

Abstract
Objective
To characterize spirometry and height changes in cohorts of 6-year-olds with cystic fibrosis (CF).
Study design
Global Lung Initiative (GLI) forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC) and FEV1/FVC and CDC height-for-age (HFA) Z-scores were generated for 6-year-olds in the from the CF Foundation Patient Registry (CFFPR) each year from 1994 and 2012. Z-score mean differences were analyzed by t-test and time trends of means by least squares regression for all children and for subgroups (sex, F580del mutation genotype, Medicaid insurance, and prenatal/newborn screening identification). Z-score distributions were compared by two-sample Kolmogorov-Smirnov tests.
Results
11,670 children with CF were studied, of whom 50.5% were males, 50.2% had the F508del/F508del genotype, and 46.6% were insured by Medicaid. Mean HFA, FEV1 and FVC Z-scores increased significantly over the period in the entire population and all subgroups (P<0.001), but FEV1/FVC Z-scores were below normal and did not change significantly. In 2012, children identified by screening had significantly higher mean HFA (P=0.002), FEV1 (P<0.001) and FVC (P<0.001) Z-scores than those not screened, with 90% of FVC and 71.4% of FEV1 Z-scores greater than predicted by the Normal distribution; FEV1/FVC Z-scores were not different between screening groups.
Conclusions
Consistent, significant increases in HFA, FEV1, and FVC occurred from 1994–2012, but FEV1/FVC, a measure of airway obstruction, did not appreciably change. FVC and FEV1 Z-score distributions suggest that normative equation reference populations under-predict lung volumes of children with CF, but the reason(s) for this remain unclear.
Cystic fibrosis (CF) is characterized by progressive pulmonary obstruction and inflammation resulting in premature death.1,2 Spirometry and in particular forced expiratory volume in 1 second (FEV1) and FEV1 % predicted are benchmark measures used to guide CF management,3,4 evaluate intervention efficacy,5 and stratify lung disease by stage.6,7 Because spirometry requires patient cooperation, it is only sporadically performed in children under the age of 6 years; at age 6 we can first characterize CF birth cohorts with respect to measures that will become integral to their management throughout their lifetimes.
Median FEV1 % predicted has steadily increased over the past two decades for cohorts of 6-year-olds followed in the Cystic Fibrosis Foundation Patient Registry (CFFPR), increasing from ~86% predicted in 1992 to ~98% predicted by 2012.8 This improvement may be associated with the introduction of chronic respiratory therapies, expansion of CF newborn screening,9 greater awareness of markers of early lung disease in newborns with CF,10–14 greater availability of information regarding lung function outcomes across CF care centers, and the CFF Quality Improvement initiative, among other things. However, it is not clear whether lung function gains were enjoyed uniformly across the population, or instead resulted from disproportionate gains in children of a particular sex, those with milder CF mutations, those with favorable socio-economic status, or indeed, by the inclusion of children identified by newborn screening that would not have been diagnosed and included in earlier years. Thus, it is not clear how close we are to achieving the goal of “normal” lung function across the entire population of 6-year-olds with CF.
In order to better characterize the health of successive cohorts of children with CF, we have analyzed spirometric and morphometric data from 19 successive cohorts of 6-year-olds followed in the CFFPR from 1994 through 2012. We have employed Z-scores, which facilitate comparisons across subgroups and across measures. Z-scores describe the number of standard deviations (SD) an observation is from the population mean, and are derived by dividing differences between observed values and reference mean values by the SD for the measure from the reference population.
Methods
Children followed in the CFFPR who were 6 years of age between 1994 and 2012 were studied. For each child, FEV1, forced vital capacity (FVC), height, and age were collected from the encounter at which his or her highest FEV1% predicted (Wang normative equations)15 had been recorded during the 365 days between 6th and 7th birthdays. Demographic data were also collected, including sex, race, identification by newborn or prenatal screening, CFTR genotype, and Medicaid insurance. Medicaid status was used as the socioeconomic status (SES) indicator in part to avoid the missing data for mother’s education and the inaccuracy of using the median income by zip code resulting from infrequent updating of zip codes. Only children for whom both FEV1 and FVC values were available were included in analyses. When available, forced expiratory flow 25%-75% (FEF25-75) values were analyzed. FEF25-75, FEV1, FVC, and FEV1/FVC % predicted and associated Z-scores were calculated using child sex, height, age, and race by Global Lung Initiative (GLI) reference equations.16 Height-for-age (HFA) Z-scores were calculated using Centers for Disease Control and Prevention (CDC) growth charts (http://www.cdc.gov/growthcharts/2000growthchart-us.pdf). Mean and SD values for spirometric variables and for height were calculated for each review year.
Z-score distributions were compared over time and with the normal distribution. Within-year and between-year mean values for males and females, children insured and not insured by Medicaid, and children identified by prenatal or newborn screening were studied to identify subgroup differences. Finally, values for F508del homozygotes were compared with those of the entire population as well as to children with other recorded CFTR genotypes. Within- and between-year means were compared by t-test with alpha = 0.05. Time trends in annual mean values from 1994 to 2012 were studied by least squares regression. Differences in Z-score distributions between groups were compared by plotting cumulative frequencies along with the Normal distribution and by two-sample Kolmogorov-Smirnov tests of the null hypothesis that the two sample populations were drawn from the same distribution.
As an alternate approach to characterizing changes in spirometry among cohorts, changes in FVC values (in liters) within narrow height ranges were analyzed across the period. For each year, males and females were grouped by encounter height rounded down to the nearest centimeter and then annual changes in median FVC values from 1994 to 2012 for each height group were analyzed by least squares regression. Only height groups in which at least at least two children were represented in each calendar year were studied.
Guardians of children followed in the CFFPR began providing consent for collected data beginning in 2003. Data from patients in the CFFPR were de-identified prior to analysis and analyses were approved prospectively by the University Hospitals Case Medical Center IRB. Analyses were performed with Microsoft Excel 2013, a macro downloaded from http://www.lungfunction.org,16 and an online statistical applet (http://scistatcalc.blogspot.co.uk/2013/11/kolmogorov-smirnov-test-calculator.html). All P values are two-sided and no adjustment was made for multiple comparisons.
Results
A total of 11,670 children, 5,893 (50.5%) males and 5,777 (49.5%) females, were studied. The racial background of most children was white (94.8%), with 4.3% African, 0.7%, American Indian or Alaska Native, 0.6% Asian, and 1.2% of ‘other’ descent; about 1.5% of children were identified as being of more than one race. Among the 11,310 children (89.0%) for which Hispanic heritage information was available, 8.1% were identified as Hispanic. Racial composition changed somewhat over the period, with the proportion of white children declining (from 95.3% in 1994 to 89.5% in 2012) accompanied by corresponding increases in representation of children of other racial and ethnic groups. The proportion of children identified as being of Hispanic descent nearly doubled, from 5.2% in 1994 to 9.9% in 2012. Slightly more than half of children (50.2%) were homozygous for the F508del CFTR mutation, and less than half (46.6%) were reported to be insured by Medicaid during their review year. Between 1994 and 2012, the proportion of children with CF identified by prenatal or newborn screening increased from 4.0% to 30.9%.
Mean HFA, FEV1, FVC and FEV1/FVC Z-scores in 1994 were lower than predicted for their peers without CF (Table I and Figure 1). Z-scores measure the distance (in SD) between an individual’s value and the reference population mean, with negative scores representing values below the mean and positive scores above. The mean GLI FEV1 in 1994 was 86.8% predicted (SD = 20.8) and the mean FVC was 92.1% predicted (SD = 19.6). Mean HFA, FEV1 and FVC Z-scores increased between 1994 and 2012 (Table I) and annual increases in means were consistent across the period (Figure 1). Broad shifts in HFA, FEV1, and FVC distributions towards improvement occurred between 1994 and 2012 (Figure 2). Cumulative Z-score plots such as those in Figure 2 allow for comparison of variable distributions among subpopulations with different variances. In Figure 2, a random sampling of a variable measured among 6-year-olds without CF should theoretically follow the normal distribution (eg, 68.2% of children should have Z-scores between −1.0 and + 1.0). In contrast, children with consistently subnormal variable values, such as HFA in children with CF, have distributions shifted to the left of the Normal distribution. “Movement” of a cumulative Z-score plot from the left to the right suggests that a variable’s distribution has become more like that of the general population, or “more normal.”
Table 1.
Mean Z-scores among CF 6-year-olds, 1994 to 2012
| 1994 | 2012 | Regression of Means, 1994–2012 | ||||
|---|---|---|---|---|---|---|
| Mean (SD) | Mean (SD) | P | Slope | R Square | P | |
| All 6-year-olds | N = 624 | N = 680 | ||||
| HFA Z-score | −0.69 (0.96) | −0.36 (1.04) | <0.001 | 0.0202/yr | 0.7861 | <0.001 |
| FEV1 Z-score | −1.03 (1.63) | −0.36 (1.45) | <0.001 | 0.0377/yr | 0.9321 | <0.001 |
| FVC Z-score | −0.65 (1.57) | −0.02 (1.42) | <0.001 | 0.0342/yr | 0.8932 | <0.001 |
| FEV1/FVC Z-score | −0.68 (1.33) | −0.58 (1.17) | 0.17 | 0.0058/yr | 0.1759 | 0.074 |
| F508del/F508del | N = 319 | N = 316 | ||||
| HFA Z-score | −0.73 (0.94) | −0.34 (1.02) | <0.001 | 0.0190/yr | 0.7339 | <0.001 |
| FEV1 Z-score | −1.03 (1.64) | −0.34 (1.45) | <0.001 | 0.0349/yr | 0.8150 | <0.001 |
| FVC Z-score | −0.66 (1.54) | 0.02 (1.44) | <0.001 | 0.0337/yr | 0.7921 | <0.001 |
| FEV1/FVC Z-score | −0.66 (1.32) | −0.62 (1.16) | 0.66 | 0.0009/yr | 0.0030 | 0.83 |
| Males | N = 330 | N = 345 | ||||
| HFA Z-score | −0.58 (0.96) | −0.29 (1.04) | <0.001 | 0.0172/yr | 0.6597 | <0.001 |
| FEV1 Z-score | −0.93 (1.60) | −0.40 (1.33) | <0.001 | 0.0338/yr | 0.8206 | <0.001 |
| FVC Z-score | −0.48 (1.54) | −0.00 (1.28) | <0.001 | 0.0288/yr | 0.7783 | <0.001 |
| FEV1/FVC Z-score | −0.74 (1.30) | −0.64 (1.15) | 0.29 | 0.0073/yr | 0.1815 | 0.069 |
| Females | N = 294 | N = 335 | ||||
| HFA Z-score | −0.82 (0.95) | −0.43 (1.03) | <0.001 | 0.0235/yr | 0.7836 | <0.001 |
| FEV1 Z-score | −1.14 (1.66) | −0.30 (1.56) | <0.001 | 0.0423/yr | 0.9170 | <0.001 |
| FVC Z-score | −0.83 (1.59) | −0.03 (1.54) | <0.001 | 0.0403/yr | 0.8778 | <0.001 |
| FEV1/FVC Z-score | −0.61 (1.36) | −0.52 (1.19) | 0.41 | 0.0043/yr | 0.0953 | 0.20 |
| Insured by Medicaid | N = 284 | N = 315 | ||||
| HFA Z-score | −0.88 (0.95) | −0.53 (1.02) | <0.001 | 0.0203/yr | 0.6790 | <0.001 |
| FEV1 Z-score | −1.33 (1.65) | −0.58 (1.52) | <0.001 | 0.0436/yr | 0.8326 | <0.001 |
| FVC Z-score | −0.89 (1.59) | −0.20 (1.49) | <0.001 | 0.0408/yr | 0.7926 | <0.001 |
| FEV1/FVC Z-score | −0.80 (1.39) | −0.64 (1.24) | 0.14 | 0.0054/yr | 0.0656 | 0.29 |
| Not Insured by Medicaid | N = 340 | N = 365 | ||||
| HFA Z-score | −0.54 (0.94) | −0.21 (1.04) | <0.001 | 0.0206/yr | 0.8096 | <0.001 |
| FEV1 Z-score | −0.78 (1.57) | −0.16 (1.35) | <0.001 | 0.0337/yr | 0.8783 | <0.001 |
| FVC Z-score | −0.44 (1.52) | 0.14 (1.33) | <0.001 | 0.0292/yr | 0.8106 | <0.001 |
| FEV1/FVC Z-score | −0.57 (1.27) | −0.53 (1.10) | 0.63 | 0.0064/yr | 0.2450 | 0.031 |
Figure 1.
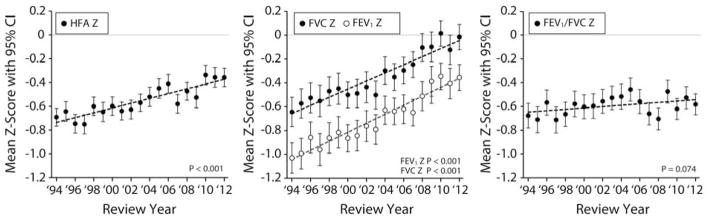
Mean HFA, FEV1, FVC, and FEV1/FVC Z-scores among CF 6-year-old Cohorts from 1994 to 2012. Least squares linear regressions (dashed lines) of mean Z-scores by review year are shown. Bars represent 95% confidence intervals; P values are for test of non-zero slope.
Figure 2.

Cumulative frequencies of HFA, FVC, FEV1, and FEV1/FVC Z-Scores in 1994 and 2012. Plots show the proportions of children (Y axis) with Z-scores less than or equal to a given value (X axis). P values are for two-sample Kolmogorov-Smirnov tests for differences between 1994 and 2012 distributions.
In contrast to HFA, FEV1, and FVC, the difference in mean FEV1/FVC between 1994 and 2012 did not reach statistical significance (P = 0.172), nor did the modest trend of annual improvement in FEV1/FVC means across the period (P = 0.074) (Table I and Figure 1). Although 2012 FEV1/FVC distributions were somewhat improved over 1994 distributions, changes were largely limited to children with Z-scores below −1; there was essentially no change in the distribution of FEV1/FVC Z-scores > −1.0 (Figure 2, D). The CFFPR began collecting FEF25-75 measures in 2010. In 2012, the mean FEF25-75 Z-score among 652 children with values was −0.58 (SD = 1.32).
Essentially identical results were obtained when analyses were limited to F508del homozygotes, with statistically significant changes in HFA, FEV1, and FVC accompanied by essentially unchanged FEV1/FVC (Table I). In 1994, mean HFA, FEV1, and FVC Z-scores of females were lower than those of males (Table I). However, both groups experienced increases in mean HFA, FEV1, and FVC during the period (Table I), with differences between groups failing to reach statistical significance in 2012. As before, mean FEV1/FVC Z-scores were not different between males and females in 1994 and neither sex experienced significant changes over the study period (Table I).
Mean Z-scores were consistently lower among Medicaid-insured children than among children not insured by Medicaid (Table I). Despite these differences, the magnitudes of mean HFA, FEV1 and FVC improvements from 1994 to 2012 were not markedly different between the two groups (Table I). Changes (or lack thereof) in FEV1/FVC followed the general trends reported for other subgroups (Table I). However, the modest 0.0063/yr rate of change in FEV1/FVC Z-score means among children not insured by Medicaid was statistically significant (P=0.037), whereas the slightly smaller 0.0054/yr rate among Medicaid-insured children did not reach statistical significance (P=0.29). Identification by prenatal or newborn screening was associated with greater HFA, FEV1, and FVC (but not FEV1/FVC) means in the cohorts since 2008 (Table II and Figure 2).
Table 2.
Effect of Prenatal or Newborn Screening CF Diagnosis on Mean Z-scores, 2008–2012
| CF Diagnosis | 2008 (N=647) | 2009 (N=586) | 2010 (N=634) | 2011 (N=610) | 2012 (N=680) |
|---|---|---|---|---|---|
| Not Screened, N (%) | 535 (82.7) | 450 (76.7) | 491 (77.4) | 450 (73.8) | 470 (69.0) |
| Screened, N (%) | 112 (17.3) | 136 (23.4) | 143 (22.6) | 160 (26.2) | 210 (31.0) |
| HFA Z-score | |||||
| Not Screened, Mean (SD) | −0.53 (0.97) | −0.61 (1.08) | −0.43 (1.03) | −0.44 (1.05) | −0.44 (1.03) |
| Screened, Mean (SD) | −0.21 (0.95) | −0.26 (1.04) | −0.01 (1.08) | −0.12 (1.02) | −0.16 (1.03) |
| P-valuea | 0.002 | 0.001 | <0.001 | 0.001 | 0.001 |
| FEV1 Z-score | |||||
| Not Screened, Mean (SD) | −0.55 (1.61) | −0.46 (1.53) | −0.38 (1.41) | −0.52 (1.60) | −0.52 (1.46) |
| Screened, Mean (SD) | −0.34 (1.51) | −0.15 (1.56) | −0.22 (1.50) | −0.08 (1.62) | 0.01 (1.35) |
| P-value | 0.202 | 0.043 | 0.23 | 0.003 | <0.001 |
| FVC Z-score | |||||
| Not Screened, Mean (SD) | −0.15 (1.54) | −0.17 (1.54) | 0.00 (1.33) | −0.25 (1.54) | −0.16 (1.45) |
| Screened, Mean (SD) | 0.13 (1.38) | 0.13 (1.52) | 0.08 (1.52) | 0.23 (1.57) | 0.31 (1.29) |
| P-value | 0.073 | 0.048 | 0.52 | 0.001 | <0.001 |
| FEV1/FVC Z-score | |||||
| Not Screened, Mean (SD) | −0.68 (1.20) | −0.47 (1.21) | −0.65 (1.15) | −0.51 (1.20) | −0.61 (1.22) |
| Screened, Mean (SD) | −0.84 (1.14) | −0.49 (1.18) | −0.52 (1.11) | −0.56 (1.10) | −0.52 (1.07) |
| P-value | 0.19 | 0.89 | 0.23 | 0.65 | 0.39 |
| FEF25-75 Z-score | |||||
| Not Screened, Mean (SD) | −0.60 (1.37)b | −0.58 (1.40)c | −0.67 (1.34)d | ||
| Screened, Mean (SD) | −0.60 (1.24)e | −0.52 (1.39)f | −0.39 (1.27)g | ||
| P-value | 0.99 | 0.63 | 0.012 | ||
T-test of means;
N = 467;
N = 429;
N = 446;
N = 127;
N = 150;
N = 206
When changes in median FVC (in liters) from 1994 to 2012 were analyzed by least squares regression within narrow height ranges, most male and female height subgroups had a positive slope in FVC change over the study period (Figures 3 and 4; available at www.jpeds.com). Of 20 male height subgroups, 18 had a positive slope for median FVC change over the period (of which 8 had P <0.05), and 16 of 18 female slopes were similarly positive (10 with P <0.05).
Discussion
We have compared height and spirometric Z-score distributions for successive cohorts of 6-year-olds followed in the CFFPR both to each other and to the GLI reference population to characterize health improvements between 1994 and 2012. Children experienced substantial shifts toward normal values in HFA, FVC and FEV1 from 1994 to 2012, most notably among children identified by prenatal or newborn screening (Table II and Figure 2). Mean FEV1 improved from 86.8% predicted (SD = 20.8) to 95.3% predicted (SD = 18.4) and mean FVC improved from 92.1% predicted (SD = 19.6) to 100.0% predicted (SD = 17.7). Improvements were not limited to children with milder CFTR mutation phenotypes or attributable to a change in the mix of mutations represented in the registry, as distributions among F508del homozygotes were indistinguishable from those of the remainder of the 2012 cohort. Although Medicaid-insured children had lower (ie, less normal) Z-scores, they also experienced substantial HFA, FEV1, and FVC increases over the study period. In contrast, surprisingly little change in FEV1/FVC, a measurement of airway obstruction, was observed between 1994 and 2012, with distributions remaining below normal, even among the children identified by screening (Figure 2). Modest shifts towards normality were only observed for FEV1/FVC Z-scores <-1, suggesting some reduction in the prevalence of children with more severe obstruction.
Our analyses have inherent limitations. First, we selected spirometry data from a single encounter to describe the lung health of individual children, when measures among stable children are somewhat variable and acute disease-related declines are common. By choosing the best FEV1 % predicted recorded between the 6th and 7th birthday, we reduced the probability of capturing data associated with acute declines in lung function, but as a result may have slightly overestimated lung health within cohorts. However, because this effect would have been consistent across cohorts, we believe the changes observed over the study period are real and not artifacts of data collection. Second, serial cross-sectional analyses of successive cohorts can be affected by demographic differences between cohorts. Ethnic/racial compositions of our cohorts were not constant, and to the extent that CF disease phenotypes differ among racial or ethnic subpopulations, analyses across cohorts may have been affected. As an example, increasing proportions of Hispanic children in latter cohorts presumably had the effect of reducing improvements in mean FVC and FEV1 Z-scores observed across the period relative to what would have been observed among non-Hispanic white children alone. However, these demographic changes were modest and we believe are unlikely to have substantially influenced our results. Finally, there may be discrepancies between the genetic backgrounds of children with CF and those of GLI reference populations not accounted for by the stated race of the children.
In 2012 189 FVC Z-scores from screened children (90%) exceeded those predicted by the Normal distribution, as did 150 FEV1 Z-scores (71.4%) (Figure 2). This suggests that the reference population is not perfectly representative, as we would not expect measures from children with CF (even with ‘normal’ lung function) to substantially exceed those predicted by a representative reference population. Although a recent study identified subgroups of children with CF where GLI and Wang reference equations provide different FEV1 and FVC percent predicted results,17 we did not find appreciable differences depending on equation used within our narrow age-range cohorts (Figure 5; available at www.jpeds.com), suggesting that the above normal FEV1 and FVC distributions we observed in screened children are not peculiar to GLI reference equations.
Why 6-year-olds with CF appear imperfectly matched with their peers without CF with respect to FVC and FEV1 is not clear. It could be that the two groups have never been well-matched, with differences only becoming apparent as the lung health of successive cohorts of children with CF have improved. For example, it may be that the emphasis placed on spirometry for children with CF has produced more accomplished spirometric test behavior by children with CF. Alternatively, changes in nutritional management of infants and children with CF may have resulted in overall greater relative lung growth than that of the population without CF. The nutritional status of 3-year-olds with CF has been correlated with their FEV1 % predicted at age 6,18 but the distinction between improved function and anatomically larger lungs has not been addressed. We believe that it is unlikely that increased heights between 1994 and 2012 alone can account for apparent differences between 6-year-olds with CF and reference populations for three reasons: (1) GLI reference equations adjust for height16; (2) the height distribution of the population with CF remains below that of the reference population (Figure 2); and (3) there is a trend of increasing FVC across the period among both boys and girls with CF of the same height (Figures 3 and 4).
It is unclear whether subnormal FEV1/FVC distributions in children with CF are clinically relevant. It may be that the imperfect FEV1 and FVC representation by reference populations also extends to FEV1/FVC, such that even healthy children with CF may have sub-normal FEV1/FVC. The phenomenon of near-normal FEV1 and above-normal FVC contributing to a low FEV1/FVC has been recently described in subgroups of children without CF.19 Alternatively, recent improvements in FEV1 and FVC among children with CF may have been realized by better lung growth but not accompanied by substantial reduction of obstruction. In 2012, FEF25-75 Z-score means were below normal, even among screened children (Table II), suggesting relatively reduced expiratory flow in a population with exceptional FEV1 and FVC Z-scores. Interestingly, recent observations in piglets with CF suggest that airflow obstruction may be a congenital abnormality associated with CF.20
Larger lungs would presumably benefit a population for which the primary cause of death is loss of lung function,8 but reduced lung obstruction would also be beneficial. If subnormal FEV1/FVC is the result of obstruction, younger children might benefit from an expansion of the systematic approach previously applied to their nutrition21 to include chronic respiratory therapies shown to slow obstruction and spare FEV1 in older patients. Use of chronic CF respiratory therapies in young children has increased: <10% of North American children with CF <6 years of age received dornase alfa at least once in 1995, compared with about 50% in 2005.4 In 2012, 40% of children <3 years of age and 67.5% of those 3 to 5 years of age were treated at least once with dornase alfa, with proportionally fewer treated with hypertonic saline or inhaled corticosteroids.8 However, US guidelines for infants with CF suggest that chronic respiratory therapies be considered for that minority of infants and children who display symptoms of lung disease,21 and studies of newborns with CF have shown that lung inflammation, reduced flow, ventilation anomalies, early bronchiectasis, and infection occur in a majority of infants, most of whom do not present with signs and symptoms of disease.10–14
With recent expansion of CF newborn screening to every US state, almost all children in future cohorts will have been identified by screening. Our results suggest that these children will have FEV1 and FVC values exceeding those of reference populations, but with below normal FEV1/FVC. Investigation is warranted to determine if reduced FEV1/FVC at age 6 years is associated with poorer outcomes later in life and whether increased early intervention with respiratory therapies has the potential to increase FEV1/FVC in these children.
Acknowledgments
The authors thanks the children, families, and CF caregivers who have contributed data to the CFFPR, and the CFFPR support staff for their assistance. We also thank Stefanie Millar (ICON Clinical Research) for assistance with HFA Z-scores, Alijah Ahmed for his Kolmogorov-Smirnov test calculator applet, and Drs Patrick A. Flume (served as an advisor/consultant to Aptalis, Bayer, Boehringer Ingelheim, CF Foundation, Enanta, Genentech, Gilead Sciences, Insmed, Kalobios, NIH, Novartis, Pharmaxis, Savara, and Vertex) and Wayne J. Morgan (consultant to The CF Foundation and Genentech).
M.K. was supported by the National Institutes of Health (P30 DK27651) and the Cystic Fibrosis Foundation (CFFT KONSTA09Y0), and has served as an advisor/investigator for Aradigm, Celtaxsys, Digestive Care Inc, Genentech, Gilead Sciences, Insmed, Novartis, Savara, and Vertex. D.vD. has served in the past year as an advisor/consultant for Affinium Pharmaceuticals, Aptalis Pharma, Aradigm, Baxter Healthcare, the US CF Foundation, CURx Pharma, Forest Pharmaceuticals, Genentech, Gilead Sciences, ICON Clinical Sciences, Kalobios, and MedImmune. D.P. is employed by ICON Clinical Research, which provides clinical research services to pharmaceutical, biotechnology, and device companies.
Abbreviations
- CDC
Centers for Disease Control and Prevention
- CF
cystic fibrosis
- CFFPR
Cystic Fibrosis Foundation Patient Registry
- CI
confidence interval
- GLI
Global Lung Initiative
- HFA
height-for-age
- FEV1
forced expiratory volume in 1 second
- FEF25-75
forced expiratory flow 25%–75%
- FVC
forced vital capacity
- SD
standard deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med. 1996;154:1229–56. doi: 10.1164/ajrccm.154.5.8912731. [DOI] [PubMed] [Google Scholar]
- 2.Chmiel JF, Berger M, Konstan MW. The role of inflammation in the pathophysiology of CF lung disease. Clin Rev Allergy Immunol. 2002 Aug;23:5–27. doi: 10.1385/CRIAI:23:1:005. [DOI] [PubMed] [Google Scholar]
- 3.Johnson C, Butler SM, Konstan MW, Morgan W, Wohl ME. Factors influencing outcomes in cystic fibrosis: a center-based analysis. Chest. 2003 Jan;123:20–7. doi: 10.1378/chest.123.1.20. [DOI] [PubMed] [Google Scholar]
- 4.Konstan MW, VanDevanter DR, Rasouliyan L, Pasta DJ, Yegin A, Morgan WJ, et al. Trends in the use of routine therapies in cystic fibrosis: 1995–2005. Pediatr Pulmonol. 2010 Dec;45:1167–72. doi: 10.1002/ppul.21315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.VanDevanter DR, Konstan MW. Outcome measures for clinical trials assessing treatment of cystic fibrosis lung disease. Clin Invest. 2012;2:163–175. doi: 10.4155/cli.11.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schluchter MD, Konstan MW, Drumm ML, Yankaskas JR, Knowles MR. Classifying severity of cystic fibrosis lung disease using longitudinal pulmonary function data. Am J Respir Crit Care Med. 2006;174:780–6. doi: 10.1164/rccm.200512-1919OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konstan MW, Wagener JS, VanDevanter DR. Characterizing aggressiveness and predicting future progression of CF lung disease. J Cyst Fibros. 2009 Jun;8(Suppl 1):S15–9. doi: 10.1016/S1569-1993(09)60006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cystic Fibrosis Foundation Patient Registry. 2012 Annual Data Report to the Center Directors. Bethesda, MD: Cystic Fibrosis Foundation; 2013. [Google Scholar]
- 9.Grosse SD, Boyle CA, Botkin JR, Comeau AM, Kharrazi M, Rosenfeld M, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep. 2004;53:1–36. Review. [PubMed] [Google Scholar]
- 10.Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. 2009;180:146–152. doi: 10.1164/rccm.200901-0069OC. [DOI] [PubMed] [Google Scholar]
- 11.Stick SM, Brennan S, Murray C, Douglas T, von Ungern-Sternberg BS, Garratt LW, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. 2009;155:623–628. doi: 10.1016/j.jpeds.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Hall GL, Logie KM, Parsons F, Schulzke SM, Nolan G, Murray C, et al. Air Trapping on Chest CT Is Associated with Worse Ventilation Distribution in Infants with Cystic Fibrosis Diagnosed following Newborn Screening. PLoS ONE. 2011;6:e23932. doi: 10.1371/journal.pone.0023932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoo AF, Thia LP, Nguyen TT, Bush A, Chudleigh J, Lum S, et al. Lung function is abnormal in 3-month-old infants with cystic fibrosis diagnosed by newborn screening. Thorax. 2012;67:874–881. doi: 10.1136/thoraxjnl-2012-201747. [DOI] [PubMed] [Google Scholar]
- 14.Mott LS, Park J, Murray CP, Gangell CL, de Klerk NH, Robinson PJ, et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax. 2012;67:509–16. doi: 10.1136/thoraxjnl-2011-200912. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Dockery DW, Wypij D, Fay ME, Ferris BG., Jr Pulmonary function between 6 and 18 years of age. Pediatr Pulmonol. 1993;15:75–88. doi: 10.1002/ppul.1950150204. [DOI] [PubMed] [Google Scholar]
- 16.Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40:1324–43. doi: 10.1183/09031936.00080312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanojevic S, Stocks J, Bountziouka V, Aurora P, Kirkby J, Bourke S, et al. The impact of switching to the new global lung function initiative equations on spirometry results in the UK CF Registry. J Cyst Fibros. 2014;13:319–27. doi: 10.1016/j.jcf.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, et al. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr. 2003;142:624–30. doi: 10.1067/mpd.2003.152. [DOI] [PubMed] [Google Scholar]
- 19.Quanjer PH, Weiner DJ. Interpretative consequences of adopting the Global Lungs 2012 reference equations for spirometry for children and adolescents. Pediatr Pulmonol. 2014;49:118–25. doi: 10.1002/ppul.22876. [DOI] [PubMed] [Google Scholar]
- 20.Adam RJ, Michalski AS, Bauer C, Abou Alaiwa MH, Gross TJ, Awadalla MS, et al. Air trapping and airflow obstruction in newborn cystic fibrosis piglets. Am J Respir Crit Care Med. 2013;188:1434–41. doi: 10.1164/rccm.201307-1268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borowitz D, Robinson KA, Rosenfeld M, Davis SD, Sabadosa KA, et al. Cystic Fibrosis Foundation. Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. 2009;155:S73–93. doi: 10.1016/j.jpeds.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]