
Figure 2. Identification and validation of microbe-derived gene product functions.
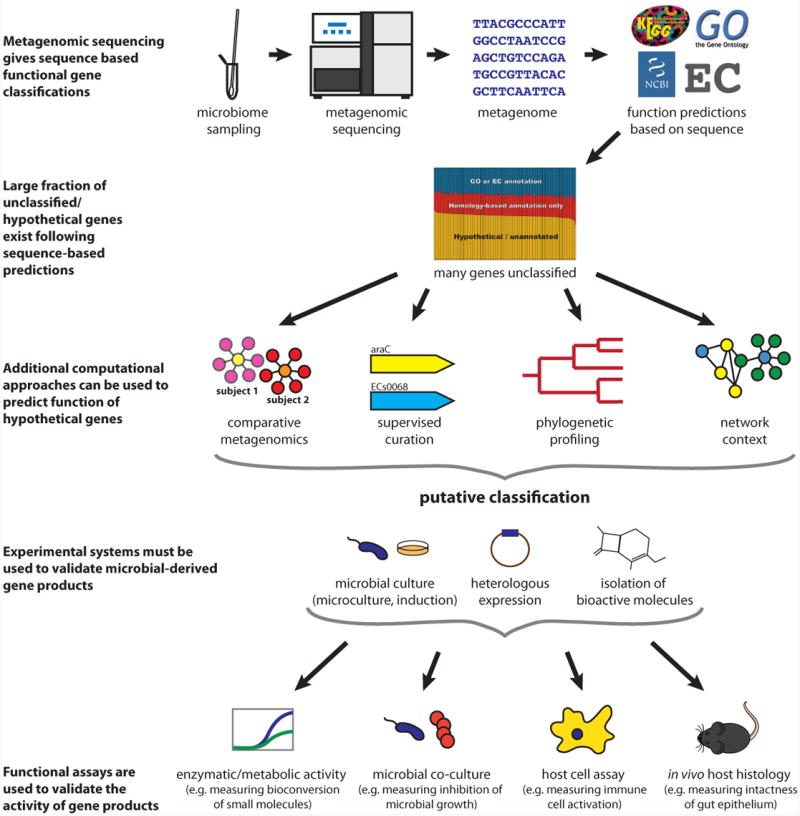
An overview of the process of microbial gene functional annotation and validation. In microbial isolate genomes and metagenomes alike, gene function is typically first assigned using standard sequence analysis methods (homology-based assignment (Loewenstein et al., 2009) and domain profiling (Finn et al., 2013)). These predictions can be further refined by additional bioinformatic approaches, such as comparative metagenomics (“guilt by association” of uncharacterized microbial products with characterized genes across samples through the use of data integration), supervised curation (manual determination of a consensus among multiple complementary automated annotations (Richardson and Watson, 2013)), phylogenic profiling (analysis of co-occurrence of genes across isolates (Eisen and Fraser, 2003)), and network context (“guilt by association” in isolate coexpression, interaction, or functional linkage networks (Sharan et al., 2007)). Following putative classification, bioactivity must be validated and further characterized by experimental methods. When standard culture is challenging (as is common for the microbiome), microculture and induction culture, as well as heterologous expression of genes and direct isolation of products are particularly useful. Functional assays for investigating the activity of microbial products include enzymatic/metabolic activity assays (Craciun and Balskus, 2012), microbial co-culture (Yan et al., 2013), host cell profiling (Wieland Brown et al., 2013), and in vivo host phenotype assessments (Olle, 2013).