
Figure 2. A schematic diagram of the possible mechanisms of presynaptic Gi/o coupled GPCR activation-induced LTD.
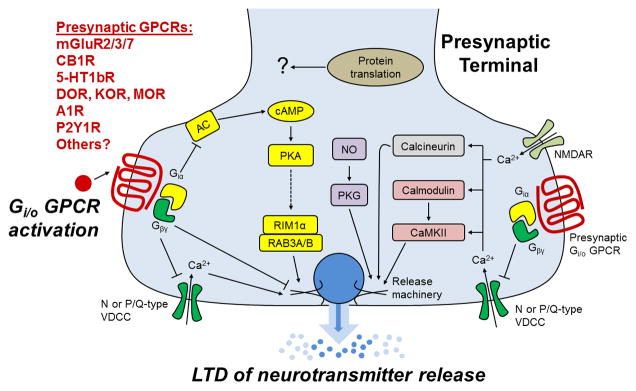
Activation of presynaptic Gi/o-coupled GPCRs results in the dissociation of the Gαi and Gβγ subunits from the receptor complex. Gβγ subunits directly and negatively couple to voltage-dependent Ca2+ channels (VDCCs) resulting in reduced Ca2+ entry into the presynaptic terminal. Reduced Ca2+ influx decreases vesicle fusion and neurotransmitter release probability. In addition, Gβγ may directly inhibit components of the vesicular release machinery (e.g. SNAP25). The Gαi subunits negatively couple to adenylyl cyclase (AC) resulting in decreased cAMP levels. A decrease in cAMP levels results in dampened PKA activity, which is associated with reduced functionality of RIM1α. RIM1α complexes with the vesicular proteins RAB3A and RAB3B to enhance neurotransmitter release. Therefore, reduced PKA activity directly or indirectly inhibits this complex’s ability to promote neurotransmission. In addition, Ca2+ entry through VDCCs or NMDARs activates the presynaptic kinases calmodulin and CaMKII and the protein phosphatase calcineurin. GPCR modulation of presynaptic Ca2+ levels likely influences the activity of these proteins, which subsequently alters neurotransmitter release. Although not directly modulated by presynaptic Gi/o GPCRs, nitric oxide (NO) signaling intersects with GPCR-mediated signaling to promote LTD. NO signaling results in PKG activation that influences release machinery function. Finally, at some synapses protein translation appears to be a necessary component of LTD maintenance. The mechanisms that engage the protein translation apparatus as well as the proteins that are expressed are currently unknown.