB site was significantly altered, or CuB was lacking, showed no evidence of CO binding saturation up to 21 mM CO. This was interpreted as CuB acting as a way-station for CO moving to the heme. In contrast, in Tt ba3, the observed rate constant for CO rebinding to the heme a3 appears to be independent of CO over a large concentration range, 25 μM–3 mM [47]. This indicates that the pre-equilibrium of CO with a non-heme site, i.e. CuB, in ba3 is saturated at the lowest CO concentration used.
The TRIR and transient UV–visible results of the kinetics of CO rebinding following its photodissociation from the high-spin heme have been explained by the kinetics model shown in Scheme 1, which includes an obligatory binding of CO to to and from the high-spin heme. In Scheme 1, k1 represents CO binding to CuB from solution prior to the rate-limiting CO transfer to , represented by k2. The thermal dissociation of CO from is represented by k−2. The equilibrium constants are represented by K1 = k1/k−1 and K2 = k2/k−2. Based on our CO recombination kinetics of the bovine enzyme as a function of CO concentration, we calculated a value for k−2 of 0.027 s−1, which is in excellent agreement with the value of 0.023 s−1, reported previously by Gibson and Greenwood [36]. In Tt ba3, this rate is much faster or 0.8 s−1 [59].
Scheme 1.
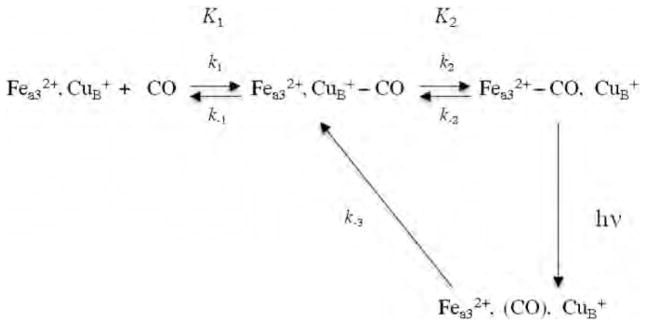
Proposed mechanism for CO photodissociation and rebinding in heme-copper oxidases.
3. O2 and NO binding in heme–copper oxidases in the absence and presence of CO
If the binding of CO to CuB in the heme-copper oxidases precedes the binding of CO to the high-spin heme, does this binding affect the access of other ligands, such as O2 and NO, to the active site in CO/NO and CO/O2 flow-flash experiments? As shown above, the complex decays with a half-life of ~1.5 μs in the bovine enzyme [32,43] while in Tt ba3 the complex decays on a millisecond time scale (Fig. 6 and [53]), much slower than that of O2 binding to heme in the aa3 oxidases (~10–20 μs at ~0.5–1 mM O2). This long lifetime of the complex in ba3 raises questions whether O2 binding to heme a3 may be impeded by the binding of the photodissociated CO to in CO/O2 flow-flash experiments on this enzyme, and secondly, whether the binding of O2 and NO to on route to heme a3 is a general feature of the ligand binding dynamics of the heme–copper oxidases as observed for CO. Results from studies aimed at answering these questions are summarized below.
3.1. Does CO impede O2 and NO access to the active site in heme-copper oxidases?
To investigate whether CO impedes access of O2 and NO to the active site in the heme-copper oxidases, we monitored the reactions of O2 and NO with bovine aa3 and Tt ba3 under more physiological conditions, namely, in the absence of CO using time-resolved optical absorption spectroscopy in combination with photolabile O2 and NO carriers [40, 60]. This technique eliminates the possible interferences from the photodissociated CO in typical CO flow-flash experiments as well as circumvents the low NO quantum yield in NO flash-photolysis studies. The results from these studies were compared to those obtained in the presence of CO using a double-laser approach in which the O2 and NO were generated by photolyzing the respective O2 and NO photolabile carrier with a 355 nm laser pulse and the CO-bound enzyme was photolyzed simultaneously using a second 532 nm laser pulse [40,60]. Time-resolved optical absorption spectra were recorded and the data were analyzed with a combination of singular value decomposition (SVD), global exponential fitting and an algebraic kinetic approach and the spectra of the intermediates were determined. The data show that O2 and NO bind to cytochrome a3 in Tt ba3 in the absence of CO with a second order rate constant of 1 × 109 M−1 s−1, which is 10-times faster than observed in the bovine enzyme (1 × 108 M−1 s−1) under the same conditions [40,60]. Moreover, the O2 and NO binding in Tt ba3 is 10-times slower in the presence of CO (1 × 108 M−1 s−1) while the presence of CO does not affect the rate of O2 and NO binding in the bovine enzyme (Fig. 9). These results show that the reactions of O2 and NO with reduced Tt ba3 are indeed compromised by the transient binding of the photodissociated CO to and that the CO flow-flash method does not give accurate results for “physiological” O2 and NO binding in ba3, i.e. that observed in the absence of CO.
Fig. 9.
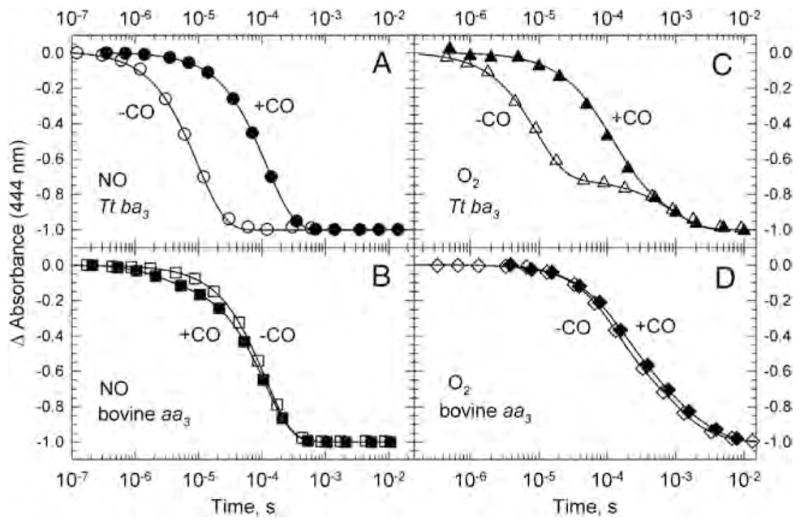
Comparison of the transient absorbance changes at 444 nm during the reaction of the fully reduced ba3 (panels A and C) and bovine aa3 (panels B and D) with photoproduced NO (A and B) and photoproduced O2 (C and D) in the presence of CO (filled symbols) and absence of CO (open symbols). The kinetics traces are from time-resolved optical absorption data recorded at multiple wavelengths and are normalized to the total absorbance change. The solid lines represent the absorbance traces at 444 nm calculated on the basis of a single exponential fit. The conditions are those reported in [40,60].
The post-photolysis structure of the bovine enzyme shows that CO is quite weakly bound to [55], while CO is bound much more tightly to CuB in Tt ba3 [49]. For example, the CuB–C distance is 2.4 Å and 1.9 Å in the bovine and ba3 structures, respectively [49,55]. Likewise, the Fe–O distance is 3.8 Å (not bound) and 2.3 Å in the bovine and ba3 post-photolysis structures, respectively. It is noteworthy that the CO in the post-photolysis bovine enzyme moves “out” of the binuclear center, i.e. away from the H2O exit channel and back toward the ligand entrance channel. This suggests that in the post-photolysis fully reduced CO-bound bovine enzyme, CO is already “escaping” the binuclear center; this is in contrast to the ba3 enzymes, in which the photodissociated CO rotates within the binuclear cavity, but remains bound to the metals. This may explain why CO impedes O2 binding in ba3 but not in the bovine enzyme.
3.2. Differences in ligand access to the active site in bovine aa3 and Tt ba3
The 10-fold faster rate of O2 and NO binding in Tt ba3 compared to bovine aa3 in the absence of CO indicates inherent structural differences between ligand access in the two enzymes, which may reflect their different physiological requirements. In the aa3 oxidases, a constriction point defined by conserved tryptophan and phenylalanine residues was identified [24,27,28], while in Tt ba3 these sites are occupied by smaller residues, tyrosine (Y133) and threonine (T231), respectively [26]. Recent experiments in our laboratory have shown that O2 and NO binding to heme a3 in the absence of CO in the Y133W and Y133W/T231F mutants of Tt ba3 is ~5 times slower than in the wild type enzyme [29]. This suggests that the significantly slower ligand binding in the bovine enzyme (1 × 108 M−1 s−1) compared to that in Tt ba3 (1 × 109 M−1 s−1) in the absence of CO is in part due to the tryptophan constriction residue in the ligand channel of the bovine aa3 (W126) impeding O2 and NO access to the active site [29]. Interestingly, mutation of the T231 residue in Tt ba3 to the corresponding phenylalanine in the aa3 oxidases did not have any effect on the ligand binding rate [29]. Classical molecular dynamics simulations of Xe and O2 diffusion to the active sites in ba3 and bovine aa3 showed that the native bovine F238 residue and the F231 side chain of the Y133W/T231F mutant, which in the crystal structures extend into the ligand channels, rotate out of the channels, resulting in no effect on ligand access in the T231F mutant and, by extension, in the bovine enzyme [29]. The rate of O2 and NO binding in the Y133W and Y133W/T231F mutants of Tt ba3 in the presence of CO was also 10-times slower than in the corresponding mutants in the absence of CO and 50 times slower than in the wild-type enzyme in the absence of CO. This demonstrates that the photodissociated CO directly or indirectly slows down ligand binding in the mutants to the same extent as in the wild type Tt ba3.
3.3. How does CO impede access of O2 and NO to the active site in Tt ba3?
In the mitochondrial enzyme, the photoproduct decays with a half-life of ~1.5 μs based on the TRIR transient at 2062 cm−1 [32,43], significantly faster than the ~10 μs binding of O2 to heme a3 (at 625 μM [O2]) observed in either the absence or presence of CO. Thus the dissociation of CO from CuB does not limit O2 or NO binding at the active site (CuB or heme a3) in the bovine enzyme at this O2 concentration.
In the absence of CO, the rate of O2 and NO binding to heme a3 in Tt ba3 is close to the diffusion-controlled limit. Therefore, if O2 and NO were to bind to prior to heme a3 in Tt ba3, this obligatory binding would not appear to limit access of the ligands to heme a3. Even in the presence of CO, the O2 and NO binding in Tt ba3 is 1 × 108M−1 s−1 (~ 10 μs at 625 μM O2), significantly faster than the millisecond CO dissociation from CuB observed in the absence of O2.
If CO remains on in Tt ba3 on millisecond time scale in the presence of O2 and the active site is only able to accommodate one ligand at the active site, O2 or NO would not be able to bind to the heme, which is clearly not the case. On the other hand, if the active site is able to accommodate two ligands, albeit transiently, and if the photodissociated CO stays bound to in Tt ba3 for tens of milliseconds in the presence of O2, then CuB would not be an obligatory way-station for O2 (or NO) binding to heme a3. In this case, O2 (NO) would presumably bind to heme a3 with CO remaining on . However, a prolonged presence of CO on CuB would not allow CuB to act as an electron donor for the rapid O–O bond cleavage (4–5 μs) observed during O2 reduction in ba3 both in the absence [40,60] and presence of CO [61]. This leads us to the conclusion that the photodissociated CO in ba3 does not remain on CuB for a prolonged period of time (hundred of microseconds) when O2 is present but still long enough to decrease the O2 binding rate to the high-spin heme by an order of magnitude. The binding of CO to CuB in Tt ba3 could either impede the binding of O2 to or if O2 does not bind to prior to heme a3, the complex could sterically restrict access of O2 to the heme. The two scenarios will be discussed in more detail below.
3.4. Is an obligatory gate for O2 binding to the high-spin heme in bovine aa3 and Tt ba3?
The binding of O2 in the heme–copper oxidases has been proposed to follow Scheme 1, namely, that the obligatory path of O2 to and from the high-spin heme involves the transient binding of O2 to , as observed for CO. In early flow-flash studies of the reaction of the reduced bovine enzyme with O2, the fast phase was found to increase proportionally with O2 concentration at low concentrations but saturate at higher concentration (~1 mM) [37,62]. Later studies used two lasers to study the kinetics of O2 binding in the bovine enzyme as a function of O2 concentration, in which the first pulse photolyzed the heme a3–CO complex and the second pulse photolyzed the early transient species in the O2 reaction, presumably the heme complex [63]. The authors found that the fast component of the reaction displayed a rate limit at higher O2 concentration (up to 700 μM). However, there was no evidence of rate limitation in the flow-flash experiment with NO, and the authors suggested that if the escape of CO from the active site in the bovine enzyme was the cause of the O2 saturation kinetics, such a mechanism would not be in place for NO; these types of measurements have not been carried out on Tt ba3. It should be noted that the limited O2 concentration in these experiments of 1 mM or less makes it difficult to ascertain unequivocally saturation kinetics behavior at high O2 concentrations as well as separate the redox processes following O2 binding. Subsequent high O2 pressure flow-flash studies of the reaction of dioxygen with the bovine enzyme (up to 16 mM O2) reported that the rate of O2 binding showed saturation kinetics at high O2 con- centration with a limiting rate constant of 1 × 106 s−1 [64]. It was suggested that this rate (~1 μs) was set by the dissociation of CO from , which occurs at approximately the same rate. The authors concluded that either the complex forms prior to transfer of O2 to the high-spin heme or that the directly blocks access of the incoming O2 to the heme. However, as discussed above, at ~625 μM O2 concentration the lifetime of the photoproduct of the bovine enzyme is too short (1.5 μs) to interfere with the ~10 μs O2 binding. High O2 pressure flow-flash measurements have not been carried out on the reaction of O2 with Tt ba3. In an FTIR study of the steady-state Tt ba3 CO complex in the presence of limited amount of O2 (70 μM), the C–O stretching frequency of the CuB–CO complex was found to shift from the usual 2053 cm−1 to 2045 cm−1 [65]. The latter peak belonged to CO as demonstrated by the appropriate shift in the 13CO spectrum. These results were interpreted as being the result of structural changes at CuB caused by O2 being close to but not bound to CuB.
3.5. Does the complex directly block access of O2 to heme a3 in Tt ba3?
Alternatively, O2 may not bind to in the heme–copper oxidases, in particular in Tt ba3, prior to being transferred to the high-spin heme. In the case of ba3, the direct binding of O2 to heme a3 may give rise to changes in the geometry around CuB from tetrahedrally-distorted square planar to more square planar, which in a concerted manner could give rise to the dissociation of CO from , thus allowing CuB to act as an electron donor to the bound dioxygen. This would require the active site in Tt ba3 to be able to transiently accommodate two ligand molecules, CO on CuB and O2 on heme a3. The relatively short distance between heme and in Tt ba3 (4.4 Å) [66] compared to that in bovine aa3 (5.1 Å) [67] would likely preclude the simultaneous binding of CO to and either O2 (or NO) to heme without some conformational changes [68]. Such structural changes as a result of ligand binding to heme a3 are supported by time-resolved magnetic circular dichroism and circular dichroism measurements of the unligated Tt ba3 formed after photodissociation of its CO complex [69]. These measurements showed spectral differences between the photolyzed enzyme and the steady-state unliganded enzyme, which were explained in terms of CO binding to heme a3 inducing a global conformational change at the active site, which persisted on a time scale comparable to that of CO rebinding. Both the “dark” heme a3–CO and “light” structures [49] show a structural distortion in heme a3, with somewhat greater distortion (the porphyrin being not planar) in the dark structure. The Fea3–CuB distance increases from 4.7 Å in the “dark” structure to 5.1 Å in the “light” structure, which may be due to tighter binding of CO to CuB than to Fea3. It should also be noted that the crystal structure of Tt ba3 shows that the high spin heme a3 is tilted away from CuB, thus providing a larger surface area to the incoming ligand than in the aa3 oxidases. This might allow a second ligand to be accommodated at the active site, an essential requirement of the NO reductase activity of this enzyme [12]. Based on theoretical studies, Blomberg and coworkers have proposed that two NO molecules bind at the active site of Tt ba3, one to heme a3 and one to CuB (through the two oxygen atoms) during the conversion of NO to N2O [70]. Low-temperature FTIR photolysis experiments of the ba3–NO complex led to the proposal of a Fea3–NO NO–CuB intermediate during the NO reductase reaction, suggesting that the ba3 binuclear center is able to transiently bind two ligands [71]. Previous spectroscopic measurements also show that two CN− molecules can be accommodated simultaneously at the active site of Tt ba3, one bound to and the other one to [72]. Several studies have reported that the A-type oxidases can simultaneously accommodate two ligands at the active site [14,15,73–76].
4. Conclusions
Several important conclusions can be drawn from the results described here. 1) Our infrared experiments, as well as UV–vis saturation CO rebinding studies as a function of CO pressure provide strong evidence that the obligatory path of CO to and from the high-spin heme in the heme–copper oxidases involves the transient binding of CO to . 2) Our TRIRLID measurements allowed us to determine the orientation of the C–O bond axis with respect to the heme normal in both the heme a3–CO complex and the CuB–CO product of the bovine enzyme, and the results are in good agreement with later crystallographic results. 3) Our time-resolved optical absorption measurements show superfast O2 and NO binding to Fea3 in Tt ba3 in the absence of CO, 1 × 109 M−1 s−1, which approaches the diffusion-controlled limit and is 10-times faster than in the bovine enzyme under the same conditions. The slower O2 and NO binding in the bovine enzyme compared to ba3 is partially due to the tryptophan constriction point residue in the O2 channel of the bovine enzyme impeding access of O2 and NO to the active site. The more open channel in Tt ba3, which allows easier access of O2 to the heme, likely reflects the functional requirements of the thermophilic bacterium, which is found under microaerobic conditions and grows optimally at 70 °C (at which temperature O2 solubility is half of that in water at 25 °C). 4) The rate of O2 and NO binding is 10-times slower in the presence of CO while in the bovine enzyme the O2 and NO binding rate is the same in the presence and absence of CO (1 × 108 M−1 s−1). These results indicate that the CO flow-flash method does not accurately reflect the O2 and NO binding in Tt ba3 under physiological conditions, namely, in the absence of CO.
Despite considerable progress, there are still some outstanding issues regarding ligand binding in the heme–copper oxidases, most importantly whether the route of O2 (NO) to and from the high-spin heme involves an obligatory binding to CuB, as has been proposed for CO, and whether this path is the same for all heme–copper oxidases. While the CO flow-flash UV–visible measurements carried out at high O2 pressure on the bovine enzyme reported a nonlinear dependence of the observed rate of O2 binding as a function of O2 concentration [64], no saturation limit was observed in flow-flash experiments on the binding of NO to the bovine enzyme [63], a ligand that is expected to model O2 binding. In the bovine enzyme, our TRIR experiments show that CO dissociates from CuB with a half-life of 1.5 μs, rapidly enough not to interfere with ~10 μs O2 (NO) binding to heme a3 at 1 mM O2. Moreover, the rate of O2 binding to heme a3 is the same in the presence and absence of CO, indicating that the complex is not sterically restricting access of O2 to the heme. Thus the evidence is inconclusive whether acts a way-station for O2 (NO) in the bovine enzyme.
In Tt ba3, our UV–visible results show that the binding of the photodissociated CO to slows the access of O2 (NO) to heme a3 by an order of magnitude compared to that observed in the absence of CO. The CuB in ba3 has significantly higher affinity for CO compared to the bovine enzyme [47], and the crystal structure of the CuB–CO complex shows that the CuB–C bond is significantly shorter (1.88 Å) [49] and stronger than the corresponding bond in the bovine enzyme (2.43 Å) [55]. Thus it seems unlikely that O2 would replace CO on . If CO remains bound to CuB for longer than a few microseconds, would not be able to provide one of the electrons required for the rapid (5 μs) breaking of the O–O bond. This is clearly not the case. We propose that the direct binding of O2 to heme a3 in Tt ba3 and the driving force for the breaking of the O–O bond cause CO to dissociate from in a concerted manner through steric and/or electronic effects, thereby allowing to act as an electron donor during the breaking of the O–O bond. For this to happen would require the transient presence of two ligands, one on heme a3 and the other on CuB. This proposal is supported by the NO reductase activity of Tt ba3, which would require two NO molecules to be transiently bound at the active site. FTIR studies aimed at resolving whether the active site in Tt ba3 is indeed able to accommodate two ligands are in progress.
Acknowledgments
This work was supported by the National Science Foundation Grant CHE-1158548 (ÓE) and the National Institutes of Health Grants GM068036 (BD).
Footnotes
This article is part of a Special Issue entitled: Vibrational Spectroscopies in Molecular Bioenergetics.
References
- 1.Brzezinski P, Larsson G. Redox-driven proton pumping by heme–copper oxidases. Biochim Biophys Acta. 2003;1605:1–13. doi: 10.1016/s0005-2728(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 2.Einarsdóttir Ó. Fast reactions of cytochrome oxidase. Biochim Biophys Acta. 1995;1229:129–147. doi: 10.1016/0005-2728(94)00196-c. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson-Miller S, Babcock GT. Heme/copper terminal oxidases. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 4.Wikström MKF. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 5.Chan SI, Li PM. Cytochrome c oxidase: understanding nature’s design of a proton pump. Biochemistry. 1990;29:1–12. doi: 10.1021/bi00453a001. [DOI] [PubMed] [Google Scholar]
- 6.Wikström M, Krab K, Saraste M. Cytochrome Oxidase—A Synthesis. Academic Press; New York: 1981. [Google Scholar]
- 7.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta. 2001;1504:46–57. doi: 10.1016/s0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- 8.Ignarro LJ. Nitric Oxide: Biology and Pathobiology. Academic Press; San Diego, CA: 2000. [Google Scholar]
- 9.Brunori M, Giuffrè A, Forte E, Mastronicola M, Barone BC, Sarti P. Control of cytochrome c oxidase activity by nitric oxide. Biochim Biophys Acta. 2004;1655:365–371. doi: 10.1016/j.bbabio.2003.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Butler CS, Forte E, Scandurra FM, Arese M, Giuffrè A, Greenwood C, Sarti P. Cytochrome bo3 from Escherichia coli: the binding and turnover of nitric oxide. Biochem Biophys Res Commun. 2002;296:1272–1278. doi: 10.1016/s0006-291x(02)02074-0. [DOI] [PubMed] [Google Scholar]
- 11.Forte E, Urbani A, Saraste M, Sarti P, Brunori M, Giuffrè A. The cytochrome cbb3 from Pseudomonas stutzeri displays nitric oxide reductase activity. Eur J Biochem. 2001;268:6486–6491. doi: 10.1046/j.0014-2956.2001.02597.x. [DOI] [PubMed] [Google Scholar]
- 12.Giuffrè A, Stubauer G, Sarti P, Brunori M, Zumft WG, Buse G, Soulimane T. The heme–copper oxidases of Thermus thermophilus catalyze the reduction of nitric oxide: evolutionary implications. Proc Natl Acad Sci U S A. 1999;96:14718–14723. doi: 10.1073/pnas.96.26.14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Reimann J, Lepp H, Drici N, Ädelroth P. Vectorial proton transfer coupled to reduction of O2 and NO by a heme–copper oxidase. Proc Natl Acad Sci U S A. 2008;105:20257–20262. doi: 10.1073/pnas.0805429106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stubauer G, Giuffrè A, Brunori M, Sarti P. Cytochrome c oxidase does not catalyze the anaerobic reduction of NO. Biochem Biophys Res Commun. 1998;245:459–465. doi: 10.1006/bbrc.1998.8457. [DOI] [PubMed] [Google Scholar]
- 15.Zhao XJ, Sampath V, Caughey WS. Cytochrome c oxidase catalysis of the reduction of nitric oxide to nitrous oxide. Biochem Biophys Res Commun. 1995;212:1054–1060. doi: 10.1006/bbrc.1995.2076. [DOI] [PubMed] [Google Scholar]
- 16.Hemp J, Gennis RB. Diversity of the heme–copper superfamily in Archaea: insights from genomics and structural modeling. Results Probl Cell Differ. 2008;45:1–31. doi: 10.1007/400_2007_046. [DOI] [PubMed] [Google Scholar]
- 17.Pereira MM, Santana M, Teixeira M. A novel scenario for the evolution of haem–copper oxygen reductases. Biochim Biophys Acta. 2001;1505:185–208. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]
- 18.Buschmann S, Warkentin E, Xie H, Langer JD, Ermler U, Michel H. The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science. 2010;329:327–330. doi: 10.1126/science.1187303. [DOI] [PubMed] [Google Scholar]
- 19.Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 Å resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc Natl Acad Sci U S A. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soulimane T, Buse G, Bourenkov GP, Bartunik HD, Huber R, Than ME. Structure and mechanism of the aberrant ba3-cytochrome c oxidase from Thermus thermophilus. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mitzushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1731. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 22.Abramson J, Riistama S, Larsson G, Jasaitis A, Svensson-Ek M, Laakkonen L, Puustinen A, Iwata S, Wikström M. The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat Struct Biol. 2000;7:910–917. doi: 10.1038/82824. [DOI] [PubMed] [Google Scholar]
- 23.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 24.Svensson-Ek M, Abramson J, Larsson G, Törnroth S, Brzezinski P, Iwata S. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 25.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 26.Luna VM, Chen Y, Fee JA, Stout CD. Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry. 2008;47:4657–4665. doi: 10.1021/bi800045y. [DOI] [PubMed] [Google Scholar]
- 27.Hofacker I, Schulten K. Oxygen and proton pathways in cytochrome c oxidase. Proteins Struct Funct Genet. 1998;30:100–107. [PubMed] [Google Scholar]
- 28.Salomonsson L, Lee A, Gennis RB, Brzezinski P. A single-amino-acid lid renders a gas-tight compartment within a membrane-bound transporter. Proc Natl Acad Sci U S A. 2004;101:11617–11621. doi: 10.1073/pnas.0402242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald W, Funatogawa C, Li Y, Szundi I, Chen Y, Fee JA, Stout CD, Einarsdottir Ó. Ligand access to the active site in Thermus thermophilus ba3 and bovine heart aa3 cytochrome oxidases. Biochemistry. 2013;52:640–652. doi: 10.1021/bi301358a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alben JO, Moh PP, Fiamingo FG, Altschuld RA. Cytochrome oxidase (a3) heme and copper observed by low-temperature Fourier transform infrared spectroscopy of the CO complex. Proc Natl Acad Sci U S A. 1981;78:234–237. doi: 10.1073/pnas.78.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Einarsdóttir Ó, Choc MG, Weldon S, Caughey WS. The site and mechanism of dioxygen reduction in bovine heart cytochrome c oxidase. J Biol Chem. 1988;263:13641–13654. [PubMed] [Google Scholar]
- 32.Einarsdóttir Ó, Dyer RB, Lemon DD, Killough PM, Hubig SM, Atherton SJ, López-Garriga JJ, Palmer G, Woodruff WH. hotodissociation and recombination of carbonmonoxy cytochrome oxidase: dynamics from picoseconds to kiloseconds. Biochemistry. 1993;32:12013–12024. doi: 10.1021/bi00096a011. [DOI] [PubMed] [Google Scholar]
- 33.Einarsdóttir Ó, Killough PM, Fee JA, Woodruff WH. An infrared study of the binding and photodissociation of carbon monoxide in cytochrome ba3 from Thermus thermophilus. J Biol Chem. 1989;264:2405–2408. [PubMed] [Google Scholar]
- 34.Yoshikawa S, Choc MG, O’Toole MC, Caughey WS. An infrared study of CO binding to heart cytochrome c oxidase and hemoglobin A. J Biol Chem. 1977;252:5498–5508. [PubMed] [Google Scholar]
- 35.Brzezinski P, Gennis RB. Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibson QH, Greenwood C. Reactions of cytochrome oxidase with oxygen and carbon monoxide. Biochem J. 1963;86:541–554. doi: 10.1042/bj0860541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill BC, Greenwood C. The reaction of fully reduced cytochrome c oxidase with oxygen studied by flow-flash spectrophotometry at room temperature. Biochem J. 1984;218:913–921. doi: 10.1042/bj2180913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szundi I, Funatogawa C, Cassano J, McDonald W, Ray J, Hiser C, Ferguson-Miller S, Gennis RB, Einarsdóttir Ó. Spectral identification of intermediates generated during the reaction of dioxygen with the wild-type and EQ(I-286) mutant of Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 2012;51:9302–9311. doi: 10.1021/bi301166u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szundi I, Van Eps N, Einarsdóttir Ó. pH dependence of the reduction of dioxygen to water by cytochrome c oxidase. 2. Branched electron transfer pathways linked by proton transfer. Biochemistry. 2003;42:5074–5090. doi: 10.1021/bi020483e. [DOI] [PubMed] [Google Scholar]
- 40.Szundi I, Funatogawa C, Fee JA, Soulimane T, Einarsdóttir Ó. CO impedes superfast O2 binding in ba3 cytochrome oxidase from Thermus thermophilus. Proc Natl Acad Sci U S A. 2010;107:21010–21015. doi: 10.1073/pnas.1008603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caughey WS. In: Methods for Determining Metal Ion Environments in Proteins: Structure and Function of Metallo-proteins. Darnall DW, Wilkins RG, editors. Elsevier/North-Holland; New York: 1980. pp. 95–115. [Google Scholar]
- 42.Fiamingo FG, Altschuld RA, Moh PP, Alben JO. Dynamic interactions of CO with a3Fe and CuB in cytochrome c oxidase in beef heart mitochondria studied by Fourier transform infrared spectroscopy at low temperature. J Biol Chem. 1982;257:1639–1650. [PubMed] [Google Scholar]
- 43.Dyer RB, Einarsdóttir Ó, Killough PM, López-Garriga JJ, Woodruff WH. Transient binding of photodissociated CO to of eukaryotic cytochrome oxidase at ambient temperature. Direct evidence from time-resolved infrared spectroscopy. J Am Chem Soc. 1989;111:7657–7659. [Google Scholar]
- 44.Dyer RB, López-Garriga JJ, Einarsdóttir Ó, Woodruff WH. The orientation of CO in carbonmonoxy cytochrome oxidase and its transient photoproducts. Direct evidence from time-resolved infrared linear dichroism. J Am Chem Soc. 1989;111:8962–8963. [Google Scholar]
- 45.Dyer RB, Petersen KA, Stoutland PO, Woodruff WH. Ultrafast photoinduced ligand transfer in carbonmonoxy cytochrome c oxidase. Observation by picosecond infrared spectroscopy. J Am Chem Soc. 1991;113:6276–6277. [Google Scholar]
- 46.Dyer RB, Peterson KA, Stoutland PO, Woodruff WH. Picosecond infrared study of the photodynamics of carbonmonoxy-cytochrome c oxidase. Biochemistry. 1994;33:500–507. doi: 10.1021/bi00168a015. [DOI] [PubMed] [Google Scholar]
- 47.Woodruff WH, Dyer RB, Einarsdóttir Ó. Spectroscopy, dynamics, and function of cytochrome oxidase. In: Clark RJH, Hester RE, editors. Biological Spectroscopy, Part B. John Wiley and Sons Ltd; Chichester, England: 1993. pp. 189–233. [Google Scholar]
- 48.Woodruff WH, Einarsdóttir Ó, Dyer RB, Bagley KA, Palmer G, Atherton SJ, Goldbeck RA, Dawes TD, Kliger DS. Nature and functional implications of the cyto- chrome a3 transients after photodissociation of CO-cytochrome oxidase. Proc Natl Acad Sci U S A. 1991;88:2588–2592. doi: 10.1073/pnas.88.6.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu B, Zhang Y, Sage JT, Soltis SM, Doukov T, Chen Y, Stout CD, Fee JA. Structural changes that occur upon photolysis of the Fe(II)a3–CO complex in the cytochrome ba3-oxidase of Thermus thermophilus: a combined X-ray crystallographic and infrared spectral study demonstrates CO binding to CuB. Biochim Biophys Acta. 2012;1817:658–665. doi: 10.1016/j.bbabio.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Einarsdóttir Ó, Dyer B, Killough PM, Fee JA, Woodruff WH. Fourier transform infrared and resonance Raman characterization of cytochrome ba3 from Thermus thermophilus. SPIE Proc. 1989;1055:254–262. [Google Scholar]
- 51.Oertling WA, Surerus KK, Einarsdóttir Ó, Fee JA, Dyer RB, Woodruff WH. Spectroscopic characterization of cytochrome ba3, a terminal oxidase from Thermus thermophilus: comparison of the a3/CuB site to that of bovine cytochrome aa3. Bio- chemistry. 1994;33:3128–3141. doi: 10.1021/bi00176a048. [DOI] [PubMed] [Google Scholar]
- 52.Stoutland PO, Lambry J-C, Martin J-L, Woodruff WH. Femtosecond dynamics of reduced cytochrome oxidase and its CO derivatives. J Phys Chem. 1991;1991:6406–6408. [Google Scholar]
- 53.Koutsoupakis K, Stavrakis S, Pinakoulaki E, Soulimane T, Varotsis C. Observation of the equilibrium CuB–CO complex and functional implications of the transient heme a3 propionates in cytochrome ba3–CO from Thermus thermophilus. Fourier transform infrared (FTIR) and time-resolved step-scan FTIR studies. J Biol Chem. 2002;277:32860–32866. doi: 10.1074/jbc.M204943200. [DOI] [PubMed] [Google Scholar]
- 54.Bailey JA, Tomson FL, Mecklenburg SL, MacDonald GM, Katsonouri A, Puustinen A, Gennis RB, Woodruff WH, Dyer RB. Time-resolved step-scan Fourier transform infrared spectroscopy of the CO adducts of bovine cytochrome c oxidase and of cytochrome bo3 from Escherichia coli. Biochemistry. 2002;41:2675–2683. doi: 10.1021/bi010823g. [DOI] [PubMed] [Google Scholar]
- 55.Muramoto K, Ohta K, Shinzawa-Itoh K, Kanda K, Taniguchi M, Nabekura H, Yamashita E, Tsukihara T, Yoshikawa S. Bovine cytochrome c oxidase structures en- able O2 reduction with minimization of reactive oxygens and provide a proton- pumping gate. Proc Natl Acad Sci U S A. 2010;107:7740–7745. doi: 10.1073/pnas.0910410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Birukou I, Maillett DH, Birukova A, Olson JS. Modulating distal cavities in the alpha and beta subunits of human HbA reveals the primary ligand migration pathway. Biochemistry. 2011;50:7361–7374. doi: 10.1021/bi200923k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vos MH. Ultrafast dynamics of ligands within heme proteins. Biochim Biophys Acta. 2008;1777:15–31. doi: 10.1016/j.bbabio.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Lemon DD, Calhoun MW, Gennis RB, Woodruff WH. The gateway to the active site of heme–copper oxidases. Biochemistry. 1993;32:11953–11956. doi: 10.1021/bi00096a002. [DOI] [PubMed] [Google Scholar]
- 59.Giuffrè A, Forte E, Antonini G, D’Itri E, Brunori M, Soulimane T, Buse G. Kinetic properties of ba3 oxidase from Thermus thermophilus: effect of temperature. Bio- chemistry. 1999;38:1057–1065. doi: 10.1021/bi9815389. [DOI] [PubMed] [Google Scholar]
- 60.Einarsdóttir Ó, Funatogawa C, Soulimane T, Szundi I. Kinetic studies of the reactions of O2 and NO with reduced Thermus thermophilus ba3 and bovine aa3 using photolabile carriers. Biochim Biophys Acta. 2012;1817:672–679. doi: 10.1016/j.bbabio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Siletsky SA, Belevich I, Jasaitis A, Konstantinov AA, Wikström M, Soulimane T, Verkhovsky MI. Time-resolved single-turnover of ba3 oxidase from Thermus thermophilus. Biochim Biophys Acta. 2007;1767:1383–1392. doi: 10.1016/j.bbabio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 62.Greenwood C, Gibson QH. The reaction of reduced cytochrome c oxidase with oxygen. J Biol Chem. 1967;242:1782–1787. [PubMed] [Google Scholar]
- 63.Blackmore RS, Greenwood C, Gibson QH. Studies of the primary oxygen intermediate in the reaction of fully reduced cytochrome oxidase. J Biol Chem. 1991;266:19245–19249. [PubMed] [Google Scholar]
- 64.Bailey JA, James CA, Woodruff WH. Flow-flash kinetics of O2 binding to cytochrome c oxidase at elevated [O2]: observations using high-pressure stopped- flow for gaseous reactants. Biochem Biophys Res Commun. 1996;220:1055–1060. doi: 10.1006/bbrc.1996.0531. [DOI] [PubMed] [Google Scholar]
- 65.Koutsoupakis K, Stavrakis S, Soulimane T, Varotsis C. Oxygen-linked equilibrium CuB–CO species in cytochrome ba3 oxidase from Thermus thermophilus. Implications for an oxygen channel ar the CuB site. J Biol Chem. 2003;278:14893–14896. doi: 10.1074/jbc.M210293200. [DOI] [PubMed] [Google Scholar]
- 66.Hunsicker-Wang LM, Pacoma RL, Chen Y, Fee JA, Stout CD. A novel cryoprotection scheme for enhancing the diffraction of crystals of recombinant cytochrome ba3 oxidase from Thermus thermophilus. Acta Crystallogr. 2005;D61:340–343. doi: 10.1107/S0907444904033906. [DOI] [PubMed] [Google Scholar]
- 67.Muramoto K, Hirata K, Shinzawa-Itoh K, Yoko-o S, Yamashita E, Aoyama H, Tsukihara T, Yoshikawa S. A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc Natl Acad Sci U S A. 2007;104:7881–7886. doi: 10.1073/pnas.0610031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu B, Chen Y, Doukov T, Soltis SM, Stout CD, Fee JA. Combined microspectrophotometric and crystallographic examination of chemically reduced and X-ray radiation-reduced forms of cytochrome ba3 oxidase from Thermus thermophilus: structure of the reduced form of the enzyme. Biochemistry. 2009;48:820–826. doi: 10.1021/bi801759a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goldbeck RA, Einarsdóttir Ó, Dawes TD, O’Connor DB, Surerus KK, Fee JA, Kliger DS. Magnetic circular dichroism study of cytochrome ba3 from Thermus thermophilus: spectral contributions from cytochromes b and a3 and nanosecond spectroscopy of CO photodissociation intermediates. Biochemistry. 1992;31:9376–9387. doi: 10.1021/bi00154a008. [DOI] [PubMed] [Google Scholar]
- 70.Blomberg LM, Blomberg MR, Siegbahn PE. A theoretical study on nitric oxide reductase activity in a ba3-type heme–copper oxidase. Biochim Biophys Acta. 2006;1757:31–46. doi: 10.1016/j.bbabio.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 71.Hayashi T, Lin IJ, Chen Y, Fee JA, Moënne-Loccoz P. Fourier transform infrared characterization of a CuB–nitrosyl complex in cytochrome ba3 from Thermus thermophilus: relevance to NO reductase activity in heme–copper terminal oxidase. J Am Chem Soc. 2007;129:14952–14958. doi: 10.1021/ja074600a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Surerus KK, Oertling WA, Fan C, Gurbiel RJ, Einarsdottir Ó, Antholine WE, Dyer RB, Hoffman BM, Woodruff WH, Fee JA. Reaction of cyanide with cytochrome ba3 from Thermus thermophilus: spectroscopic characterization of the Fe(II)a3–CN·Cu(II)B–CN complex suggests four 14N atoms are coordinated to CuB. Proc Natl Acad Sci U S A. 1992;89:3195–3199. doi: 10.1073/pnas.89.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brudvig GW, Stevens TH, Chan SI. Reactions of nitric oxide with cytochrome c oxidase. Biochemistry. 1980;19:5275–5285. doi: 10.1021/bi00564a020. [DOI] [PubMed] [Google Scholar]
- 74.Young LJ, Caughey WS. Oxygenation of carbon monoxide by bovine heart cytochrome c oxidase. Biochemistry. 1986;25:152–161. doi: 10.1021/bi00349a022. [DOI] [PubMed] [Google Scholar]
- 75.Pilet E, Nitschke W, Rappaport F, Soulimane T, Lambry JC, Liebl U, Vos MH. NO binding and dynamics in reduced heme–copper oxidases aa3 from Paracoccus denitrificans and ba3 from Thermus thermophilus. Biochemistry. 2004;43:14118–14127. doi: 10.1021/bi0488808. [DOI] [PubMed] [Google Scholar]
- 76.Hayashi T, Lin MT, Ganesan K, Chen Y, Fee JA, Gennis RB, Moenne-Loccoz P. Accommodation of two diatomic molecules in cytochrome bo: insights into NO reductase activity in terminal oxidases. Biochemistry. 2009;48:883–890. doi: 10.1021/bi801915r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharrock M, Yonetani T. Low-temperature flash photolysis studies of cytochrome oxidase and its environment. Biochim Biophys Acta. 1977;462:718–730. doi: 10.1016/0005-2728(77)90113-x. [DOI] [PubMed] [Google Scholar]