
Fig. 1.
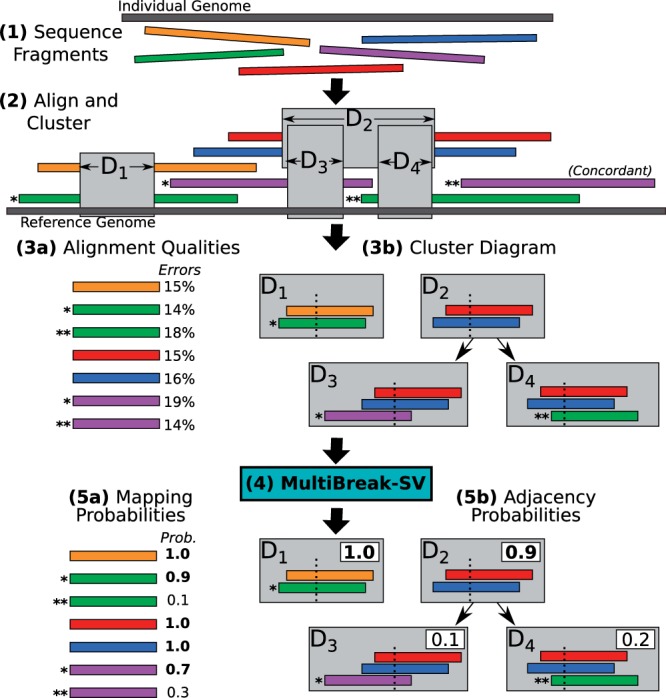
Overview of MultiBreak-SV. (1) Five long reads are sequenced from an individual genome. (2) The reads are aligned to the reference genome, producing seven distinct multi-breakpoint-mappings. When clustered, the multi-breakpoint-mappings indicate four novel adjacencies (). (3a) The quality of the read alignments (e.g. the edit distance) is noted for each multi-breakpoint-mapping. (3b) The set of all possible novel adjacencies is represented as a cluster diagram G, where the nodes are novel adjacencies and the directed edges represent overlapping novel adjacencies. (4) The cluster diagram and alignment qualities are input to MultiBreak-SV. (5a) MultiBreak-SV assigns probabilities to each multi-breakpoint-mapping. (5b) From these mappings, the probability of each novel adjacency is computed. A solution to the Multi-Read Mapping Problem is a selection of at most one alignment for each multi read and a selection of at most one novel adjacency for each connected component in G (bold)