

Abstract
Transcriptional activation throughout the eukaryotic lineage has been tightly linked with disruption of nucleosome organization at promoters, enhancers, silencers, insulators and locus control regions due to transcription factor binding. Regulatory DNA thus coincides with open or accessible genomic sites of remodeled chromatin. Current chromatin accessibility assays are used to separate the genome by enzymatic or chemical means and isolate either the accessible or protected locations. The isolated DNA is then quantified using a next-generation sequencing platform. Wide application of these assays has recently focused on the identification of the instrumental epigenetic changes responsible for differential gene expression, cell proliferation, functional diversification and disease development. Here we discuss the limitations and advantages of current genome-wide chromatin accessibility assays with especial attention on experimental precautions and sequence data analysis. We conclude with our perspective on future improvements necessary for moving the field of chromatin profiling forward.
Electronic supplementary material
The online version of this article (doi:10.1186/1756-8935-7-33) contains supplementary material, which is available to authorized users.
Keywords: Chromatin, MNase, DNase, ATAC, FAIRE, Sequencing, Library, Epigenome, Histone, Nucleosome
Introduction: chromatin accessibility
Eukaryotic chromatin is tightly packaged into an array of nucleosomes, each consisting of a histone octamer core wrapped around by 147 bp of DNA and separated by linker DNA [1–3]. The nucleosomal core consists of four histone proteins [1] that can be post-translationally altered by at least 80 known covalent modifications [4, 5] or replaced by histone variants [6–8]. Positioning of nucleosomes throughout a genome has a significant regulatory function by modifying the in vivo availability of binding sites to transcription factors (TFs) and the general transcription machinery and thus affecting DNA-dependent processes such as transcription, DNA repair, replication and recombination [9]. Experiments designed to decipher how nucleosome positioning regulates gene expression have led to the understanding that transcriptional activation coincides with nucleosome perturbation, whereas transcriptional regulation requires the repositioning of nucleosomes throughout the eukaryotic lineage [10–18].
Nucleosome eviction or destabilization at promoters and enhancers results from the binding of specific regulatory factors responsible for transcriptional activation in eukaryotes [19, 20]. Open or accessible regions of the genome are, thus, regarded as the primary positions for regulatory elements [21] and have been historically characterized by nuclease hypersensitivity in vivo[22]. Notably, changes in chromatin structure have been implicated with many aspects of human health, as a result of mutations in chromatin remodelers that affect nucleosome positioning [23–25]. Therefore, current interest is placed on collecting and comparing genome-wide chromatin accessibility, to locate instrumental epigenetic changes that accompany cell differentiation, environmental signaling and disease development. Large collaborative projects such as ENCODE [26] have become part of this major effort.
Low-throughput experiments in Drosophila using DNase I and MNase treatment, provided the first demonstration that active chromatin coincides with nuclease hypersensitivity, that is chromatin accessibility [27–30]. Currently, all chromatin accessibility assays separate the genome by enzymatic or chemical means and isolate either the accessible or protected locations. Isolated DNA is then quantified using a next-generation sequencing (NGS) platform. In this review, we focus on the latest methods for identifying chromatin accessibility genome-wide, and discuss the considerations for experimental design and data analysis. We conclude with current limitations that need to be overcome for this field to move forward.
Review
Assays for genome-wide chromatin accessibility
General considerations
Chromatin accessibility approaches measure directly the effect of chromatin structure modifications on gene transcription, in contrast to histone chromatin immunoprecipitation with NGS (ChIP-seq) (for a thorough review on ChIP-seq read [31–33]) where such effects must be inferred by presence or absence of overlapping histone tail modifications. Also, chromatin accessibility assays do not require antibodies or epitope tags that can introduce potential bias. An important limitation with all chromatin accessibility experiments is the lack of a standard for the number of replicates required to achieve accurate and reproducible results. This is because replicate number depends on the achieved signal-to-noise ratio, which can vary depending on the assay used, the assay conditions, and the cell or tissue type. In addition, replicate number is a function of technical variance, which is also experiment-specific and difficult to model in a generalized format. Following we discuss chromatin accessibility assays that directly (DNase-seq, FAIRE-seq and ATAC-seq) isolate accessible locations of a genome separate from MNase-seq, which indirectly evaluates chromatin accessibility, and present their principal mode of action, examples of application and main experimental considerations (Table 1).
Table 1.
Current genome-wide high-throughput chromatin accessibility assays
| Cell type/Number | Sequencing type | Traditional approach | Genomic target | Experimental considerations | Key references | |
|---|---|---|---|---|---|---|
| MNase-seq | Any cell type 1 to 10 million cells | Paired-end or Single-end | MNase digests unprotected DNA | Maps the total nucleosome population in a qualitative and quantitative manner | 1. Requires many cells. | [37, 46, 49] |
| 2. Laborious enzyme titrations. | ||||||
| 3. Probes total nucleosomal population, not active regulatory regions only. | ||||||
| 4. Degrades active regulatory regions, making their detection possible only indirectly. | ||||||
| 5. Requires 150 to 200 million reads for standard accessibility studies of the human genome. | ||||||
| DNase-seq | Any cell type 1 to 10 million cells | Paired-end or Single-end | DNase I cuts within unprotected DNA | Maps open chromatin | 1. Requires many cells. | [61, 75, 76] |
| 2. Time-consuming and complicated sample preparations. | ||||||
| 3. Laborious enzyme titrations. | ||||||
| 4. Requires 20 to 50 million reads for standard accessibility studies of the human genome. | ||||||
| FAIRE-seq | Any cell type 100,000 to 10 million cells | Paired-end or Single-end | Based on the phenol-chloroform separation of nucleosome-bound and free sonicated areas of a genome, in the interphase and aqueous phase respectively | Maps open chromatin | 1. Low signal-to-noise ratio, making computational data interpretation very difficult. | [86–90] |
| 2. Results depend highly on fixation efficiency. | ||||||
| 3. Requires 20 to 50 million reads for standard accessibility studies of the human genome. | ||||||
| ATAC-seq | 500 to 50,000 freshly isolated cells | Paired-end | Unfixed nuclei are tagged in vitro with adapters for NGS by purified Tn5 transposase. Adapters are integrated into regions of accessible chromatin | Maps open chromatin, TF and nucleosome occupancy | 1. Contamination of generated data with mitochondrial DNA. | [103] |
| 2. Immature data analysis tools. | ||||||
| 3. Requires 60 to 100 million reads for standard accessibility studies of the human genome. |
ATAC: assay for transposase-accessible chromatin; DNase I: deoxyribonuclease I; FAIRE: formaldehyde-assisted isolation of regulatory elements; MNase: micrococcal nuclease.
MNase-seq: an indirect chromatin accessibility assay
MNase is commonly reported as a single-strand-specific endo-exonuclease, although its exonuclease activity appears to be limited to only a few nucleotides on a single strand before cleavage of the antiparallel strand occurs [34–36]. Since the early 1970s MNase digestion has been applied to study chromatin structure in a low-throughput manner [37–40] and later in combination with tiled microarrays [41–44]. Currently, MNase digestion is used with NGS (MNase-seq or MAINE-seq [45]) for genome-wide characterization of average nucleosome occupancy and positioning in a qualitative and quantitative manner. In a typical MNase-seq experiment, mononucleosomes are extracted by extensive MNase treatment of chromatin that has been crosslinked with formaldehyde (Figure 1) [46]. The nucleosomal population is subsequently submitted to single-end (identifies one end of template) or paired-end (identifies both ends of template) NGS with a varying level of coverage depending on the exact goal of the experiment [31].
Figure 1.
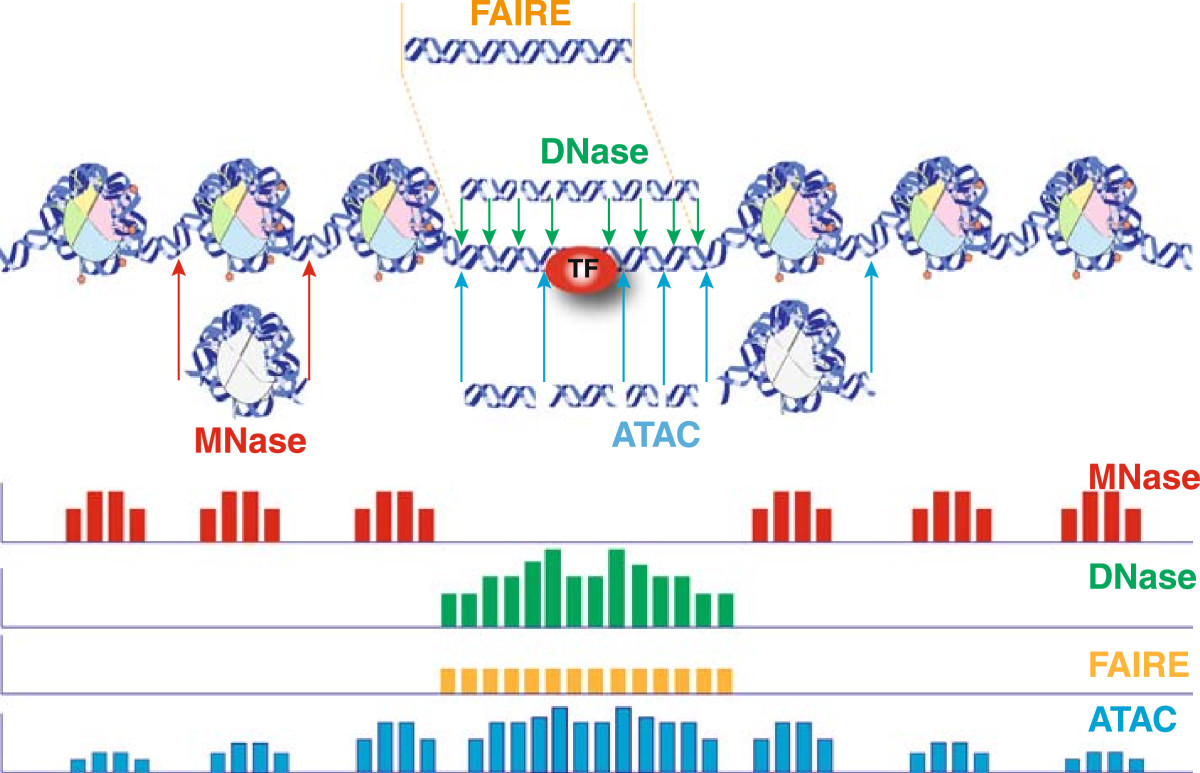
Schematic diagram of current chromatin accessibility assays performed with typical experimental conditions. Representative DNA fragments generated by each assay are shown, with end locations within chromatin defined by colored arrows. Bar diagrams represent data signal obtained from each assay across the entire region. The footprint created by a transcription factor (TF) is shown for ATAC-seq and DNase-seq experiments.
MNase-seq thus probes chromatin accessibility indirectly by unveiling the areas of the genome occupied by nucleosomes and other regulatory factors. Commonly referred to as a nucleosome occupancy assay, it shares same principal mode of action (enzymatic cleavage) and can provide information on TF occupancy as other chromatin accessibility assays. MNase-seq has been implemented in a number of organisms, ranging from yeast to humans, for the mapping of chromatin structure [47–49]. In addition, MNase digestion has been successfully combined with ChIP-seq for enrichment of regulatory factors or histone-tail modifications and variants. Henikoff et al. [50] have also introduced a modified MNase-seq protocol for library preparation of fragments down to 25 bp, allowing the mapping of both nucleosomes and non-histone proteins with high resolution.
Important considerations in the design of MNase-seq experiments include extent of chromatin crosslinking and level of digestion. Traditionally, chromatin accessibility experiments have been conducted with formaldehyde as a crosslinking agent to capture in vivo protein-nucleic acid and protein-protein interactions [51]. It has been observed that in the absence of crosslinking, nucleosome organization can change during regular chromatin preparation steps and thus use of formaldehyde is recommended for accurate characterization of chromatin structure [31]. Also, MNase has been shown to have a high degree of AT-cleavage specificity in limiting enzyme concentrations [52–54] and comparisons between different experiments will vary for technical reasons unless MNase digestion conditions are tightly controlled [55–57]. MNase titration experiments specifically support differential digestion susceptibility of certain nucleosome classes, with nucleosomes within promoter and ‘nucleosome-free’ regions being highly sensitive [50, 58, 59]. Thus, it has been suggested that combination of templates from different levels of MNase digestion may alleviate biased sampling of mononucleosome populations [58].
However, the cause of differences in MNase-seq output across differential levels of enzymatic digestion is difficult to assess due to the effect of inter-nucleosomal linker length on the recovered signal [55]. MNase digestion simulation experiments have provided evidence that nucleosome configurations with or near long linkers are sampled easier compared to nucleosomes with normal linkers at low levels of MNase digestion and this sampling bias dissipitates with increased levels of enzymatic cleavage (80 or 100% monos) [55]. Comparison of in vivo experimental data of two distinct nucleosome configurations from different MNase-seq technical preparations supports the same conclusions, and underscores the importance of standardized collection of mononucleosomes for accurate and reproducible comparisons [55]. Specifically, extensive (approximately 95 to 100% mononucleosomes) digestion of a standardized initial amount of crosslinked chromatin is considered ideal for comparisons of different MNase-seq experiments, since at that level of digestion all linkers are cut and the recovered signal is not confounded by nucleosome configuration [31, 55].
Overall, MNase-seq is a superior method for probing genome-wide nucleosome distributions and also provides an accurate way for assessing TF occupancy in a range of cell types [60]. However, it requires a large number of cells and careful enzymatic titrations for accurate and reproducible evaluation of differential substrates.
Direct chromatin accessibility assays
DNase-seq
Historically, open chromatin has been identified by the hypersensitivity of genomic sites to nuclease treatment with MNase and the non-specific double-strand endonuclease DNase I [61]. In a typical experiment, low concentrations of DNase I liberate accessible chromatin by preferentially cutting within nucleosome-free genomic regions characterized as DNase I hypersensitive sites (DHSs) (Figure 1). Early low-throughput experiments, provided the first demonstration that active genes have an altered chromatin conformation that makes them susceptible to digestion with DNase I [61]. Further research in Drosophila and other eukaryotes, supported the conserved observation that chromatin structure is disrupted during gene activation and that DHSs are the primary sites of active chromatin rendering access of trans-factors to regulatory elements [14, 27, 28, 62–65]. It has later been shown that DHSs result during gene activation [17], due to loss or temporal destabilization of one or more nucleosomes from cis-regulatory elements with the combinatorial action of ATP-dependent nucleosome- and histone-remodelers [20, 66, 67].
Traditionally, identification of DHSs has been based on Southern blotting with indirect end-labeling [28] and involves laborious and time-consuming steps that limit the applicability of the method to a narrow extent of the genome. Further attempts to improve the efficiency and resolution of the method have used low-throughput sequencing, real-time PCR strategies and later hybridization to tiled microarrays [68–74]. The advent of NGS gave rise to DNase-seq allowing the genome-wide identification of DHSs with unparalleled specificity, throughput and sensitivity in a single reaction. In recent times the drop of sequencing costs and the increased quality of the data have made DNase-seq the ‘golden standard’, for probing chromatin accessibility. During a typical DNase-seq experiment, isolated nuclei are submitted to mild DNase I digestion according to the Crawford or Stamatoyannopoulos protocol [75, 76]. In the Crawford protocol, DNase I digested DNA is embedded into low-melt gel agarose plugs to prevent further shearing. Optimal digestions are selected by agarose pulsed field gel electrophoresis, with an optimal smear range from 1 MB to 20 to 100 KB, and are blunt-end ligated to a biotinylated linker. After secondary enzymatic digestion with MmeI, ligation of a second biotinylated linker and library amplification, the digested population is assayed using NGS [75]. In the Stamatoyannopoulos protocol, DNA from nuclei is digested with limiting DNase I concentrations and assessed by q-PCR and/or agarose gel electrophoresis. Optimal digestions are purified with size selection of fragments smaller than 500 bp using sucrose gradients, and are submitted for high-throughput sequencing after library construction [76]. The main difference between the two protocols is that the first one depends on the single enzymatic cleavage of chromatin, whereas the latter requires double cleavage events in close proximity to each other. The Stamatoyannopoulos protocol has been preferentially used by the ENCODE consortium.
DNase-seq has been extensively used by the ENCODE consortium [26] and others to unveil cell-specific chromatin accessibility and its relation to differential gene expression in various cell lines [21, 77–79]. It has also been modified to study rotational positioning of individual nucleosomes [80] based on the inherent preference of DNase I to cut within the minor groove of DNA at approximately every ten bp around nucleosomes [79, 81, 82]. In addition, binding of sequence-specific regulatory factors within DHSs can affect the intensity of DNase I cleavage and generate footprints (digital genomic footprinting (DGF) or DNase I footprinting) that have been used to study TF occupancy at nucleotide resolution in a qualitative and quantitative manner [83]. DGF with deep sequencing has been implemented to uncover cell-specific TF binding motifs in humans, yielding expansive knowledge on regulatory circuits and the role of TF binding in relation to chromatin structure, gene expression, and cellular differentiation [19, 78]. Due to its high resolution, DGF has also allowed the probing of functional allele-specific signatures within DHSs [78].
The main controversy over DNase-seq is the ability for DNase I to introduce cleavage bias [31, 79, 81, 82, 84], thus affecting its use as a reliable TF footprint detection assay. Two recent publications clearly demonstrate that cleavage signatures traditionally attributed to protein protection of underlying nucleotides, are detected even in the absence of TF binding as a result of DNase I inherent sequence preferences that span over more than two orders of magnitude [84, 85]. This observation is strongly supported by frequent lack of correspondence between TF binding events detected with ChIP-seq versus DGF [85]. Also, TFs with transient DNA binding times in living cells leave minimal to no detectable footprints at their sites of recognition, making the quality of footprinting highly factor-dependent [84, 85]. Collectively, these findings challenge previous DGF research on TF footprinting and its applicability as a reliable recognition assay of complex factor-chromatin interactions in a dynamic timescale.
Less concerning limitations of DNase-seq are that it requires many cells and involves many sample preparation and enzyme titration steps. Success of this assay depends on the quality of nuclei preparations and small-scale preliminary experiments are essential to ascertain the exact amount of detergent needed for cell lysis [76]. Also, DNase I concentrations may need to be adjusted empirically depending on initial type and number of cells, the lot of DNase I used and the exact purpose of the experiment [84]. Overall, DNase-seq represents a reliable and robust way to identify active regulatory elements across the genome and in any cell type from a sequenced species, without a priori knowledge of additional epigenetic information. Its reliability as a TF footprint detection assay in a temporal scale is questionable and needs to be investigated further in detail.
FAIRE-seq
One of the easiest methods for directly probing nucleosome-depleted areas of a genome is FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) (Figure 1), although the high background in the output data limits its usefulness [15, 86–89]. FAIRE is based on the phenol-chloroform separation of nucleosome-bound and free areas of a genome in the interphase and aqueous phase respectively. The procedure involves the initial crosslinking of chromatin with formaldehyde to capture in vivo protein-DNA interactions, and subsequent shearing of chromatin with sonication. Following phenol-chloroform extraction, nucleosome-depleted areas of the genome are released to the aqueous phase of the solution due to much higher crosslinking efficiency of histones to DNA, compared to other regulatory factors [87, 90]. The chromatin-accessible population of fragments can then be detected by quantitative PCR, tiling DNA microarrays [15, 86] or more recently with paired-end or single-end NGS (FAIRE-seq) [87, 91].
Initially demonstrated to identify accessible regulatory elements in Saccharomyces cerevisiae[90], FAIRE has been extended to a wide range of eukaryotic cells and tissues, consistently demonstrating a negative relationship with nucleosome occupancy and an overlap with various cell type-specific marks of active chromatin [15, 45, 86, 87, 92, 93]. This assay has been instrumental for the identification of active regulatory elements in a number of human cell lines by ENCODE [26]. It has been used widely to detect open chromatin in normal and diseased cells [86, 91, 94, 95], to associate specific chromatin states with known sequence variants of disease susceptibility [91] or allele-specific signatures [96], and to decipher the effects of TF binding to chromatin structure [97, 98].
Overall, FAIRE enriches directly for areas of active chromatin, with the added benefit that the nucleosome-depleted regions are not degraded, it can be applied to any type of cells or tissue and that there is no requirement for initial preparation of cells and laborious enzyme titrations [15, 86, 89, 94]. FAIRE has been shown to identify additional distal regulatory elements not recovered by DNase-seq, although it remains unclear what these sites represent [94]. In addition, FAIRE overcomes the sequence-specific cleavage bias observed with MNase and DNase I, and thus represents an ancillary approach for these assays [52–54, 60, 99].
Success of any FAIRE-seq experiment heavily depends on adequate fixation efficiency that can alter depending on cell permeability, composition and a variety of other physiological factors. For most mammalian cells, 5 minutes of fixation time is usually ample [89]. Fungi and plants may require a much higher fixation time [15, 93] or improved fixation solutions [100] and optimization is necessary to avoid inconsistent results. Also, FAIRE has lower resolution in identifying open-chromatin at promoters of highly expressed genes compared to DNase-seq [94]. FAIRE’s major limitation, that far outweighs all benefits, is that it has a lower signal-to-noise ratio compared to the other chromatin accessibility assays. This high background makes computational data interpretation very difficult, with only strong recovered signal being informative.
ATAC-seq
ATAC-seq is the most current method for probing open chromatin, based on the ability of hyperactive Tn5 transposase [101, 102] to fragment DNA and integrate into active regulatory regions in vivo (Figure 1) [103]. During ATAC-seq, 500–50,000 unfixed nuclei are tagged in vitro with sequencing adapters by purified Tn5 transposase. Due to steric hindrance the majority of adapters are integrated into regions of accessible chromatin that are subsequently submitted to PCR for library construction followed by paired-end NGS. This method has been recently used in a eukaryotic line to uncover open chromatin, nucleosome positioning and TF footprints genome-wide [103]. Despite its limited application so far, ATAC-seq is attracting a growing interest due to its simple and fast two-step protocol, its high sensitivity with a low starting cell number (500 to 50,000 cells) and the ability to study multiple aspects of chromatin architecture simultaneously at high resolution.
The sensitivity and specificity of ATAC-seq is similar to DNase-seq data obtained from approximately three to five orders of magnitude more cells, and it diminishes only for really small numbers of cells [103]. The ATAC-seq protocol does not involve any size-selection steps and can thus identify accessible locations and nucleosome positioning simultaneously. However, its ability to map nucleosomes genome-wide is limited to regions in close proximity to accessible sites [103]. The most challenging aspect of ATAC-seq is the analysis of the sequence data, since generalized methods are unavailable or limited. With the additional demonstrated ability for analyzing a patient’s epigenome on a clinical timescale [103], we foresee ATAC-seq to become the preferred method for the study of chromatin structure in the near future.
Chromatin accessibility high-throughput sequence data analysis
Detection of chromatin accessibility genome-wide with all the above methods requires initial library construction and use of NGS [31, 104]. Resulting data represents an average in vivo snapshot of chromatin accessibility, as represented in the constructed sequencing libraries. Normally, a specialized sequencing facility performs library construction and sequencing using the appropriate kits for the operated sequencer. Otherwise, a research laboratory can use in-house instrumentation and manufacturer or custom library protocols, with the latter being more cost efficient.
Although a number of sequencers are currently available for deep sequencing, most researchers use Illumina next-generation platforms due to the high number of molecules (tag count) that can be sequenced per sample. Tag count represents the most instrumental parameter of output sequencing quality. The number of tags that need to be sequenced depends on the goal of the specific experiment, with nucleosome mapping and TF footprinting experiments requiring higher coverage compared to standard chromatin accessibility detection. To obtain a target coverage depth per sample, the researcher should take into account the minimal number of mappable tags delivered by the instrument in use and adjust accordingly the number of multiplexed samples per lane of flow cell (for details read [31]). A secondary parameter of sequencing quality is tag length, which is mainly a function of the applied sequencing chemistry and currently varies between approximately 36 to 300 bp. Generally speaking, paired-end and longer-read sequencing provides the most accurate results and is recommended whenever possible, especially for areas of the genome with low-complexity or many repetitive elements [31, 104]. However, in most experimental cases chromatin accessibility can be accurately determined with single-end, shorter-length reads without the unnecessary additional expense.
The vast amount of generated sequencing data is subsequently analyzed using a variety of analytical tools, with progressively increased level of difficulty and advanced requirements for computational and genomics expertise. As a result, data analysis along with computing power and storage capacity, are often regarded the current bottleneck in chromatin accessibility experiments. Below we discuss each stage of analysis with separate references to specific chromatin accessibility assays, and more specialized reviews whenever necessary, in an attempt to provide a comprehensive analysis workflow for the novice chromatin accessibility researcher (Figure 2 and Table 2). We mainly discuss analysis of sequence data generated with Illumina-based chemistry since this is the currently most preferred approach.
Figure 2.
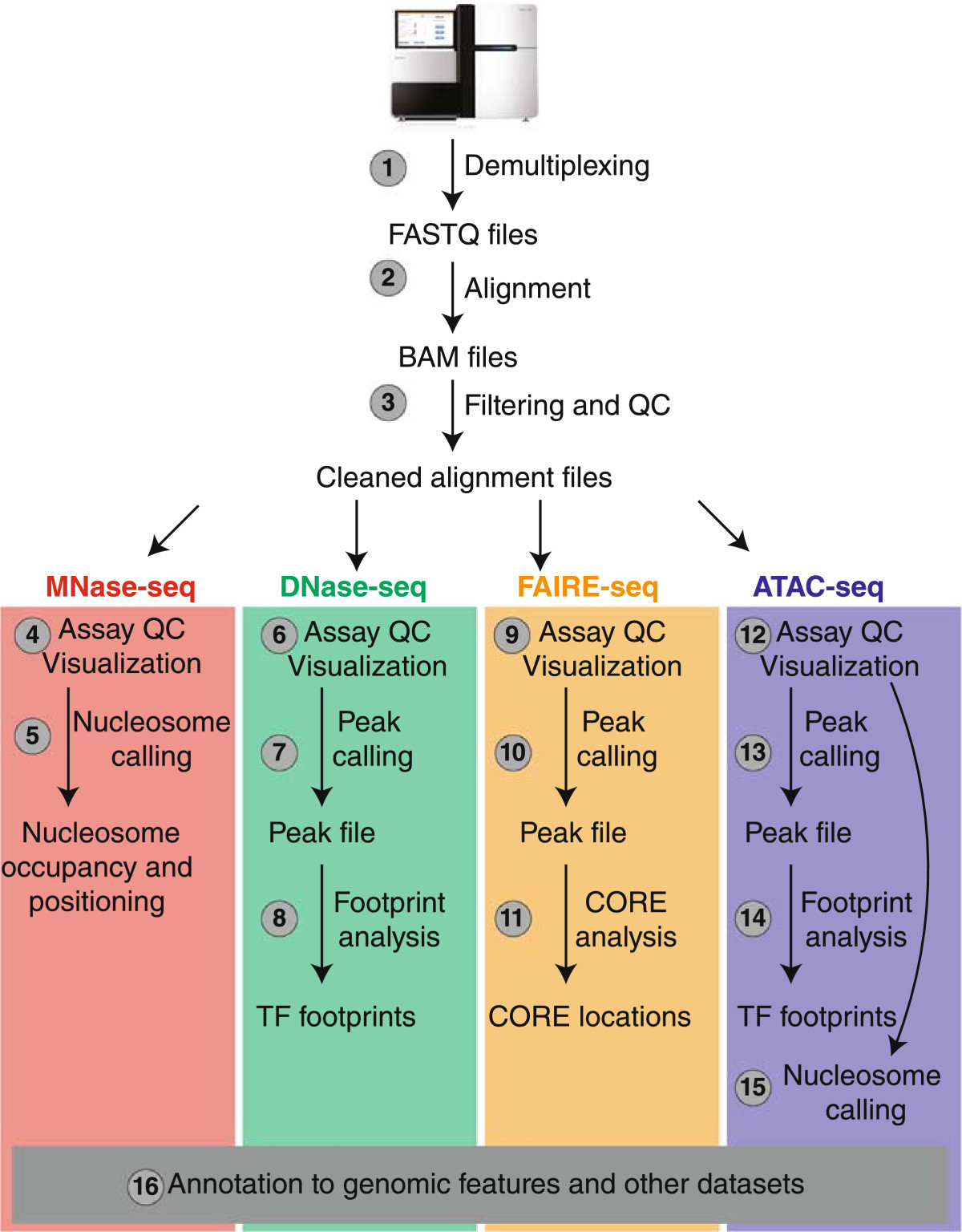
Chromatin accessibility high-throughput data analysis workflow. Chromatin accessibility data analysis involves a number of stages with progressively increased level of difficulty and advanced requirements for computational and genomics expertise. All major steps of analyses, from sequence tags to data annotation/integration are shown in a comprehensive workflow format (read text for additional details).
Table 2.
Chromatin accessibility high-throughput sequence data analysis
| Detection of enriched regions | Estimation of nucleosome organization and TF occupancy metrics | |
|---|---|---|
| MNase-seq | 1. GeneTrack [126] | 1. Nucleosome positioning algorithms [48, 58, 111, 144] |
| 2. Template filtering algorithm [58] | 2. Nucleosome occupancy algorithms [48, 145] | |
| 3. DANPOS [109] | 3. V-plots for TF occupancy [50] | |
| 4. iNPS [127] | ||
| DNase-seq | 1. F-Seq [129] | 1. Digital genomic footprinting algorithms [19, 78, 83, 85, 128, 146–149]. |
| 2. Hotspot, DNase2Hotspots [21, 130] | 2. Nucleosome and TF occupancy algorithms [150] | |
| 3. ZINBA [131] | 3. CENTIPEDE [151] | |
| 4. MACS [132] | ||
| FAIRE-seq | 1. MACS2; https://github.com/taoliu/MACS/, [132] | Not available |
| 2. ZINBA [131] | ||
| ATAC-seq | 1. ZINBA [131] | 1. Digital genomic footprinting algorithms [19, 78, 83, 85, 128, 146, 149] |
| 2. MACS2; https://github.com/taoliu/MACS/, [132] | 2. CENTIPEDE [151] | |
| 3. Hotspot, DNase2Hotspots [21, 130] |
Stage 1 analysis
Overall, most initial data analysis steps are the same for all chromatin accessibility assays discussed above and are normally done by the NGS facility performing the sequencing reactions. These steps include demultiplexing, alignment to a reference genome, tag filtering and measurement of sequencing quality control (QC) (Figure 2). The goal of this stage of analysis is to determine if the sequencing was done with the required depth of coverage and to prepare BAM alignment files for downstream assay-specific analyses.
Initially raw sequencing reads are demultiplexed (Step 1) based on index information into FASTQ files with CASAVA (Illumina) and aligned (Step 2) to a user-defined reference genome (that is human, mouse, and so on) [105]. A number of aligning software is available, such as Maq, RMAP, Cloudburst, SOAP, SHRiMP, BWA and Bowtie [106]. The last two represent the most popular aligning software packages currently. During the alignment process data is filtered (Step 3) to remove overrepresented areas of the genome due to technical bias. Tag filtering is often performed with SAMtools [107] or Picard tools (http://broadinstitute.github.io/picard). For ATAC-seq data specifically, mapped fragments below 38 bp are removed since that is the minimum spacing of transposition events due to steric hindrance [102]. Also, ATAC-seq reads mapping to the mitochondrial genome are discarded as unrelated to the scope of the experiment. Sequencing performance QC (Step 3) is performed during the alignment process, by estimating specific statistical metrics (that is total number of reads, % of unpaired reads, % of reads aligned 0 times, % reads aligned exactly once, % of reads aligned more than once, and overall alignment rate) for each sequenced sample.
Stage 2 analysis
This is the stage where most researchers begin their analysis and includes assay QC, data visualization, and detection of genomic regions of enrichment (nucleosome or peak calling; Figure 2).
Assay QC and data visualization
The goal of this analysis is to determine if the experiment was successful and is often performed by constructing composite plots and by visualization (Steps 4, 6, 9, and 12). Multiple tools are available for generating composite plots including ArchTEX [108], DANPOS-profile [109], and CEAS [110]. For example, TSSs have been shown to be chromatin accessible on average across all eukaryotic genomes [48, 111, 112]. A drop of composite plot signal intensity is expected at this feature when analyzing MNase-seq data, whereas DNase-seq, FAIRE-seq and ATAC-seq data will exhibit an overall increase at the same sites. ArchTEX can also be used to assess the cross-correlation of MNase-seq data, with successful experiments exhibiting enrichment at nucleosomal banding sizes [113]. ATAC-seq QC can be further performed by estimating the percentage of sequence reads that map to the mitochondrial genome and by generating ‘insert size metric plots’ using Picard tools. High quality ATAC-seq data will coincide with a low percentage of mitochondrial reads, and a distribution of insert sizes that depicts a five to six nucleosomal array along with ten bp periodicity of insert sizes.
A number of publicly available stand-alone genome browser tools [114], including Artemis [115], EagleView [116], MapView [117], Tablet [118], Savant [119], and Apollo [120], can be used to visualize raw tag density profiles (and enriched genomic regions, see below) in relation to available annotation tracks. The University of California Santa Cruz (UCSC) [121] and the Integrative Genomics Viewer (IGV) [122] represent some of the most powerful options currently. UCSC provides a plethora of information on whole-genome and exome sequencing, epigenetic and expression data, single nucleotide polymorphisms (SNPs), repeat elements and functional information from the ENCODE and other research projects. It supports incorporation of personally generated data as BED, BedGraph, GFF, WIG and BAM files, so that a researcher can compare his/her own data directly with the publicly available one. IGV represents another efficient, high-performance and intuitive genomics visualization and exploration tool, characterized by its ability to handle large and diverse datasets on a desktop computer. The user can input a variety of data types to compare them with publicly available data from the ENCODE, Cancer Genome Atlas [123], 1000 Genomes [124] and other projects.
Detection of enriched regions
MNase-seq data
In a typical MNase-seq experiment, chromatin accessibility is probed indirectly by deciphering areas of the genome that are occluded by nucleosomes (Figure 1). The location of each mapped tag is identified by the genomic coordinate of the 5′ end in the forward or reverse strand and represents the strand-corresponding nucleosome border (unshifted tag) [125]. Tags can also be shifted 73 bp [111] or extended for 120 to 147 bp [48, 113] towards the 3′ direction to represent the midpoint or full nucleosome length respectively. For organisms with short linkers a 120 bp extension provides better nucleosome resolution and reduces overlaps between neighboring nucleosomes [113]. With paired-end sequencing, the nucleosome midpoint is assumed to coincide with the midpoint of the forward and reverse reads. To map consensus nucleosome positions representative of the average cell population, overlapping reads have to be clustered over genomic regions (Step 5).
Current popular nucleosome calling methods are GeneTrack [126], template filtering [58], DANPOS [109], and iNPS [127]. GeneTrack implements a Gaussian smoothing and averaging approach to convert measurements at each genomic coordinate into a continuous probabilistic landscape. Nucleosomes are then detected as the maximal data subset from all local maxima with a user-defined exclusion zone that represents the steric exclusion between neighboring nucleosomes (that is 147 bp) and is centered over each assigned peak. The template filtering algorithm was developed to control for the variable MNase cut patterns observed at different concentrations of MNase digestion. This method uses a set of templates, which match frequently found distributions of sequence tags at MNase-generated nucleosome ends, to extract information about nucleosome positions, sizes and occupancies directly from aligned sequence data. However, the current version of template filtering is only suitable for small genomes (approximately 12 MB) due to memory limitations. iNPS differs from other nucleosome callers in that it uses the wave-like structure of nucleosome datasets as part of its smoothing approach. iNPS detects nucleosomes with various shapes from the first derivative of the Gaussian smoothed profile. DANPOS differs from all above approaches in that it allows the comparison of MNase-seq datasets and identifies dynamic nucleosomes based on fuzziness change, occupancy change and position shift. In addition, DANPOS performs well in assigning nucleosomes from a single experiment, and should prove an invaluable analysis tools for deciphering underlying chromatin perturbations responsible for various disease and cellular phenotypes.
DNase-seq data
Scientists have traditionally applied algorithms developed for ChIP-seq, without an input DNA control, to detect enriched DHSs although peculiarities of DNase-seq data render this approach unsuitable without adjustment of default settings at minimal [128]. Currently, the most widely used peak-calling algorithms for DNase-seq data analysis are the publicly available F-Seq [129], Hotspot [130], ZINBA [131] and MACS [132–135] (Step 7). F-Seq and Hotspot represent the only tools specifically developed for handling the unique characteristics of DNase-seq data. ZINBA can be applied as a general peak-calling algorithm for many types of NGS data and MACS, although initially developed for the model-based analysis of ChIP-seq data, has been successfully used as a peak-caller for DNase-seq data in many instances [136]. All these tools are based on different algorithms, parameters and background evaluation metrics (for details read [135]).
Briefly, F-Seq [129] is a parametric density estimator of sequence tag data, developed to overcome the bin-boundary effects of histogram metrics for peak enrichment [129]. F-seq implements a smooth Gaussian kernel density estimation that takes into account the estimated center of each sequence read. F-seq has been implemented in a number of studies [17, 19, 79, 94] for the identification of chromatin accessibility and the evaluation of TF footprints in relation to ChIP-seq data [17, 19, 79, 94]. However, it requires time-consuming designing for statistical testing [137]. The Hotspot algorithm [21, 130] has been widely used by the ENCODE consortium to identify regions of chromatin accessibility and represents, to our knowledge, the only DNase-seq-specific algorithm that reports statistical significance for identified DHSs [128]. The algorithm isolates localized DHS peaks within areas of increased nuclease sensitivity (‘hotspots’). Results are evaluated with false discovery rate analysis for statistical significance, employing generation of a random dataset with the same number of reads as the analyzed dataset. The newest version of Hotspot, DNase2hotspots, merges the two-pass detection in the original algorithm into a single-pass [130].
ZINBA, is a statistical pipeline characterized by its flexibility to process recovered signals with differential characteristics [131]. Following data preprocessing, the algorithm classifies genomic regions as background, enriched or zero-inflated using a mixture regression model, without a priori knowledge of genomic enrichment. In turn, identified proximal enriched regions are combined within a defined distance using the broad setting, and the shape-detection algorithm is implemented to discover sharp signals within broader areas of enrichment. The advantage of ZINBA is that it can accurately identify enriched regions in the absence of an input control. In addition, the software uses a priori or modeled covariate information (for example G/C content) to represent signal components, which improves detection accuracy especially when the signal-to-noise ratio is low or in analysis of complex datasets (for example DNA copy number amplifications). MACS a model-based analysis algorithm with wide applicability for the analysis of ChIP-seq data [138–140], has also been effectively applied for DHS detection. The algorithm empirically models the shift size of sequence reads, and employs a Poisson distribution as a background model to capture local biases attributed to inherent differential sequencing and mapping genomic properties.
A recent comparison of the above four peak callers demonstrated that F-Seq and ZINBA have the highest and lowest sensitivity respectively [135]. F-Seq has also been shown to perform better than window-clustering approaches in a separate study [129], and its accuracy can be significantly increased by reducing the peak signal threshold setting from the default value of four to a value between 2 and 3 [135].
FAIRE-seq data
For FAIRE-seq data the algorithm MACS [132] has been further extended to MACS2 (https://github.com/taoliu/MACS/) and performs reliably in identifying genomic regions of open chromatin (Step 10). This application is invoked by using the command macs2 callpeak and can be combined with the options broad, broad cutoff, no model, no lambda (unless a control file is given) and shift size. The algorithm uses default peak calling (q = 0.05) and broad (q = 0.10) cutoff values, but these settings can be adjusted or converted to P-values empirically. Once the peak-calling cutoff is set as a P-value, the broad cutoff value is automatically perceived as P also. The shift size parameter should be set as the midpoint of the average sonication fragment length in the analyzed dataset. In addition, upon availability a matched control sample can be used as input to increase detection confidence. In this case, command line parameters should be adjusted accordingly. FAIRE enrichment can also be detected using ZINBA [131]. As mentioned above, this software improves detection accuracy when the signal-to-noise ratio is low or in complex datasets. However, for high signal-to-noise datasets it performs equally well with MACS, although it is much more computationally intensive.
Identified FAIRE-seq enriched regions residing in proximity to each other, have been traditionally merged together using BedTools [141] (for detailed instructions read [142]) to form Clusters of Open Regulatory Elements (COREs) (Step 11) [91, 94, 95]. Formation of COREs allows the identification of chromatin accessibility and gene regulation patterns that may have otherwise remained undetectable in a smaller genomic scale. COREs can be also generated from all other chromatin accessibility datasets.
ATAC-seq data
ATAC-sec peak calling (Step 13) can be performed also by using ZINBA [103]. Alternatively, our group has found that MACS2 and Hotspot [130] perform equally well with ZINBA at identifying accessible locations (unpublished data).
Stage 3 analysis
This stage of analysis involves estimation of various parameters of the epigenomic landscape, including nucleosome spacing, positioning and occupancy [31], and TF binding for footprinting experiments (Figure 2).
MNase-seq data
Nucleosome or translational positioning indicates the position of a population of nucleosomes in relation to DNA, and considers a specific reference nucleosome point like its start, dyad or end [143]. Translational positioning is reflected in the standard deviation of the population positioning curve, and is used to distinguish between strongly and poorly positioned nucleosomes [143]. Translational positioning can be further characterized as absolute, based on the probability of a nucleosome starting at a specific base x, and conditional, based on the probability of a nucleosome starting within an extended region with center base pair x[56]. Nucleosome occupancy on the other hand, measures density of nucleosome population and is reflected in the area under the population positioning curve [143]. Nucleosome occupancy is tightly linked to chromatin accessibility, and depends on the degree a genomic site is occupied by nucleosomes in all genomic configurations [56]. A number of methods have been applied to measure nucleosome positioning [48, 58, 111, 144] and occupancy [48, 145] from MNase-seq data based on the number of sequence reads that start at each base pair, assessed for a consensus nucleosome position or in a per base pair basis [56]. In addition high-resolution MNase-seq data generated using a modified paired-end library construction protocol can be analyzed using V-plots to detect TF binding. V-plots are two dimensional dot-plots that display each fragment’s length in the Y-axis versus the corresponding fragment midpoint position in the X-axis [50].
DNase-seq data
Stable binding of TFs in the vicinity of DHSs protects DNA from nuclease cleavage and generates DNase I footprints that at high-sequencing depth can unveil occupancy of TFs with long DNA residence times (for example CTCF and Rap1) [84, 85]. Thus, high-coverage DNase-seq data can be analyzed with specialized algorithms to detect long-standing TF binding (Step 8). Previously specialized algorithms developed for DGF have identified hundreds of TF binding sites at genome-wide resolution, by comparing the depth of DNase I digestion at TF binding sites to adjacent open chromatin and taking into account only raw counts of 5′ ends of sequencing tags [19, 78, 83, 128, 146–149]. However, some of these algorithms are inefficient for mammalian genomes [130] or publicly unavailable. The latest publicly available footprinting algorithm, DNase2TF, allows fast evaluation of TF occupancy in large genomes with better or comparable detection accuracy to previous algorithms [85]. However, it still suffers from detection inaccuracies stemming from transient TF DNA residence time and the inherent cutting preferences of DNase I like all currently available footprinting algorithms [85].
The recently reported modified approach DNase I-released fragment-length analysis of hypersensitivity (DNase-FLASH) [150] allows simultaneous probing of TF occupancy, interactions between TFs and nucleosomes and nucleosome occupancy at individual loci, similar to ATAC-seq. The method is based on the concurrent quantitative analysis of different size fragments released from DNase I digestion of genomic DNA, with microfragments (<125 bp) depicting TF occupancy, and larger fragments (126 to 185 bp) representative of nucleosomal elements.
ATAC-seq data
Analysis of ATAC-seq paired-end data can reveal indispensable information on nucleosome packing and positioning, patterns of nucleosome-TF spacing, and TF occupancy simultaneously at genome-wide resolution similar to DNase-FLASH [103]. Analysis is based on the distribution of insert lengths and the positions of insertions after Tn5 transposition within open chromatin of active regulatory elements (Step 15). For TF foot printing (Step 14) our laboratory uses CENTIPEDE [151] (see below), although other footrprinting algorithms are also available [19, 78, 83, 85, 128, 146–149]. For footprinting analysis, cleavage sites have to be adjusted four to five bp upstream or downstream due to the biophysical characteristics of Tn5 transposase, which inserts two adaptors separated by nine bp [102]. It is not known if footprinting detection with ATAC-seq data is factor-dependent or affected by Tn5 cleavage preferences.
Stage 4 analysis
Data annotation and integration represents the final and most informative stage of analysis and requires computational and genomics background on genomic organization and structure (Step 16). After identification of enriched regions and estimation of metrics of nucleosome organization and TF occupancy, it is often desirable to evaluate this data in light of relevant information from other experiments. For example, a researcher can evaluate the overlap or association of the sequence data with genomic features (that is promoters, introns, intergenic regions, TSSs, TTSs) and ontological entities (that is molecular functions, biological processes, cellular components, disease ontologies, and so on). For that purpose, BedTools (documentation is available at http://bedtools.readthedocs.org) and its sister PyBEDTools represent a versatile suite of utilities for a variety of comparative and exploratory operations on genomic features such as identifying overlap between two datasets, extracting unique features, and merging enriched regions using a predefined distance value [141, 142, 152]. Also the UCSC genome browser offers a suite of similar utilities specifically tailored for data file conversions (http://genome.ucsc.edu/util.html). Identified chromatin accessible locations can be compared against functional annotations with GREAT, to identify significantly enriched pathways or ontologies and direct future hypotheses [153].
One can also inspect enriched regions of interest for discovery of putative TF binding events using two approaches. The first approach is straightforward and is based on comparing sequence data against a database of known TF motifs. The second type of analysis can be computationally intensive and involves the de novo discovery of novel TF binding sites. A number of available software (MEME [154, 155], DREME [156], Patser (http://stormo.wustl.edu/software.html), Matrix Scan [157], LASAGNA [158], CompleteMOTIFs [159], and MatInspector (Genomatix) [160]), and TF motif databases (MatBase Genomatix; http://www.genomatix.de/online_help/help_matbase/matbase_help.html), JASPAR [161], TRANSFAC [162] and UniPROBE [163]) can arrogate TF motif identification and de novo discovery within enriched regions.
For DNase-seq and ATAC-seq experiments TF footprints can be analyzed with CENTIPEDE [151]. CENTIPEDE is an integrative algorithm for rapid profiling of many TFs simultaneously that combines known information on TF motifs and positional weight matrices, with DNase-seq or ATAC-seq cutting patterns in one unsupervised Bayesian mixture model. Combination of all this information with publicly available expression, DNA methylation and histone modification data can be instrumental for answering questions on epigenetic regulation and inheritance and unveiling long-range patterns of gene regulation and disease development [17, 19, 137]. Finally, multistep sequential data analysis can be generated and stored using Galaxy [164] or Cistrome [165].
Conclusions
Each of the chromatin accessibility assays discussed here has inherent limitations in identifying regions of enrichment, based on the fragmentation method used and the involvement of any size selection steps. MNase-seq, DNase-seq and ATAC-seq are all based on the double enzymatic cleavage of DNA fragments and are sensitive to the excision-ability of a fragment. As shown in MNase-seq and ATAC-seq experiments, this sensitivity represents an issue only when mapping larger fragments (>100 bp) because the data is heavily biased by the overall nucleosome configuration at the region [55, 103]. In MNase-seq experiments, it was specifically shown that nucleosomes flanked by hypersensitive sites or long linkers are excised easier at low enzymatic concentrations and exhibit artificially higher nucleosome occupancy compared to nucleosomes without these characteristics, thus leading to biased results [55].
Functional annotation of accessible regions is factor-dependent and relies highly on the availability of accurate TF binding motifs and their relevant information content as well as the spatial and temporal interaction of TFs with DNA [84, 85]. Recent research supports that DNase I cleavage patterns are affected by the time of interaction of TF with their recognition sites, with depth of cleavage being proportional to residence time [85]. Consequently, transient TFs leave minimal or no detectable cut signatures and their binding cannot be identified with any of the current footprinting algorithms. In addition, cleavage signatures appear in genomic sites with no apparent protein binding, providing further support that footprint profiles may arise as a result of inherent DNase I cleavage bias instead of protein protection from enzymatic activity. Thus, to accurately characterize gene regulatory networks from accessibility data, we need comprehensive TF motif databases generated using in vivo/in vitro assays or computationally based de novo motif discovery algorithms. More importantly there is an imminent need to further investigate the applicability of DNase-seq, and ATAC-seq for that matter, to accurately detect factor-chromatin interactions in dynamic cellular settings. It is possible that future footprinting algorithms will be able to accurately identify only a subset of TF binding events based solely on analysis of footprints with high depth (above a statistically validated threshold), and not on generic analysis of all cleavage profiles.
Currently, most researchers compare their chromatin accessibility data to other published datasets. Although, this approach is advantageous when public datasets are available, it does not explain the cause of identified differences. In the absence of a ‘golden standard’, experimental and computational approaches need to be compared against independently generated data. For example, active regulatory regions identified by chromatin segmentation of histone modification ChIP-seq data, can serve as an independent control for experimental and computational accuracy of current chromatin accessibility assays. Finally, development of specialized statistically supported peak-calling algorithms for DNase-seq and ATAC-seq data will be instrumental in the identification of active regulatory elements genome-wide. We foresee that future applications of chromatin accessibility will include the detection of allele-specific effects to identify functionally important SNPs, use of accessibility in eQTL studies to link regulatory regions with disease phenotypes, and assessment of clinical samples for epigenetic biomarkers of disease.
Authors’ information
MJB is an associate professor at the Department of Biochemistry at the State University of New York at Buffalo, a director of the WNYSTEM Stem Cell Sequencing/Epigenomics Facility, a co-director of the UB Genomics and Bioinformatics Core, and an adjunct faculty for the Cancer Genetics Roswell Park Cancer Institute and the Department of Biomedical Informatics. He has extensive experience with chromatin accessibility assays and bioinformatics analysis of related data. He is currently the head of an active laboratory focused on epigenomic profiling and the detection of key determinants for gene regulation, disease development and progression. MT is a senior research scientist/project leader in MJB’s laboratory involved in a number of studies on epigenetic regulation.
Acknowledgments
The authors would like to thank J Bard, B Marzullo and S Valiyaparambil for their input with the NGS and data analysis section. This work was supported by funds from the NY State Department of Health grant C026714 to MJB.
Abbreviations
- ATAC
assay for transposase-accessible chromatin
- bp
base pair
- ChIP-seq
chromatin immunoprecipitation with deep sequencing
- COREs
Clusters of Open Regulatory Elements
- DGF
digital genomic footprinting
- DHS
DNase I hypersensitive site
- DNase I
deoxyribonuclease I
- FAIRE
Formaldehyde-Assisted Isolation of Regulatory Elements
- MNase
micrococcal nuclease
- NGS
next-generation sequencing
- PCR
polymerase chain reaction
- QC
quality control
- SNPs
single nucleotide polymorphisms
- TF
transcription factor
- TSSs
transcription start sites
- UCSC
University of California Santa Cruz.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MT and MJB have been involved in drafting the manuscript and revising it critically for important intellectual content. MJB has given final approval of the version to be published. Both authors read and approved the final manuscript.
Contributor Information
Maria Tsompana, Email: tsompana@buffalo.edu.
Michael J Buck, Email: mjbuck@buffalo.edu.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423(6936):145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 3.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184(4139):868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 4.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henikoff S, Ahmad K. Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol. 2005;21:133–153. doi: 10.1146/annurev.cellbio.21.012704.133518. [DOI] [PubMed] [Google Scholar]
- 7.Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21(3):421–434. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the ‘H3 barcode hypothesis’. Proc Natl Acad Sci U S A. 2006;103(17):6428–6435. doi: 10.1073/pnas.0600803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radman-Livaja M, Rando OJ. Nucleosome positioning: how is it established, and why does it matter? Dev Biol. 2010;339(2):258–266. doi: 10.1016/j.ydbio.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shivaswamy S, Bhinge A, Zhao Y, Jones S, Hirst M, Iyer VR. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008;6(3):e65. doi: 10.1371/journal.pbio.0060065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36(8):900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 13.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell. 2003;11(6):1587–1598. doi: 10.1016/S1097-2765(03)00231-4. [DOI] [PubMed] [Google Scholar]
- 14.Wallrath LL, Lu Q, Granok H, Elgin SC. Architectural variations of inducible eukaryotic promoters: preset and remodeling chromatin structures. Bioessays. 1994;16(3):165–170. doi: 10.1002/bies.950160306. [DOI] [PubMed] [Google Scholar]
- 15.Hogan GJ, Lee CK, Lieb JD. Cell cycle-specified fluctuation of nucleosome occupancy at gene promoters. PLoS Genet. 2006;2(9):e158. doi: 10.1371/journal.pgen.0020158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korber P, Luckenbach T, Blaschke D, Horz W. Evidence for histone eviction in trans upon induction of the yeast PHO5 promoter. Mol Cell Biol. 2004;24(24):10965–10974. doi: 10.1128/MCB.24.24.10965-10974.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shu W, Chen H, Bo X, Wang S. Genome-wide analysis of the relationships between DNaseI HS, histone modifications and gene expression reveals distinct modes of chromatin domains. Nucleic Acids Res. 2011;39(17):7428–7443. doi: 10.1093/nar/gkr443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buck MJ, Lieb JD. A chromatin-mediated mechanism for specification of conditional transcription factor targets. Nat Genet. 2006;38(12):1446–1451. doi: 10.1038/ng1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyle AP, Song L, Lee BK, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res. 2011;21(3):456–464. doi: 10.1101/gr.112656.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet. 2008;9(1):15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- 21.John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43(3):264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 23.Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho-Santos J, McManus MT, Plath K, Meshorer E, Ramalho-Santos M. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460(7257):863–868. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21(3):396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 26.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu C, Wong YC, Elgin SC. The chromatin structure of specific genes: II. Disruption of chromatin structure during gene activity. Cell. 1979;16(4):807–814. doi: 10.1016/0092-8674(79)90096-5. [DOI] [PubMed] [Google Scholar]
- 28.Wu C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature. 1980;286(5776):854–860. doi: 10.1038/286854a0. [DOI] [PubMed] [Google Scholar]
- 29.Keene MA, Elgin SC. Micrococcal nuclease as a probe of DNA sequence organization and chromatin structure. Cell. 1981;27(1 Pt 2):57–64. doi: 10.1016/0092-8674(81)90360-3. [DOI] [PubMed] [Google Scholar]
- 30.Levy A, Noll M. Chromatin fine structure of active and repressed genes. Nature. 1981;289(5794):198–203. doi: 10.1038/289198a0. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Pugh BF. High-resolution genome-wide mapping of the primary structure of chromatin. Cell. 2011;144(2):175–186. doi: 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wal M, Pugh BF. Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol. 2012;513:233–250. doi: 10.1016/B978-0-12-391938-0.00010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Axel R. Cleavage of DNA in nuclei and chromatin with staphylococcal nuclease. Biochemistry. 1975;14(13):2921–2925. doi: 10.1021/bi00684a020. [DOI] [PubMed] [Google Scholar]
- 35.Sulkowski E, Laskowski M., Sr Action of micrococcal nuclease on polymers of deoxyadenylic and deoxythymidylic acids. J Biol Chem. 1969;244(14):3818–3822. [PubMed] [Google Scholar]
- 36.Williams EJ, Sung SC, Laskowski M., Sr Action of venom phosphodiesterase on deoxyribonucleic acid. J Biol Chem. 1961;236:1130–1134. [PubMed] [Google Scholar]
- 37.Noll M. Subunit structure of chromatin. Nature. 1974;251(5472):249–251. doi: 10.1038/251249a0. [DOI] [PubMed] [Google Scholar]
- 38.Reeves R, Jones A. Genomic transcriptional activity and the structure of chromatin. Nature. 1976;260(5551):495–500. doi: 10.1038/260495a0. [DOI] [PubMed] [Google Scholar]
- 39.Lohr D, Van Holde KE. Yeast chromatin subunit structure. Science. 1975;188(4184):165–166. doi: 10.1126/science.1090006. [DOI] [PubMed] [Google Scholar]
- 40.Lohr D, Kovacic RT, Van Holde KE. Quantitative analysis of the digestion of yeast chromatin by staphylococcal nuclease. Biochemistry. 1977;16(3):463–471. doi: 10.1021/bi00622a020. [DOI] [PubMed] [Google Scholar]
- 41.Hartley PD, Madhani HD. Mechanisms that specify promoter nucleosome location and identity. Cell. 2009;137(3):445–458. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganapathi M, Palumbo MJ, Ansari SA, He Q, Tsui K, Nislow C, Morse RH. Extensive role of the general regulatory factors, Abf1 and Rap1, in determining genome-wide chromatin structure in budding yeast. Nucleic Acids Res. 2011;39(6):2032–2044. doi: 10.1093/nar/gkq1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39(10):1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 44.Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309(5734):626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 45.Ponts N, Harris EY, Prudhomme J, Wick I, Eckhardt-Ludka C, Hicks GR, Hardiman G, Lonardi S, Le Roch KG. Nucleosome landscape and control of transcription in the human malaria parasite. Genome Res. 2010;20(2):228–238. doi: 10.1101/gr.101063.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rizzo JM, Sinha S. Analyzing the global chromatin structure of keratinocytes by MNase-Seq. Methods Mol Biol. 2014;1195:49–59. doi: 10.1007/7651_2014_77. [DOI] [PubMed] [Google Scholar]
- 47.Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, Pritchard JK. Controls of nucleosome positioning in the human genome. PLoS Genet. 2012;8(11):e1003036. doi: 10.1371/journal.pgen.1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, Segal E. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458(7236):362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cui K, Zhao K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol Biol. 2012;833:413–419. doi: 10.1007/978-1-61779-477-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S. Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci U S A. 2011;108(45):18318–18323. doi: 10.1073/pnas.1110731108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem Sci. 2000;25(3):99–104. doi: 10.1016/S0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- 52.Cockell M, Rhodes D, Klug A. Location of the primary sites of micrococcal nuclease cleavage on the nucleosome core. J Mol Biol. 1983;170(2):423–446. doi: 10.1016/S0022-2836(83)80156-9. [DOI] [PubMed] [Google Scholar]
- 53.Dingwall C, Lomonossoff GP, Laskey RA. High sequence specificity of micrococcal nuclease. Nucleic Acids Res. 1981;9(12):2659–2673. doi: 10.1093/nar/9.12.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horz W, Altenburger W. Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res. 1981;9(12):2643–2658. doi: 10.1093/nar/9.12.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rizzo JM, Bard JE, Buck MJ. Standardized collection of MNase-seq experiments enables unbiased dataset comparisons. BMC Mol Biol. 2012;13:15. doi: 10.1186/1471-2199-13-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaplan N, Hughes TR, Lieb JD, Widom J, Segal E. Contribution of histone sequence preferences to nucleosome organization: proposed definitions and methodology. Genome Biol. 2010;11(11):140. doi: 10.1186/gb-2010-11-11-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rizzo JM, Mieczkowski PA, Buck MJ. Tup1 stabilizes promoter nucleosome positioning and occupancy at transcriptionally plastic genes. Nucleic Acids Res. 2011;39(20):8803–8819. doi: 10.1093/nar/gkr557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res. 2010;20(1):90–100. doi: 10.1101/gr.098509.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rando OJ. Genome-wide mapping of nucleosomes in yeast. Methods Enzymol. 2010;470:105–118. doi: 10.1016/S0076-6879(10)70005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zentner GE, Henikoff S. Surveying the epigenomic landscape, one base at a time. Genome Biol. 2012;13(10):250. doi: 10.1186/gb4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weintraub H, Groudine M. Chromosomal subunits in active genes have an altered conformation. Science. 1976;193(4256):848–856. doi: 10.1126/science.948749. [DOI] [PubMed] [Google Scholar]
- 62.Keene MA, Corces V, Lowenhaupt K, Elgin SC. DNase I hypersensitive sites in Drosophila chromatin occur at the 5′ ends of regions of transcription. Proc Natl Acad Sci U S A. 1981;78(1):143–146. doi: 10.1073/pnas.78.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garel A, Zolan M, Axel R. Genes transcribed at diverse rates have a similar conformation in chromatin. Proc Natl Acad Sci U S A. 1977;74(11):4867–4871. doi: 10.1073/pnas.74.11.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stalder J, Larsen A, Engel JD, Dolan M, Groudine M, Weintraub H. Tissue-specific DNA cleavages in the globin chromatin domain introduced by DNAase I. Cell. 1980;20(2):451–460. doi: 10.1016/0092-8674(80)90631-5. [DOI] [PubMed] [Google Scholar]
- 65.McGhee JD, Wood WI, Dolan M, Engel JD, Felsenfeld G. A 200 base pair region at the 5′ end of the chicken adult beta-globin gene is accessible to nuclease digestion. Cell. 1981;27(1 Pt 2):45–55. doi: 10.1016/0092-8674(81)90359-7. [DOI] [PubMed] [Google Scholar]
- 66.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421(6921):448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 67.Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20(3):267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giresi PG, Lieb JD. How to find an opening (or lots of them) Nat Methods. 2006;3(7):501–502. doi: 10.1038/nmeth0706-501. [DOI] [PubMed] [Google Scholar]
- 69.Crawford GE, Davis S, Scacheri PC, Renaud G, Halawi MJ, Erdos MR, Green R, Meltzer PS, Wolfsberg TG, Collins FS. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods. 2006;3(7):503–509. doi: 10.1038/nmeth888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, Weaver M, Shafer A, Lee K, Neri F, Humbert R, Singer MA, Richmond TA, Dorschner MO, McArthur M, Hawrylycz M, Green RD, Navas PA, Noble WS, Stamatoyannopoulos JA. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3(7):511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- 71.Crawford GE, Holt IE, Mullikin JC, Tai D, Blakesley R, Bouffard G, Young A, Masiello C, Green ED, Wolfsberg TG, Collins FS, C. National Institutes Of Health Intramural Sequencing Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites. Proc Natl Acad Sci U S A. 2004;101(4):992–997. doi: 10.1073/pnas.0307540100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sabo PJ, Hawrylycz M, Wallace JC, Humbert R, Yu M, Shafer A, Kawamoto J, Hall R, Mack J, Dorschner MO, McArthur M, Stamatoyannopoulos JA. Discovery of functional noncoding elements by digital analysis of chromatin structure. Proc Natl Acad Sci U S A. 2004;101(48):16837–16842. doi: 10.1073/pnas.0407387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dorschner MO, Hawrylycz M, Humbert R, Wallace JC, Shafer A, Kawamoto J, Mack J, Hall R, Goldy J, Sabo PJ, Kohli A, Li Q, McArthur M, Stamatoyannopoulos JA. High-throughput localization of functional elements by quantitative chromatin profiling. Nat Methods. 2004;1(3):219–225. doi: 10.1038/nmeth721. [DOI] [PubMed] [Google Scholar]
- 74.Crawford GE, Holt IE, Whittle J, Webb BD, Tai D, Davis S, Margulies EH, Chen Y, Bernat JA, Ginsburg D, Zhou D, Luo S, Vasicek TJ, Daly MJ, Wolfsberg TG, Collins FS. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS) Genome Res. 2006;16(1):123–131. doi: 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protocols. 2010;2010(2):pdb prot5384. doi: 10.1101/pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.John S, Sabo PJ, Canfield TK, Lee K, Vong S, Weaver M, Wang H, Vierstra J, Reynolds AP, Thurman RE, Stamatoyannopoulos JA. Genome-scale mapping of DNase I hypersensitivity. Curr Protoc Mol Biol. 2013;Chapter 27:Unit 21 27. doi: 10.1002/0471142727.mb2127s103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, Maurano MT, Humbert R, Rynes E, Wang H, Vong S, Lee K, Bates D, Diegel M, Roach V, Dunn D, Neri J, Schafer A, Hansen RS, Kutyavin T, Giste E, Weaver M, Canfield T, Sabo P, Zhang M, Balasundaram G, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489(7414):83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132(2):311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Winter DR, Song L, Mukherjee S, Furey TS, Crawford GE. DNase-seq predicts regions of rotational nucleosome stability across diverse human cell types. Genome Res. 2013;23(7):1118–1129. doi: 10.1101/gr.150482.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Noll M. Internal structure of the chromatin subunit. Nucleic Acids Res. 1974;1(11):1573–1578. doi: 10.1093/nar/1.11.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cousins DJ, Islam SA, Sanderson MR, Proykova YG, Crane-Robinson C, Staynov DZ. Redefinition of the cleavage sites of DNase I on the nucleosome core particle. J Mol Biol. 2004;335(5):1199–1211. doi: 10.1016/j.jmb.2003.11.052. [DOI] [PubMed] [Google Scholar]
- 83.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, Fields S, Stamatoyannopoulos JA. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6(4):283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, Liu XS, Brown M. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods. 2014;11(1):73–78. doi: 10.1038/nmeth.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sung MH, Guertin MJ, Baek S, Hager GL. DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol Cell. 2014;56(2):275–285. doi: 10.1016/j.molcel.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17(6):877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Giresi PG, Lieb JD. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements) Methods. 2009;48(3):233–239. doi: 10.1016/j.ymeth.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Simon JM, Giresi PG, Davis IJ, Lieb JD. A detailed protocol for formaldehyde-assisted isolation of regulatory elements (FAIRE) Curr Protoc Mol Biol. 2013;Chapter 21:Unit21 26. doi: 10.1002/0471142727.mb2126s102. [DOI] [PubMed] [Google Scholar]
- 89.Simon JM, Giresi PG, Davis IJ, Lieb JD. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat Protoc. 2012;7(2):256–267. doi: 10.1038/nprot.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nagy PL, Cleary ML, Brown PO, Lieb JD. Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc Natl Acad Sci U S A. 2003;100(11):6364–6369. doi: 10.1073/pnas.1131966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, Berney T, Montanya E, Mohlke KL, Lieb JD, Ferrer J. A map of open chromatin in human pancreatic islets. Nat Genet. 2010;42(3):255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Louwers M, Bader R, Haring M, van Driel R, de Laat W, Stam M. Tissue- and expression level-specific chromatin looping at maize b1 epialleles. Plant Cell. 2009;21(3):832–842. doi: 10.1105/tpc.108.064329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Omidbakhshfard MA, Winck FV, Arvidsson S, Riano-Pachon DM, Mueller-Roeber B. A step-by-step protocol for formaldehyde-assisted isolation of regulatory elements from Arabidopsis thaliana. J Integr Plant Biol. 2014;56(6):527–538. doi: 10.1111/jipb.12151. [DOI] [PubMed] [Google Scholar]
- 94.Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, Liu Z, London D, McDaniell RM, Shibata Y, Showers KA, Simon JM, Vales T, Wang T, Winter D, Zhang Z, Clarke ND, Birney E, Iyer VR, Crawford GE, Lieb JD, Furey TS. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21(10):1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Buck MJ, Raaijmakers LM, Ramakrishnan S, Wang D, Valiyaparambil S, Liu S, Nowak NJ, Pili R. Alterations in chromatin accessibility and DNA methylation in clear cell renal cell carcinoma. Oncogene. 2014;33(41):4961–4965. doi: 10.1038/onc.2013.455. [DOI] [PubMed] [Google Scholar]
- 96.Yang CC, Buck MJ, Chen MH, Chen YF, Lan HC, Chen JJ, Cheng C, Liu CC. Discovering chromatin motifs using FAIRE sequencing and the human diploid genome. BMC Genomics. 2013;14:310. doi: 10.1186/1471-2164-14-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43(1):27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eeckhoute J, Lupien M, Meyer CA, Verzi MP, Shivdasani RA, Liu XS, Brown M. Cell-type selective chromatin remodeling defines the active subset of FOXA1-bound enhancers. Genome Res. 2009;19(3):372–380. doi: 10.1101/gr.084582.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McGhee JD, Felsenfeld G. Another potential artifact in the study of nucleosome phasing by chromatin digestion with micrococcal nuclease. Cell. 1983;32(4):1205–1215. doi: 10.1016/0092-8674(83)90303-3. [DOI] [PubMed] [Google Scholar]
- 100.Haring M, Offermann S, Danker T, Horst I, Peterhansel C, Stam M. Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Methods. 2007;3:11. doi: 10.1186/1746-4811-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goryshin IY, Reznikoff WS. Tn5 in vitro transposition. J Biol Chem. 1998;273(13):7367–7374. doi: 10.1074/jbc.273.13.7367. [DOI] [PubMed] [Google Scholar]
- 102.Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, Shendure J. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11(12):R119. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rizzo JM, Buck MJ. Key principles and clinical applications of ‘next-generation’ DNA sequencing. Cancer Prev Res. 2012;5(7):887–900. doi: 10.1158/1940-6207.CAPR-11-0432. [DOI] [PubMed] [Google Scholar]
- 105.Flicek P, Birney E. Sense from sequence reads: methods for alignment and assembly. Nat Methods. 2009;6(11 Suppl):S6–S12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- 106.Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform. 2010;11(5):473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lai WK, Bard JE, Buck MJ. ArchTEx: accurate extraction and visualization of next-generation sequence data. Bioinformatics. 2012;28(7):1021–1023. doi: 10.1093/bioinformatics/bts063. [DOI] [PubMed] [Google Scholar]
- 109.Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013;23(2):341–351. doi: 10.1101/gr.142067.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shin H, Liu T, Manrai AK, Liu XS. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009;25(19):2605–2606. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
- 111.Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446(7135):572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 112.Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, Gilmour DS, Albert I, Pugh BF. Nucleosome organization in the Drosophila genome. Nature. 2008;453(7193):358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Givens RM, Lai WK, Rizzo JM, Bard JE, Mieczkowski PA, Leatherwood J, Huberman JA, Buck MJ. Chromatin architectures at fission yeast transcriptional promoters and replication origins. Nucleic Acids Res. 2012;40(15):7176–7189. doi: 10.1093/nar/gks351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nielsen CB, Cantor M, Dubchak I, Gordon D, Wang T. Visualizing genomes: techniques and challenges. Nat Methods. 2010;7(3 Suppl):S5–S15. doi: 10.1038/nmeth.1422. [DOI] [PubMed] [Google Scholar]
- 115.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 116.Huang W, Marth G. EagleView: a genome assembly viewer for next-generation sequencing technologies. Genome Res. 2008;18(9):1538–1543. doi: 10.1101/gr.076067.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bao H, Guo H, Wang J, Zhou R, Lu X, Shi S. MapView: visualization of short reads alignment on a desktop computer. Bioinformatics. 2009;25(12):1554–1555. doi: 10.1093/bioinformatics/btp255. [DOI] [PubMed] [Google Scholar]
- 118.Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. Tablet–next generation sequence assembly visualization. Bioinformatics. 2010;26(3):401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fiume M, Williams V, Brook A, Brudno M. Savant: genome browser for high-throughput sequencing data. Bioinformatics. 2010;26(16):1938–1944. doi: 10.1093/bioinformatics/btq332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lewis SE, Searle SM, Harris N, Gibson M, Lyer V, Richter J, Wiel C, Bayraktaroglir L, Birney E, Crosby MA, Kaminker JS, Matthews BB, Prochnik SE, Smithy CD, Tupy JL, Rubin GM, Misra S, Mungall CJ, Clamp ME. Apollo: a sequence annotation editor. Genome Biol. 2002;3(12):RESEARCH0082. doi: 10.1186/gb-2002-3-12-research0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39(Database issue):D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cancer Genome Atlas Research N Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA, Genomes Project C A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 126.Albert I, Wachi S, Jiang C, Pugh BF. GeneTrack - a genomic data processing and visualization framework. Bioinformatics. 2008;24(10):1305–1306. doi: 10.1093/bioinformatics/btn119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen W, Liu Y, Zhu S, Green CD, Wei G, Han JD. Improved nucleosome-positioning algorithm iNPS for accurate nucleosome positioning from sequencing data. Nat Commun. 2014;5:4909. doi: 10.1038/ncomms5909. [DOI] [PubMed] [Google Scholar]
- 128.Madrigal P, Krajewski P. Current bioinformatic approaches to identify DNase I hypersensitive sites and genomic footprints from DNase-seq data. Front Genet. 2012;3:230. doi: 10.3389/fgene.2012.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Boyle AP, Guinney J, Crawford GE, Furey TS. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics. 2008;24(21):2537–2538. doi: 10.1093/bioinformatics/btn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baek S, Sung MH, Hager GL. Quantitative analysis of genome-wide chromatin remodeling. Methods Mol Biol. 2012;833:433–441. doi: 10.1007/978-1-61779-477-3_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rashid NU, Giresi PG, Ibrahim JG, Sun W, Lieb JD. ZINBA integrates local covariates with DNA-seq data to identify broad and narrow regions of enrichment, even within amplified genomic regions. Genome Biol. 2011;12(7):R67. doi: 10.1186/gb-2011-12-7-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Feng J, Liu T, Zhang Y. Using MACS to identify peaks from ChIP-Seq data. Curr Protoc Bioinformatics. 2011;Chapter 2:Unit 2 14. doi: 10.1002/0471250953.bi0214s34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7(9):1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Koohy H, Down TA, Spivakov M, Hubbard T. A comparison of peak callers used for DNase-Seq data. PLoS One. 2014;9(5):e96303. doi: 10.1371/journal.pone.0096303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang YM, Zhou P, Wang LY, Li ZH, Zhang YN, Zhang YX. Correlation between DNase I hypersensitive site distribution and gene expression in HeLa S3 cells. PLoS One. 2012;7(8):e42414. doi: 10.1371/journal.pone.0042414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang W, Zhang T, Wu Y, Jiang J. Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell. 2012;24(7):2719–2731. doi: 10.1105/tpc.112.098061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods. 2009;6(11 Suppl):S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim H, Kim J, Selby H, Gao D, Tong T, Phang TL, Tan AC. A short survey of computational analysis methods in analysing ChIP-seq data. Human Genomics. 2011;5(2):117–123. doi: 10.1186/1479-7364-5-2-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rye MB, Saetrom P, Drablos F. A manually curated ChIP-seq benchmark demonstrates room for improvement in current peak-finder programs. Nucleic Acids Res. 2011;39(4):e25. doi: 10.1093/nar/gkq1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Quinlan AR. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;47:11 12 11–11 12 34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pugh BF. A preoccupied position on nucleosomes. Nat Struct Mol Biol. 2010;17(8):923. doi: 10.1038/nsmb0810-923. [DOI] [PubMed] [Google Scholar]
- 144.Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, Struhl K. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol. 2009;16(8):847–852. doi: 10.1038/nsmb.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18(7):1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen X, Hoffman MM, Bilmes JA, Hesselberth JR, Noble WS. A dynamic Bayesian network for identifying protein-binding footprints from single molecule-based sequencing data. Bioinformatics. 2010;26(12):i334–i342. doi: 10.1093/bioinformatics/btq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS. The human mitochondrial transcriptome. Cell. 2011;146(4):645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012;150(6):1274–1286. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Piper J, Elze MC, Cauchy P, Cockerill PN, Bonifer C, Ott S. Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res. 2013;41(21):e201. doi: 10.1093/nar/gkt850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vierstra J, Wang H, John S, Sandstrom R, Stamatoyannopoulos JA. Coupling transcription factor occupancy to nucleosome architecture with DNase-FLASH. Nat Methods. 2014;11(1):66–72. doi: 10.1038/nmeth.2713. [DOI] [PubMed] [Google Scholar]
- 151.Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011;21(3):447–455. doi: 10.1101/gr.112623.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dale RK, Pedersen BS, Quinlan AR. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics. 2011;27(24):3423–3424. doi: 10.1093/bioinformatics/btr539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings/International Conference on Intelligent Systems for Molecular Biology; ISMB International Conference on Intelligent Systems for. Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 156.Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27(12):1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Thomas-Chollier M, Sand O, Turatsinze JV, Janky R, Defrance M, Vervisch E, Brohee S, van Helden J. RSAT: regulatory sequence analysis tools. Nucleic Acids Res. 2008;36(Web Server issue):W119–W127. doi: 10.1093/nar/gkn304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lee C, Huang CH. LASAGNA: a novel algorithm for transcription factor binding site alignment. BMC Bioinformatics. 2013;14:108. doi: 10.1186/1471-2105-14-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kuttippurathu L, Hsing M, Liu Y, Schmidt B, Maskell DL, Lee K, He A, Pu WT, Kong SW. CompleteMOTIFs: DNA motif discovery platform for transcription factor binding experiments. Bioinformatics. 2011;27(5):715–717. doi: 10.1093/bioinformatics/btq707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21(13):2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 161.Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, Lim J, Shyr C, Tan G, Zhou M, Lenhard B, Sandelin A, Wasserman WW. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42(Database issue):D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34(Database issue):D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009;37(Database issue):D77–D82. doi: 10.1093/nar/gkn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Goecks J, Nekrutenko A, Taylor J, Galaxy T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, Pape UJ, Poidinger M, Chen Y, Yeung K, Brown M, Turpaz Y, Liu XS. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12(8):R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]