

Abstract
Previous studies have demonstrated that muscle mechanoreflex and metaboreflex controls are altered in heart failure (HF), which seems to be due to changes in cyclooxygenase (COX) pathway and changes in receptors on afferent neurons, including transient receptor potential vanilloid type-1 (TRPV1) and cannabinoid receptor type-1 (CB1). The purpose of the present study was to test the hypotheses: 1) exercise training (ET) alters the muscle metaboreflex and mechanoreflex control of muscle sympathetic nerve activity (MSNA) in HF patients. 2) The alteration in metaboreflex control is accompanied by increased expression of TRPV1 and CB1 receptors in skeletal muscle. 3) The alteration in mechanoreflex control is accompanied by COX-2 pathway in skeletal muscle. Thirty-four consecutive HF patients with ejection fractions <40% were randomized to untrained (n = 17; 54 ± 2 yr) or exercise-trained (n = 17; 56 ± 2 yr) groups. MSNA was recorded by microneurography. Mechanoreceptors were activated by passive exercise and metaboreceptors by postexercise circulatory arrest (PECA). COX-2 pathway, TRPV1, and CB1 receptors were measured in muscle biopsies. Following ET, resting MSNA was decreased compared with untrained group. During PECA (metaboreflex), MSNA responses were increased, which was accompanied by the expression of TRPV1 and CB1 receptors. During passive exercise (mechanoreflex), MSNA responses were decreased, which was accompanied by decreased expression of COX-2, prostaglandin-E2 receptor-4, and thromboxane-A2 receptor and by decreased in muscle inflammation, as indicated by increased miRNA-146 levels and the stable NF-κB/IκB-α ratio. In conclusion, ET alters muscle metaboreflex and mechanoreflex control of MSNA in HF patients. This alteration with ET is accompanied by alteration in TRPV1 and CB1 expression and COX-2 pathway and inflammation in skeletal muscle.
Keywords: heart failure, muscle sympathetic nervous system, metaboreflex, mechanoreflex, exercise training
neurohumoral excitation, including activation of the sympathetic nervous system, is prevalent in chronic heart failure (HF) patients with systolic dysfunction (11, 15, 17, 19, 54). Sympathetic nerve activity (SNA), whether measured indirectly by plasma norepinephrine levels (11), norepinephrine spillover (15), or decreased heart rate (HR) variability (17) or directly by microneurographic recordings (19), is found to be elevated. Furthermore, HF patients with the greatest sympathetic activation have the poorest prognosis (4, 11).
The mechanisms underlying this increased resting muscle sympathetic nerve activity (MSNA) in HF are likely multiple and diverse. Potential mechanisms include primary central neural dysregulation, and/or abnormal peripheral reflex regulation of central sympathetic outflow [reviewed by Zucker et al. (54)]. Not only is resting MSNA elevated in HF, but neurovascular responses during exercise are also augmented and may contribute to exercise dysfunction (13, 18, 23, 24, 32, 34). HR and blood pressure (BP) are regulated during exercise by the exercise pressor reflex, in which signals from muscle afferents, including muscle metaboreceptors and mechanoreceptors, reflexively control MSNA. Muscle metaboreceptors are unmyelinated group IV fibers, which are sensitive to ischemic metabolites generated during exercise, and muscle mechanoreceptors are thinly myelinated, group III fibers sensitive to mechanical stimuli, such as stretch (16, 27). The group of Sinoway (41) was the first to report that muscle metaboreflex control of MSNA during exercise is blunted in patients with chronic HF. Conversely, the muscle mechanoreflex is augmented in patients with HF and is sensitized by cyclooxygenase (COX) products, but not lactic acid or adenosine, generated during exercise (21, 22).
Investigations in the rat-infarct model of HF, in which it is possible to make direct electrophysiologic recordings from group III and IV afferents, have confirmed and extended these findings in humans with HF (45). Interestingly, COX-2 expression is augmented in skeletal muscle of HF rats, and COX-2 inhibition attenuates the exaggerated mechanoreflex control, confirming the findings in humans with HF (21, 29). Recent findings suggest that miRNAs, including miRNA-16 and miRNA-143, may modulate the COX-2 protein expression (28). In addition, the miRNA-146 has been predicted to target nuclear factor-κB (NF-κB), an important modulator of COX-2 expression (28, 37). Conversely, with the use of direct electrophysiologic recordings from group IV muscle metaboreceptors in HF rats, afferent neuronal firing in response chemical agonists was blunted, confirming the findings in humans with HF (45). Transient receptor potential vanilloid type-1 (TRPV1) receptors and cannabinoid type-1 (CB1) receptors are colocalized on group IV afferent neurons and, when stimulated, increase efferent MSNA. In animal models of HF, decreased expression of TRPV1 in dorsal root ganglia (DRG), accompanied by decreased afferent responsiveness to TRPV1 agonists, has been reported (40, 45, 49). The mechanisms underlying these multiple abnormalities of the primary afferent neurons in the rat-infarct model of HF remain speculative, but importantly, they are preventable with exercise training (46, 47).
Exercise training has been shown to improve exercise capacity and quality of life in humans with HF (31, 33). In addition, exercise training leads to a remarkable reduction in resting MSNA in HF patients regardless of age, gender, and presence of sleep apnea (1, 2, 36, 43). Furthermore, the impact of exercise training on sympathetic responses during exercise in HF has not been studied. The mechanisms underlying the reduction in resting MSNA following training in humans with HF remain poorly understood. The purpose of these studies in patients with chronic HF was to test the following hypotheses: 1) exercise training alters the muscle metaboreflex and the mechanoreflex control of MSNA in patients with chronic HF. 2) The alteration in metaboreflex control is accompanied by increased expression of TRPV1 and CB1 receptors in skeletal muscle biopsy [vastus lateralis (VL)]. In addition, 3) the alteration in mechanoreflex control is accompanied by changes in the COX-2 pathway in VL muscle biopsy from exercise-trained patients with HF compared with untrained control HF patients.
METHODS
Study Population
Consecutive HF outpatients [age 30 to 65 yr, functional class II to III of New York Heart Association, ejection fraction ≤40%, and peak oxygen uptake (V̇o2) <20 ml·kg−1·min−1] were invited to participate in the study. The exclusion criteria were recent myocardial infarction or unstable angina (<3 mo), HF duration <3 mo, permanent pacemaker dependence, atrial fibrillation, participation in a regular exercise program, or musculoskeletal disorder (e.g., arthritis) prohibiting participation in exercise training. The study was approved by Human Subject Protection Committee of the Heart Institute (InCor; SDC-3083/07/158) and Clinical Hospital, University of Sao Paulo Medical School (CAPPesq-0020/08). All subjects gave written informed consent to participate in the study. The trial is registered at www.ClinicalTrials.gov (NCT01884142).
Measures and Procedures
Cardiopulmonary exercise testing.
Cardiopulmonary exercise testing was conducted at baseline and after 4 mo of exercise training or untrained control period, as previously described (2, 10, 43). Maximal exercise capacity was determined during a maximal progressive exercise test on cycle ergometer (Ergoline, Spirit 150), using a ramp protocol with work rate increments of 5–10 W every minute until exhaustion. V̇o2 and carbon dioxide production were determined by means of gas exchange on a breath-by-breath basis in a computerized system (Model Vmax 229; Sensor Medics, Buena Vista, CA). Peak V̇o2 was defined as the maximum attained V̇o2 at the end of the exercise period in which the subject could no longer maintain the cycle ergometer velocity at 60 rpm. Anaerobic threshold was identified at the breakpoint between the increase in the carbon dioxide output and V̇o2 (V slope) or at the point in which the ventilatory equivalent for oxygen and end-tidal oxygen partial pressure curves reached their respective minimum values and began to rise. Respiratory compensation was identified at the point at which ventilatory equivalent for carbon dioxide was lowest before a systematic increase and when end-tidal carbon dioxide partial pressure reaches a maximum value and begins to decrease (38).
Muscle sympathetic nerve activity.
MSNA was directly measured from the peroneal nerve in the nonexercising limb (right leg) using the technique of microneurography, as previously described (44a). In brief, signals were amplified by a factor of 50,000 to 100,000 and band-passed filtered (700 to 2,000 Hz). For recordings and analysis, nerve activity was rectified and integrated (time constant: 0.1 s) to obtain a mean voltage display. Muscle sympathetic bursts were identified by visual inspection by the principal investigator and by two other investigators (C. E. Negrao and M. U. Rondon) blinded to the study protocol. MSNA was expressed as total activity (arbitrary units), burst frequency (bursts/min), and burst incidence (bursts/100 heartbeats).
Muscle mechanoreflex sensitivity.
Passive leg exercise, which eliminates activation of central command and the metaboreceptors, was used to activate muscle mechanoreceptors. Passive exercise was performed by flex/extension of the knee joint on the left leg, 30 times/min by the principal investigator. Visual and manual inspection of the VL muscle was conducted to ensure that this maneuver resulted in muscle length changes to stimulate mechanoreceptors. Mechanoreflex sensitivity was calculated as the difference in percentage change in MSNA, BP, and HR between peak passive exercise and rest periods.
Muscle metaboreflex sensitivity.
Postexercise regional circulatory arrest (PECA), which eliminates the activation of central command and muscle mechanoreceptors, was used to isolate muscle metaboreceptors. The maximum voluntary contraction (MVC) was calculated by averaging the maximum load achieved in three consecutive repetitions, measured using a dynamometer (DLC-model, Dynamometer; Kratos, Sao Paulo, SP, Brazil). Just before release of 3 min of 30% MVC exercise, the circulation to the exercising leg was arrested by inflating the thigh occlusion cuff (240 mmHg) for 2 min, followed by 3 min of recovery. The isometric contraction of the quadriceps stimulates the production of metabolites and muscle changes similar to those caused by moderate physical exercise. Metaboreflex sensitivity was calculated as the difference in percentage change in MSNA, BP, and HR between peak exercise or the first minute of PECA in relation to rest period.
Other measurements.
Left ventricular ejection fraction (LVEF) was determined from the two-dimensional echocardiography by Simpson method (IE33; Philips Medical Systems, Andover, MA). HR, systolic, and diastolic and mean BP were monitored noninvasively with an oscillometric beat-to-beat device (FinometerPro; Finapress Medical Systems, Amsterdam, The Netherlands).
Skeletal muscle biopsy.
Percutaneous muscle biopsy procedures were performed in VL, approximately at the midway point between top edge of the patella and the greater trochanter. With the use of a sterile technique, and after ensuring adequate local anesthesia, a small incision was made in the skin and subcutaneous tissue. A 5-mm modified Allendale-Bergstrom needle was then inserted through the fascia, and an assistant immediately applied suction by using a syringe connected to a canister and attached to the top of the needle (30). The muscle sample was immediately frozen in liquid nitrogen and subsequently stored in a freezer at −80°C.
mRNA and miRNA analysis by real-time polymerase chain reaction.
Frozen muscle samples (∼30 mg) were homogenized in Trizol and RNA was isolated, according to the manufacturer's instructions (Invitrogen Life Technologies). After extraction, the total RNA concentration was quantified using a NanoDrop Spectrophotometer (Nano-Drop Technologies) and checked for integrity by EtBr-agarose gel electrophoresis. RNA were primed with 0.5 μg/μl oligo dT (Fermentas/Thermo Scientific Molecular Biology) to generate first-strand DNA. Reverse transcription (RT) was performed using Revertaid M-MuLV Reverse Transcriptase (Fermentas/Thermo Scientific Molecular Biology). cDNA for miRNA analysis was synthesized from total RNA using gene-specific primers according to the TaqMan MicroRNA Assay protocol (Applied Biosystems). Real-time quantification of the COX-1, COX-2, mPGES1, PGIS, TXAS, EP1, EP2, EP3, EP4, IP, TP, TRPV1, and CB1 mRNA was performed with a SYBRGreen PCR Master Mix, using ABI PRISM 7700 Sequence Detection System (Applied Biosystem). The expression of cyclophilin was measured as an internal control for sample variation in RT reaction. Cyclophilin was chosen as the internal control because of its homogeneous expression among different samples (efficiency% = 95.04; P = 0.28). In addition, cyclophilin showed better condition than other internal controls (GAPDH and 18S). Primers were designed using Primer 3 software (http://primer3plus.com/cgi-bin/dev/primer3plus.cgi). The DNA sequence was obtained from GenBank, and primers were made in separate exons to distinguish PCR products derived from cDNA by size from those derived from genomic DNA contaminants. To accurately detect miRNA-16 (INV 0391), miRNA-143 (INV 0466), and miRNA-146 (INV 0468), the real-time PCR quantification method was performed using TaqMan MicroRNA Assay protocol (Applied Biosystems). MiRNAs samples were normalized by evaluating U6 expression. Each muscle sample was analyzed in duplicate. Relative quantities of target gene expressions of untrained group vs. exercise-trained group were compared after normalization to the values of reference gene (ΔCT). Fold changes in mRNA and miRNA expression were calculated using the differences in ΔCT values between the two samples (ΔΔCT) and equation 2−ΔΔCT. Results are expressed as percentage of control.
Western blot.
Frozen muscles samples were homogenized in cold RIPA lysis buffer (Upstate) containing protease inhibitor cocktail (1:5,000 dilution; Sigma), sodium fluoride (100 mM), sodium pyrophosphate (10 mM), sodium orthovanadate (100 mM), and PMSF (10 mM). Then, homogenates were centrifuged (12,000 rpm for 30 min at 4°C), and supernatants were isolated. Protein extracts (50 μg) were electrophoretically separated using 7.5% SDS-PAGE and then transferred to PVDF membranes (Amersham-GE Healthcare) overnight at 4°C using a Mini Trans-Blot Cell system (Bio-Rad) containing transfer buffer (25 mM Tris, 190 mM glycine, 20% methanol, and 0.05% SDS) as described previously (8, 48). After blockade of nonspecific sites with 5% nonfat dried milk, membranes were incubated overnight at 4°C with the primary antibody against rabbit anti-COX-2 (1:1,000 dilution; Cayman Chemical), rabbit anti-NF-κB p65 (1:1,000; Santa Cruz Biotechnology), and rabbit anti-IκB-α (1:1,000; Santa Cruz Biotechnology). Membranes were washed and then incubated for 2 h at room temperature with peroxidase-conjugated anti-rabbit IgG antibody (Bio-Rad) for COX-2 (1:7,000 dilution), NF-κB p65 (1:1,500 dilution), and IκB-α (1:1,500 dilution). Membranes were thoroughly washed, and immunocomplexes were detected using an enhanced horseradish peroxidase/luminal chemiluminescence system (ECL Plus; Amersham-GE Healthcare) and subjecting the membrane to autoradiography (Hyperfilm ECL; Amersham-GE Healthcare). The same membrane was then stripped and used to determine sarcomeric actin protein expression as an internal control using a monoclonal mouse anti-sarcomeric actin antibody (1:1,000 dilution; Dako). Immunoblots signals were quantified using Scion Image (Scioncorp; National Institutes of Health).
COX-2 considerations.
It is widely recognized that COX-2 is a 72-kDa protein (48); however, in human skeletal muscle samples, the COX-2 band is not detected in 72 kDa and appears to be ∼10 kDa smaller in size than 72 kDa (6). In line with Burd et al. (6), we quantified the COX-2 protein bands near to 62 kDa.
Exercise training program.
The exercise training protocol was conducted for 4 mo, as previously described (1, 2, 9, 10, 43). In brief, it consisted of three 60-min exercise sessions/week. Each exercise session consisted of 5-min stretching exercises, 30 min of cycling on an ergometer bicycle in the first 15 days and up to 40 min in the rest of the period, 10 min of local strengthening exercises, and 5 min of cool down with stretching exercises. The exercise intensity was established by HR levels that corresponded to anaerobic threshold up to 10% below the respiratory compensation point obtained in the cardiopulmonary exercise test. This intensity corresponded to 60–72% peak V̇o2. In four patients, the respiratory compensation point was not detectable. In those patients, the exercise training was determined at the anaerobic threshold. During the exercise sessions, when a training effect was observed, as indicated by a decrease by 8 to 10% in HR, the bicycle work rate was increased by 0.25 or 0.5 kpm to return to the target HR levels. Subjects underwent exercise training under supervision at the Heart Institute. The untrained patients were instructed to avoid any regular exercise program. The patients were asked about exercise each visit to the outpatient clinic (approximately every 3 or 4 wk). After the end of the protocol, the untrained patients were invited to participate in the exercise training program.
Experimental Protocol
The experimental protocol (Fig. 1) was performed in a thermoneutral room at approximately the same time of day. The subject was placed in the supine position, and electrocardiogram leads were placed on the chest. The cuffs for BP measurement were placed on the left arm. The left leg was positioned on a support with 150° inclination, and the occlusion cuff used during PECA was placed on the left thigh. The right leg was positioned for microneurography, and an adequate nerve-recording site was obtained. Then the subjects rested for 10 min, and then mechanoreflex sensitivity evaluation was initiated. Ten minutes after the end of the mechanoreflex sensitivity evaluation, muscle metaboreflex sensitivity evaluation was initiated. The skeletal muscle biopsy was performed between 3 and 7 days after mechano- and metaboreflex studies. The experimental protocol and skeletal muscle biopsy were repeated before and after 4 mo of exercise training or clinical follow up, 24–48 h after the last exercise training session. Patients were instructed to continue cardiac medications throughout the study.
Fig. 1.
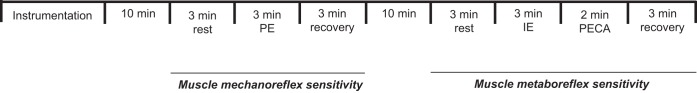
Experimental protocol. PE, passive exercise; IE, isometric exercise; PECA, postexercise regional circulatory arrest.
Statistical Analysis
The data are presented as means ± SE. The Kolmogorov-Smirnov and Levene's test were used to assess the normality of distribution and homogeneity for each variable. Parametric tests were used for variables with normal distribution and homogeneity. The baseline characteristics among groups (dropout, untrained, and exercise trained) were tested by one-way ANOVA. Differences in the resting neurovascular and hemodynamic parameters between groups before and after exercise training and untrained time control were verified by two-way ANOVA for repeated measures. The responses (percentage changes) to 30% isometric exercise and passive exercise before and after exercise training and untrained time control were tested by two-way ANOVA for repeated measures. Similarly, the responses during metaboreceptors stimulation (percentage changes; PECA vs. rest) before and after exercise training and untrained time control were tested by two-way ANOVA for repeated measures. Two-way ANOVA for repeated measures was also used to test whether the changes in the neurovascular and hemodynamic parameters during 30% isometric exercise and PECA and during passive exercise reached significance levels. This analysis was done before and after exercise training and untrained time control. In case of significance, Scheffé's post hoc multiple comparisons were conducted. For variables with no normal distribution and homogeneity, nonparametric tests were used (Mann-Whitney's test and Wilcoxon's test). A Chi-square (χ2) test was used to assess categorical data differences. Probability values of P < 0.05 were considered statistically significant.
RESULTS
Study Population
Fifty-six consecutive HF patients were randomized into two groups: 1) untrained time control group (n = 28), and 2) exercise-trained group (n = 28). In the untrained group, 11 patients did not complete the time control period because of HF decompensation or myocardial infarction (n = 4), noncardiovascular events (n = 2), death (n = 1), and drop out for personal reasons (n = 4). In the exercise-trained group, 11 patients did not complete the training protocol because of HF decompensation or myocardial infarction (n = 3), noncardiovascular events (n = 3), death (n = 1), and drop out for personal reasons (n = 4). Thus data are available for analysis in 34 subjects. Baseline characteristics of the HF patients who completed the time control period (n = 17), completed exercise training (n = 17), and the dropout patients (n = 22) are shown in Tables 1 and 2. There were no differences among the groups in any of the parameters measured.
Table 1.
Baseline characteristics in patients with heart failure selected to exercise-trained and untrained groups and dropout patients
| Dropout (n = 22) | Untrained (n = 17) | Exercise trained (n = 17) | P Value | |
|---|---|---|---|---|
| Age, yr | 56 ± 2 | 54 ± 2 | 56 ± 2 | 0.70 |
| BMI, kg/m2 | 28 ± 1 | 27 ± 1 | 27 ± 1 | 0.80 |
| Gender | ||||
| Male | 17(77%) | 15(88%) | 13(77%) | 0.62 |
| Female | 5(23%) | 2(12%) | 4(23%) | |
| Functional Class | ||||
| NYHA-II | 14(64%) | 12(70%) | 12(70%) | 0.86 |
| NYHA-III | 8(36%) | 5(30%) | 5(30%) | |
| HF etiology | ||||
| Idiopathic | 6(27%) | 6(35%) | 6(35%) | 0.78 |
| Ischemic | 12(55%) | 7(29%) | 6(35%) | |
| Hypertensive | 2(9%) | 3(18%) | 3(18%) | |
| Chagasic | 2(9%) | 3(18%) | 2(12%) | |
| Medications | ||||
| Beta-blocker | 21(95%) | 17(100%) | 17(100%) | 0.45 |
| ACEI/ARB | 22(100%) | 17(100%) | 17(100%) | 1.00 |
| Spironolactone | 18(82%) | 14(84%) | 14(84%) | 0.99 |
| Diuretics | 16(73%) | 14(82%) | 12(71%) | 0.69 |
| ASA | 12(55%) | 9(53%) | 9(53%) | 0.99 |
| Statins | 13(59%) | 10(59%) | 8(47%) | 0.71 |
| Hypertension | 15(68%) | 12(71%) | 10(59%) | 0.46 |
| Diabetes | 6(27%) | 4(24%) | 5(29%) | 0.93 |
Values are means ± SE. BMI, body mass index; NYHA, New York Heart Association; ACEI, angiotensin-converting enzyme inhibitors; ARB, angiotensin II receptor blocker; ASA, acetylsalicylic acid.
Table 2.
Neurovascular and hemodynamic parameters in heart failure patients selected to exercise-trained and untrained heart failure groups
| Dropout (n = 22) | Untrained (n = 17) |
Exercise Trained (n = 17) |
|||
|---|---|---|---|---|---|
| Pre | Pre | Post | Pre | Post | |
| LVEF, % | 33 ± 2 | 29 ± 1 | 31 ± 2 | 28 ± 2 | 32 ± 3 |
| Peak V̇o2, ml·kg−1·min−1 | 16 ± 1 | 17 ± 1 | 17 ± 1 | 18 ± 1 | 21 ± 1*† |
| HR, beats/min | 67 ± 3 | 62 ± 3 | 61 ± 2 | 63 ± 2 | 63 ± 2 |
| SBP, mmHg | 136 ± 6 | 131 ± 5 | 136 ± 6 | 130 ± 4 | 126 ± 4 |
| DBP, mmHg | 76 ± 3 | 71 ± 2 | 75 ± 3 | 69 ± 2 | 70 ± 3 |
| MBP, mmHg | 97 ± 4 | 94 ± 3 | 99 ± 4 | 92 ± 3 | 91 ± 3 |
| MSNA, total activity | 164 ± 17 | 156 ± 20 | 156 ± 17 | 135 ± 20 | 112 ± 11* |
| MSNA, burst/min | 41 ± 3 | 39 ± 2 | 38 ± 2 | 40 ± 2 | 33 ± 2*† |
| MSNA, burst/100 heartbeats | 61 ± 4 | 64 ± 4 | 64 ± 3 | 65 ± 4 | 54 ± 3*† |
Values are means ± SE. LVEF, left ventricular ejection fraction; V̇o2, oxygen uptake; HR, heart rate; SBP, systolic blood pressure; DBP, diastolic blood pressure MBP, mean blood pressure; MSNA, muscle sympathetic nerve activity.
P < 0.05 vs. pre within group.
P < 0.05 vs. untrained.
Effects of Exercise Training on resting Neurovascular and Hemodynamic Parameters
The effects of exercise training on resting neurovascular and hemodynamic parameters are shown in Table 2. LVEF and resting hemodynamic parameters were unchanged in exercise-trained group. However, exercise training significantly increased peak V̇o2 and led to a significant reduction in resting MSNA. Exercise capacity, LVEF, HR, BP, and neurovascular parameters were unchanged in the untrained time control HF patients.
Effects of Exercise Training on Neurovascular and Hemodynamic Parameters During Isometric Exercise Testing
During peak 30% MVC isometric leg exercise at baseline, MSNA increased significantly and similarly in both exercise-trained and untrained HF groups (Fig. 2). Likewise, the increase in BP and HR was similar between groups (Table 3). Significant artifact obscured the sympathetic neurogram in one exercise-trained patient during the last 30 s of 30% MVC isometric leg exercise, so only the 30 s before the motor neuron signals were analyzed. One HF patient was excluded from this study because he had arrhythmias during this study, which significantly altered the MSNA pattern.
Fig. 2.
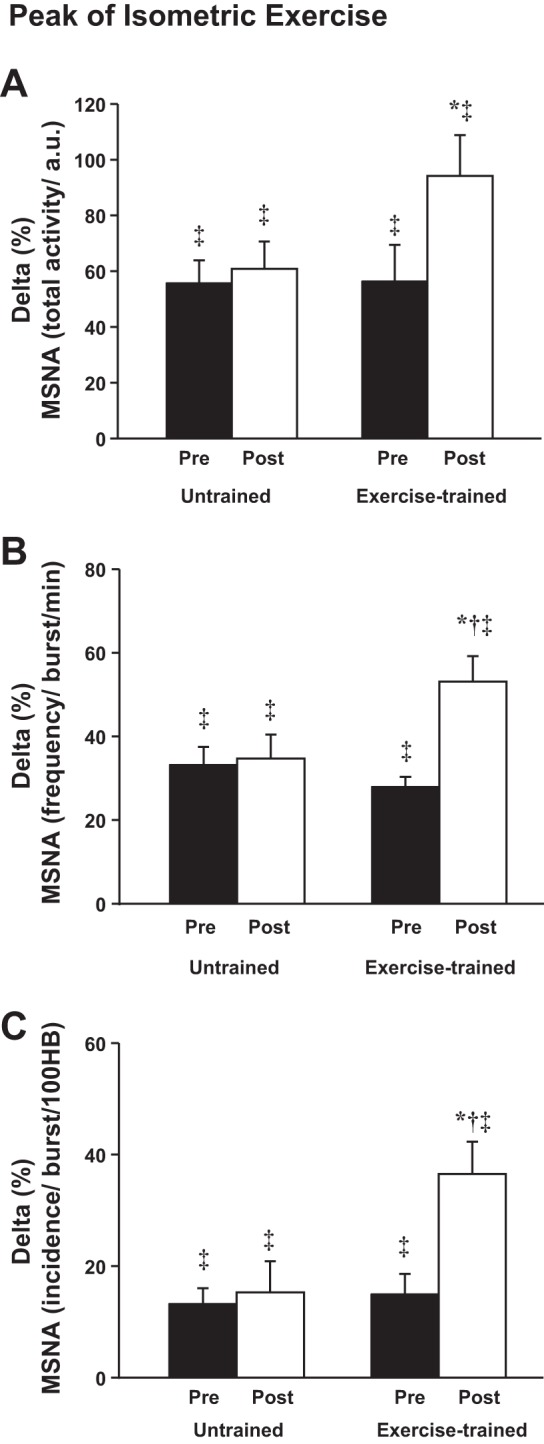
Peak of 30% isometric leg exercise in untrained and exercise-trained heart failure patients. A: muscle sympathetic nerve activity (MSNA) total activity (a.u.). B: MSNA frequency (bursts/min). C: MSNA incidence [bursts/100 heartbeats (HB)]. *P < 0.05 vs. pre within group. †P < 0.05 vs. untrained. ‡P < 0.05 vs. rest.
Table 3.
Hemodynamic responses during peak of 30% isometric leg exercise, postexercise regional circulatory arrest, and peak of passive exercise stimulation in untrained and exercise-trained heart failure patients
| Untrained |
Exercise Trained |
|||
|---|---|---|---|---|
| Pre | Post | Pre | Post | |
| Peak of 30% leg exercise | (n = 17) | (n = 16) | ||
| HR, % | 19 ± 2‡ | 17 ± 2‡ | 15 ± 3‡ | 8 ± 1*†‡ |
| SBP, % | 7 ± 2‡ | 8 ± 2‡ | 7 ± 1‡ | 7 ± 2‡ |
| DBP, % | 13 ± 3‡ | 12 ± 2‡ | 11 ± 2‡ | 7 ± 1*‡ |
| MBP, % | 11 ± 3‡ | 11 ± 2‡ | 10 ± 2‡ | 8 ± 1‡ |
| PECA | (n = 17) | (n = 16) | ||
| HR, % | 10 ± 3‡ | 9 ± 2‡ | 6 ± 1‡ | 7 ± 1‡ |
| SBP, % | 7 ± 2‡ | 7 ± 1‡ | 5 ± 1‡ | 7 ± 1‡ |
| DBP, % | 9 ± 2‡ | 7 ± 2‡ | 5 ± 1‡ | 7 ± 1‡ |
| MBP, % | 9 ± 2‡ | 8 ± 2‡ | 6 ± 1‡ | 8 ± 1‡ |
| Peak of passive exercise | (n = 17) | (n = 15) | ||
| HR, % | −1 ± 1 | 1 ± 1 | 1 ± 1 | 0 ± 1 |
| SBP, % | 1 ± 1 | 1 ± 0.5 | 1 ± 1 | −1 ± 1 |
| DBP, % | 1 ± 1‡ | 1 ± 0.5 | 2 ± 1‡ | 0 ± 1† |
| MBP, % | 1 ± 1‡ | 2 ± 1‡ | 2 ± 1‡ | 0 ± 1‡† |
Values are means ± SE. PECA, postexercise circulatory arrest.
P < 0.05 vs. pre within group.
P < 0.05 vs. untrained.
P < 0.05 vs. rest.
Following exercise training, the MSNA increased during peak 30% MVC isometric leg exercise, and the increase was significantly greater compared with the increase in MSNA in the untrained time control HF group (Fig. 2). Surprisingly, following exercise training, despite the augmented MSNA responses during 30% MVC isometric leg exercise, the increases in diastolic BP and HR were significantly blunted (Table 2).
Effects of Exercise Training on Muscle Metaboreflex Control
Examples of sympathetic neurograms during metaboreceptor stimulation pre- and postexercise training and pre- and posttime control are shown in Fig. 3A. Before exercise training or time control, MSNA returned to resting levels during PECA in both groups (Fig. 4), consistent with blunted metaboreflex control. In contrast, HR and BP were significantly increased in relation to rest period during PECA in both groups (Table 3). After exercise training, the changes in MSNA were augmented during PECA and this increase was significantly greater than the changes in MSNA in the untrained time control HF group (Fig. 4, A–C), in which, MSNA once again returned to resting levels during PECA. In contrast, the increase in HR and BP during PECA was unchanged pre- and posttime in the exercise-trained group (Table 3). There were no technical problems with the nerve recording during PECA.
Fig. 3.

Sympathetic neurograms of untrained and exercise-trained heart failure patients during metaboreceptors stimulation (A) and during mechanoreceptors stimulation (B).
Fig. 4.

Metaboreflex control of MSNA assessed by %changes between the 1st minute of postexercise regional circulatory arrest and rest period in untrained (n = 17) and exercise-trained (n = 16) heart failure patients. A: delta changes in MSNA total activity (a.u.). B: frequency (bursts/min). C: incidence (bursts/100 HB). Mechanoreflex control of MSNA assessed by difference between the peak of passive exercise and rest period in untrained (n = 17) and exercise-trained (n = 16) heart failure patients. D: delta changes in MSNA total activity (a.u.). E: frequency (bursts/min). F: incidence (bursts/100 HB). Exercise training markedly increased the MSNA responses during metaboreceptors stimulation in heart failure patients. In contrast, exercise training significantly decreased MSNA responses during mechanoreceptors stimulation. Values are mean individual response. *P < 0.05 vs. pre within group. †P < 0.05 vs. untrained.
Effects of Exercise Training on Muscle Mechanoreflex Control
Examples of sympathetic neurograms during mechanoreceptor activation pre- and postexercise training and pre- and posttime control are shown in Fig. 3B. Passive exercise, which stimulates the mechanoreflex, provoked a significant and similar increase in MSNA and diastolic and mean BP in both exercise-trained and untrained groups (Fig. 4, D–F; Table 3). There were no significant increases in systolic BP and HR in peak passive exercise. After exercise training, the increase in MSNA was reduced and significantly less than the increase in MSNA in the untrained time control group (Fig. 4, D–F), consistent with a significant decrease in mechanoreflex sensitivity. Similarly, after exercise training, the increase in diastolic and mean BP was less than the increase in the untrained time control group (Table 3). The systolic BP and HR responses pre- and posttime in exercise-trained group were unchanged (Table 3). Nerve recording amplitude abruptly decreased during the experimental protocol in one exercise-trained patient consistent with a change in microelectrode position. This patient was excluded from the data analysis.
Effects of Exercise Training on Skeletal Muscle–Muscle Metaboreflex Control
TRPV1 and CB-1 receptors.
Exercise training significantly increased gene expression of the TRPV1 receptor (Fig. 5A) and gene expression of the CB1 receptor (Fig. 5B) in the VL muscle. No changes were observed in gene expression of the TRPV1 receptor and CB1 receptor in untrained HF patients. The comparisons between groups posttraining or posttime control showed that gene expression of the TRPV1 receptor (Fig. 5A) and CB1 receptor (Fig. 5B) was significantly higher in exercise-trained HF patients when compared with untrained HF patients.
Fig. 5.
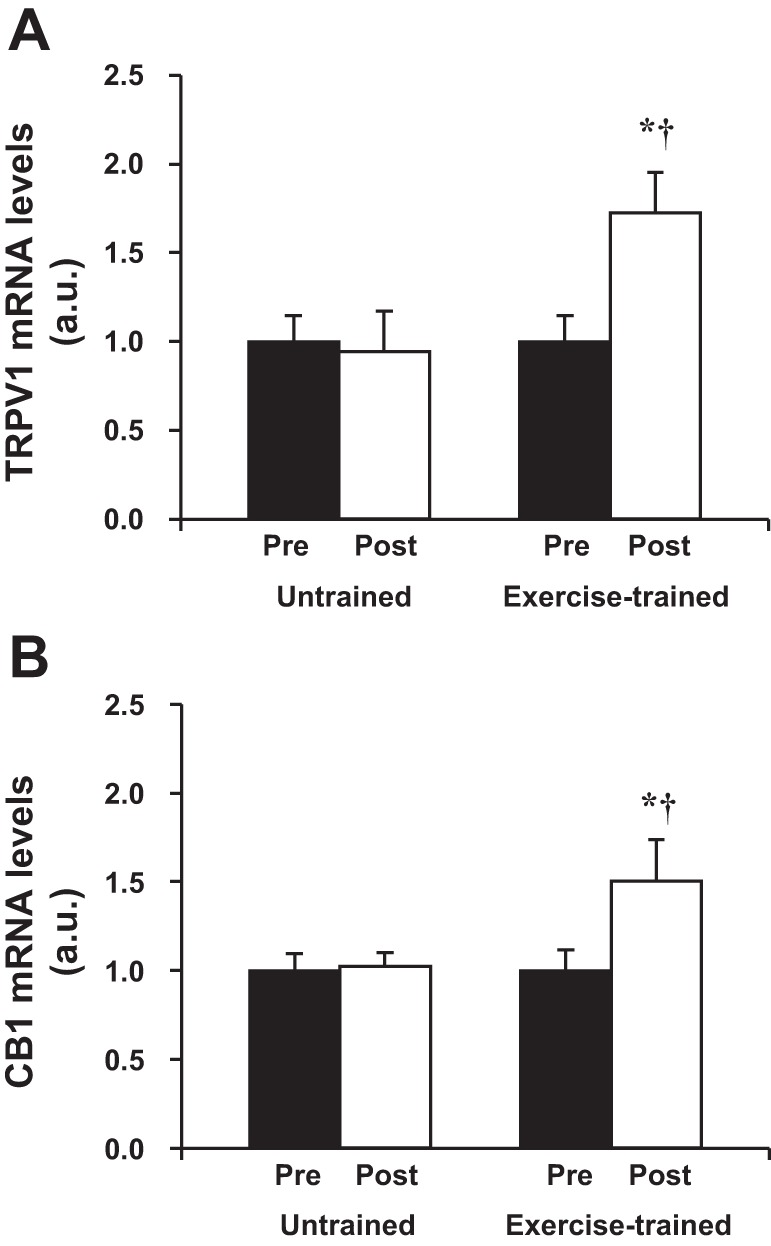
Gene expression of transient receptor potential vanilloid type-1 (TRPV1; A) and cannabinoid receptor type-1 (CB1; B) receptors in vastus lateralis in untrained (n = 11) and exercise-trained (n = 12) heart failure patients. Exercise training significantly increased gene expression of TRPV1 and CB1 receptors. Values are means ± SE. *P < 0.05 vs. pre within group. †P < 0.05 vs. untrained.
Effects of Exercise Training on Skeletal Muscle–Muscle Mechanoreflex Control
COX pathway enzymes.
COX-1 and COX-2 are rate-limiting enzymes in the formation of prostaglandins (42). Exercise training caused no significant changes in gene expression of COX-1 in the VL muscle (Fig. 6A). In contrast, exercise training tended to reduce gene expression of COX-2 in the VL muscle (P = 0.08; Fig. 6B). No significant changes in gene expression of COX-1 and COX-2 were found in the untrained patients. The comparisons between groups posttraining or posttime control showed that gene expression of COX-2 was significantly lower in exercise-trained HF patients compared with untrained HF patients (Fig. 6B). Importantly, protein levels of COX-2 were significantly reduced following exercise training compared with baseline in the exercise-trained HF group (Fig. 6C) but were unchanged pre- and posttime control period in untrained HF patients. The comparisons between groups posttraining or posttime control showed that protein levels of COX-2 were significantly lower in exercise-trained HF patients compared with untrained HF patients (Fig. 6C). COX-2 transforms arachidonic acid into prostaglandin H2, which is rapidly transformed by the downstream enzymes mPGES1, PGIS, or TXAS into the COX-2 products prostaglandin E2 (PGE2), prostacyclin (PGI2), and thromboxane A2 (TXA2), respectively (42). There were no changes in the expression of any of these downstream enzymes in the exercise-trained or untrained HF patients (data not shown).
Fig. 6.
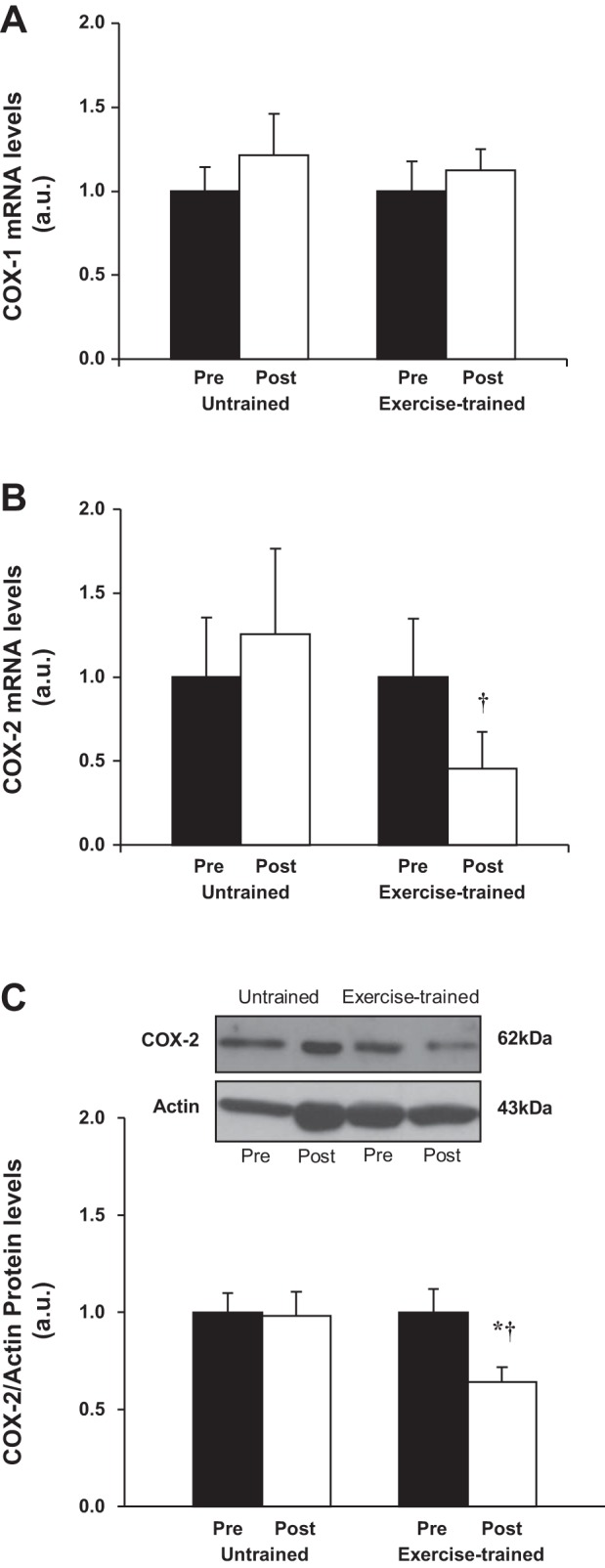
Gene expression of cyclooxygenase (COX)-1 (A) and COX-2 (B) in vastus lateralis in untrained (n = 11) and exercise-trained (n = 12) heart failure patients. C: representative Western blots (top) and densitometric analysis (bottom) of COX-2 protein expression in untrained (n = 9) and exercise-trained (n = 9) patients. Sarcomeric actin protein expression was used as internal control. Exercise training significantly decreased gene and protein expression of COX-2. Values are means ± SE. *P < 0.05 vs. pre within group. †P < 0.05 vs. untrained.
Regulation of COX-2 expression.
We then investigated possible mechanisms by which the COX-2 expression might be decreased following exercise training in HF patients. Proinflammatory cytokines induce COX-2 expression in response to inflammation (42). In this process, inactive cytoplasmic NF-κB is released from IκB-α, allowing NF-κB to enter the nucleus and increase expression of COX-2. In the present study, the ratio of NF-κB/IκB-α was increased in the untrained HF patients during time control (Fig. 7D), but this increase was not seen in the exercise-trained patients, indicative of relative decrease in inflammation and NF-κB translocation to the nucleus. The miRNA-146 expression, which acts through a negative feedback loop to decrease NF-κB (37), was significantly increased after exercise training (Fig. 7C) but not in the time control HF group. The expression of miRNA-16 and miRNA-143, which regulate COX-2, was unchanged pre- and posttraining and during the pre- and posttime control period (Fig. 7, A and B).
Fig. 7.
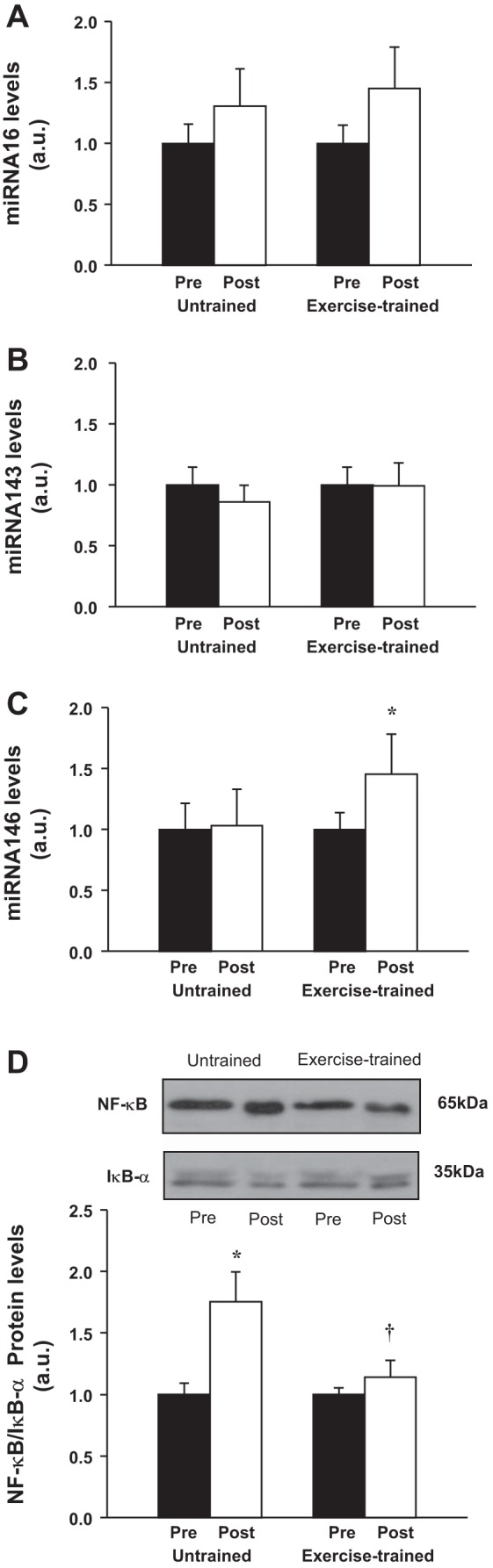
Expression of miRNA-16 (A), miRNA-143 (B), and miRNA-146 (C) in vastus lateralis in untrained (n = 11) and exercise-trained (n = 12) heart failure patients. D: representative Western blots (top) and densitometric analysis (bottom) of protein expression of NF-κB/IκB-α ratio in vastus lateralis in untrained (n = 7) and exercise-trained (n = 7) patients. Exercise training significantly increased expression of miRNA-146 and prevented the increase in protein expression of NF-κB/IκB-α ratio. Values are means ± SE. *P < 0.05 vs. pre within group. †P < 0.05 vs. untrained.
COX-2 pathway receptors.
We then compared the gene expression of receptors for the COX-2 metabolites EP1, EP2, EP3, EP4, IP, and TP. Exercise training significantly reduced gene expression of the EP4 receptor (1.0 vs. 0.70 AU) and gene expression of TP receptor (1.0 vs. 0.65 AU) in the VL muscle pre- and posttraining period but caused no significant changes in any other receptor. No changes were found in the untrained HF patients. Furthermore, the comparisons between groups posttraining or posttime control showed that gene expression of EP4 receptor was significantly lower in exercise-trained HF patients compared with untrained HF patients.
DISCUSSION
In patients with HF from systolic dysfunction and in animal models of HF, the exercise pressor reflex is augmented during exercise and has been shown to be associated with attenuated muscle metaboreflex control of MSNA and augmented muscle mechanoreflex control of MSNA (13, 18, 21, 22, 25, 39–41, 45, 49). In fact, the two seem to be inversely, and perhaps causally, related, since in healthy animal models in which the metaboreceptors are selectively destroyed, the mechanoreceptors develop heightened sensitivity (13, 39, 40). In humans with HF, the “muscle hypothesis” has been advanced, which posits that abnormalities of the skeletal muscle underlie the abnormal reflex neurovascular responses evoked during exercise and thus the diminished exercise capacity in HF (7, 32). Importantly, in animal models of HF, exercise training prevents the development of abnormal muscle afferent sensitivity and decreased exercise pressor reflex (46, 47).
The main, new findings of the present study are 1) in HF patients, exercise training increases muscle metaboreflex control of MSNA, which is accompanied by an increase in expression of TRPV1 and CB1 receptors. 2) In HF patients, exercise training decreases the muscle mechanoreflex control of MSNA, which is accompanied by a decrease in expression of COX-2, EP4 receptors, TP receptors, and the ratio of NF-κB/IκB-α and an increase in miRNA-146 in skeletal muscle. 3) The net effect of exercise training on these reflex systems is an increase in MSNA during 30% MVC isometric leg exercise, which is not accompanied by an increase in BP or HR; in fact, the changes in these hemodynamic parameters provoked by exercise tend to be decreased following exercise training. These findings support the concept, advanced by the muscle hypothesis (7, 32), that abnormalities in the skeletal muscle underlie the abnormal reflex responses during exercise in humans with HF and, most importantly, suggest that these abnormalities are reversible.
Several independent groups have reported that, in the rat-infarct model of HF, the muscle metaboreflex is attenuated and is associated with decreased expression of TRPV1 receptors (20, 40, 45, 49). Furthermore, this decreased group IV fiber responsiveness and decreased TRPV1 receptor density is not due to selective cell death or necrosis, since examination of the DRG does not reveal evidence of apoptosis or necrosis (13, 49). In our study in humans, the reversal of the attenuation of muscle metaboreflex control of MSNA accompanied by an increase in the expression of the TRPV1 receptor with exercise training further refute apoptosis and necrosis as the underlying mechanisms. Conversely, an abnormality related to the skeletal myopathy of HF is consistent with our finding of improvement with exercise training, since exercise training also reverses the skeletal myopathy (44). TRPV1 receptors depend on nerve growth factor (NGF), which is expressed in healthy skeletal muscle and binds to presynaptic terminals in IV afferents to upregulate TRPV1 receptor expression and sensitization (13, 50, 51, 53). NGF, which originates in the healthy skeletal muscle, has been found to be decreased in the DRG in the rat-infarct model of HF, reflective of abnormal skeletal muscle NGF content and potentially underlying the attenuated muscle metaboreflex (13, 50, 51, 53). The level of NGF in skeletal muscle in humans with HF before and after training has not yet been measured, but is an area of active investigation in our laboratory. Interestingly, TRPV1 receptors are colocalized with CB1 receptors and have been found to have decreased expression in the rat-infarct model of HF (49). We found that CB1 expression was increased following training. The role in NGF in CB1 expression and activity is unknown (53).
Just as several independent groups have reported attenuated muscle metaboreflex sensitivity in the rat infarct model, increased muscle mechanoreflex sensitivity in this HF model has also been widely reported (18, 20, 29, 40, 45). Similarly, we previously reported that in humans with HF, muscle mechanoreflex-mediated increases in MSNA were augmented, and this augmentation was inhibited by indomethacin, consistent with the notion that COX products sensitize muscle mechanoreceptors in humans with HF (21, 22). The present study extends these findings. The findings in the present study that, following exercise training, the muscle mechanoreflex-mediated increase in MSNA is significantly diminished, and this decrease is accompanied by a significant decrease in COX-2 expression in the VL muscle, support the notion that overexpression of COX-2 in the skeletal muscle of HF patients leads to increased prostaglandin (likely PGE2) sensitization of group III afferents. The expression of the EP4 receptor, which is present on DRG and is responsible for afferent nerve sensitization by PGE2 (52), is also decreased following exercise training. Similarly, the expression of the TP receptor, which is responsible for the afferent sensitization by TXA2 is decreased following exercise training. In summary, either the increase in COX-2 expression leading to increased PGE2 and TXA2, or the increase in afferent EP4 and TP receptors that sensitive group III afferents, or both contribute significantly to the augmented muscle mechanoreceptor sensitivity in humans with HF.
What is the mechanism increasing the COX-2 expression in VL muscle in untrained HF patients? Interestingly, the mechanism underlying the augmented COX-2 expression can also be traced back to the skeletal myopathy of HF. Expression of COX-2 is augmented by increased inflammation in skeletal muscle and mediates further inflammation (42). In this study, exercise training prevented the increase in the NF-κB/IκB ratio seen the untrained time control group, a marker of active inflammation, which may be associated with the increase in miRNA-146. In addition, exercise training decreased the expression of the COX-2, EP4, and TP receptors, consistent with improvement in the skeletal myopathy of HF. Other studies in humans with HF, or the rat-infarct model, have also demonstrated an anti-inflammatory effect of exercise training (5, 14).
In humans with HF, these findings of abnormal muscle afferent activity during exercise, accompanied by evidence of inflammation (NF-κB, COX-2) and decreased expression of afferent receptors (TRPV1, CB1), all of which reverse with exercise training, are strongly supportive of the muscle hypothesis of exercise dysfunction. Furthermore, the striking finding that resting MSNA levels are markedly decreased following exercise training is consistent with the concept that abnormal signals originating in skeletal muscle and muscle afferents are important contributors to the resting sympathetic excitation that characterizes chronic HF with systolic dysfunction in humans.
It was somewhat surprising that the MSNA response to 30% MVC isometric leg exercise was augmented following exercise training. This augmented MSNA response must, in part, be attributable to the muscle metaboreflex, since exercise training increased the muscle metaboreflex sensitivity. Our findings that expression of TRPV1 and CB1 receptors was increased following exercise training further support an important role for the muscle metaboreflex in mediating the exercise-induced increase in MSNA in trained HF patients. Interestingly, this increase in MSNA was not accompanied by exaggerated or augmented changes in BP or HR during the isometric exercise. We did not measure sympathetic activity to other organs and tissues, such as the heart or kidney. It remains possible that following training, sympathetic responses provoked by exercise directed to these organs actually decreased, explaining the minimal hemodynamic effects. Our findings suggest that the alteration in muscle metaboreflex and mechanoreflex control of MSNA may also play a role in the reduction in resting MSNA following exercise training in HF patients. However, we cannot rule out other beneficial effects of exercise training on MSNA, including effects on the arterial baroreflex and central integration on resting MSNA levels.
Limitations
We recognize limitations in our study. Although the maneuvers to isolate individual afferent limbs of the exercise pressor reflex have been used for decades, it is likely they are not pure. For example, PECA may also stimulate of the nociceptive fibers and cause pain. To address this possibility, circulatory arrest without previous exercise was conducted in three HF patients to detect the effect, if any, on efferent MSNA. We found no significant changes in MSNA under this condition (data not shown). Furthermore, no HF patient complained of pain during this PECA maneuver, which was completed in all study participants. HF patients were taking medications that could not be stopped for ethical reasons but that could theoretically alter hemodynamic and neural responses to exercise. Medications may explain the absence of significant increases in hemodynamic (e.g., HR and BP) responses during exercise, despite the increased sympathetic responses. MSNA responses may be less affected by medications; in fact, acute administration of beta-adrenergic blockers in HF patients has been reported to have no effect on MSNA (3). It is worth noting that medication doses remained stable throughout the study period, so medication changes are unlikely to underlie changes in sympathetic responses following exercise training. Approximately half the HF patients were taking aspirin, which inhibits COX-1 and COX-2 activity. Aspirin administration does not explain our findings, since aspirin use was not different between the trained and untrained HF groups (Table 1). Furthermore, when the data were reanalyzed according to aspirin use, the results were unchanged (data not shown). Although the COX pathway is an important pathway in contributing the increased SNA responses during exercise in HF, it is unlikely the only one. In the rat-infarct model, the P2X receptor is upregulated on muscle group III afferents, and infusion of the P2X antagonist attenuates the exaggerated SNA response to mechanical deformation (45). Although we did not investigate the role of the P2X receptor in mediating the exaggerated MSNA responses during mechanoreflex stimulation our HF patients, the data in the rat infarct model are strong, and the P2X receptor warrants investigation in humans with HF as well. The muscle biopsy included neuronal, vascular, muscular, and connective tissue, so the tissue of origin of the TRPV1 and CB1 receptors in the muscle homogenate, although most likely neuronal, cannot be stated unequivocally. Finally, we did not include a healthy control group. A control group of healthy age-matched volunteers would provide important information about the normal levels of TRPV1 and CB1 in the absence of HF. The literature available in animals may not reflect what occurs in humans. Furthermore, our study does not clarify how the exercise-induced increase in TRPV1 and CB1 levels in HF relates to the situation in healthy controls. Interestingly, in healthy volunteers, Ray et al. (35) reported that MSNA did not increase during 30% MVC intensity isometric leg exercise, but did increase during 30% MVC intensity isometric arm exercise. In contrast, we found that MSNA does increase significantly during 30% MVC isometric leg exercise in untrained HF patients. Ray et al. studied healthy young males, ages 19–29 yr, whereas our cohort of HF patients was ∼30 yr older and included females. Although the stated purpose of our study was to determine the effect of exercise training on neuronal reflexes in HF patients, future studies of MSNA responses during 30% MVC leg exercise in healthy middle-aged men and women would be of interest.
In conclusion, exercise training improves muscle metaboreflex and mechanoreflex control of MSNA in humans with HF. This exercise training-induced improvement in reflex control of MSNA is accompanied by augmented TRPV1 and CB1 expression, reduction of COX-2 expression, and reduced inflammation in skeletal muscle. These findings strongly support the concept that abnormalities in the skeletal muscle itself underlie the abnormal reflex control of SNA during exercise in HF, that is, the “muscle hypothesis.” Nonetheless, our results call into question the role of augmented MSNA levels during exercise in limiting exercise capacity in HF patients. Despite the increased exercise capacity following exercise training in our HF patients, MSNA responses during exercise were significantly augmented in these exercise-trained HF patients. Furthermore, augmented MSNA responses were not accompanied by greater increases in hemodynamic responses (HR and BP) during exercise. Thus in pharmacologically optimized HF patients, decreased exercise capacity may not be attributable to abnormalities of reflex sympathetic activation but may be mediated by other mechanisms, also rooted in the skeletal muscle, such as abnormalities in calcium cycling in skeletal muscle, which warrant future investigation (26). Importantly, elevated resting MSNA levels in HF, a marker of increased mortality, were markedly decreased following training. These findings provide strong support for the recommendation of exercise training in all patients with chronic HF.
GRANTS
This study was supported by Fundação de Amparo à Pesquisa do Estado de Sao Paulo (FAPESP- 2010/50048-1) and, in part, by Fundação Zerbini. L. M. Antunes-Correa, T. S. Nobre, M. U. Rondon, P. C. Brum, L. V. Rossoni, and C. E. Negrao were supported CNPq. R. V. Groehs was supported by CAPES. H. R. Middlekauff was supported by National Heart, Lung, and Blood Institute Grant RO1-HL084525.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.M.A.-C., M.-J.N.A., W.M.j., P.C.B., H.R.M., and C.E.N. conception and design of research; L.M.A.-C., T.S.N., R.V.G., M.-J.N.A., T.F., G.K.C., M.U.P.B.R., P.A.d.O., M.L., C.M., and D.R.A. performed experiments; L.M.A.-C., T.S.N., R.V.G., T.F., G.K.C., L.V.R., and E.M.d.O. analyzed data; L.M.A.-C., T.F., G.K.C., M.U.P.B.R., L.V.R., E.M.d.O., H.R.M., and C.E.N. interpreted results of experiments; L.M.A.-C. prepared figures; L.M.A.-C., M.U.P.B.R., L.V.R., E.M.d.O., and C.E.N. drafted manuscript; L.M.A.-C., H.R.M., and C.E.N. edited and revised manuscript; H.R.M. and C.E.N. approved final version of manuscript.
REFERENCES
- 1.Antunes-Correa LM, Kanamura BY, Melo RC, Nobre TS, Ueno LM, Franco FG, Roveda F, Braga AM, Rondon MU, Brum PC, Barretto AC, Middlekauff HR, Negrao CE. Exercise training improves neurovascular control and functional capacity in heart failure patients regardless of age. Eur J Prev Cardiol 19: 822–829, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Antunes-Correa LM, Melo RC, Nobre TS, Ueno LM, Franco FG, Braga AM, Rondon MU, Brum PC, Barretto AC, Middlekauff HR, Negrao CE. Impact of gender on benefits of exercise training on sympathetic nerve activity and muscle blood flow in heart failure. Eur J Heart Fail 12: 58–65, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azevedo ER, Kubo T, Mak S, Al-Hesayen A, Schofield A, Allan R, Kelly S, Newton GE, Floras JS, Parker JD. Nonselective vs. selective beta-adrenergic receptor blockade in congestive heart failure: differential effects on sympathetic activity. Circulation 104: 2194–2199, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Barretto AC, Santos AC, Munhoz R, Rondon MU, Franco FG, Trombetta IC, Roveda F, de Matos LN, Braga AM, Middlekauff HR, Negrao CE. Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int J Cardiol 135: 302–307, 2009. [DOI] [PubMed] [Google Scholar]
- 5.Batista ML, Jr, Rosa JC, Lopes RD, Lira FS, Martins E, Jr, Yamashita AS, Brum PC, Lancha AH, Jr, Lopes AC, Seelaender M. Exercise training changes IL-10/TNF-alpha ratio in the skeletal muscle of post-MI rats. Cytokine 49: 102–108, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Burd NA, Dickinson JM, Lemoine JK, Carroll CC, Sullivan BE, Haus JM, Jemiolo B, Trappe SW, Hughes GM, Sanders CE, Trappe TA. Effect of a cyclooxygenase-2 inhibitor on postexercise muscle protein synthesis in humans. Am J Physiol Endocrinol Metab 298: E354–E361, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coats AJ, Clark AL, Piepoli M, Volterrani M, Poole-Wilson PA. Symptoms and quality of life in heart failure: the muscle hypothesis. Br Heart J 72: S36–39, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davel AP, Fukuda LE, De Sá LL, Munhoz CD, Scavone C, Sanz-Rosa D, Cachofeiro V, Lahera V, Rossoni LV. Effects of isoproterenol treatment for 7 days on inflammatory mediators in the rat aorta. Am J Physiol Heart Circ Physiol 295: H211–H219, 2008. [DOI] [PubMed] [Google Scholar]
- 9.de Mello Franco FG, Santos AC, Rondon MU, Trombetta IC, Strunz C, Braga AM, Middlekauff H, Negrão CE, Pereira Barretto AC. Effects of home-based exercise training on neurovascular control in patients with heart failure. Eur J Heart Fail 8: 851–855, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Fraga R, Franco FG, Roveda F, de Matos LN, Braga AM, Rondon MU, Rotta DR, Brum PC, Barretto AC, Middlekauff HR, Negrao CE. Exercise training reduces sympathetic nerve activity in heart failure patients treated with carvedilol. Eur J Heart Fail 9: 630–636, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Francis GS, Cohn JN, Johnson G, Rector TS, Goldman S, Simon A. Plasma norepinephrine, plasma renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. The V-HeFT VA Cooperative Studies Group. Circulation 87: VI40–48, 1993. [PubMed] [Google Scholar]
- 13.Garry MG. Abnormalities of the exercise pressor reflex in heart failure. Exerc Sport Sci Rev 39: 167–176, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Gielen S, Adams V, Mobius-Winkler S, Linke A, Erbs S, Yu J, Kempf W, Schubert A, Schuler G, Hambrecht R. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol 42: 861–868, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA, Korner PI. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 73: 615–621, 1986. [DOI] [PubMed] [Google Scholar]
- 16.Kaufman MP, Longhurst JC, Rybicki KJ, Wallach JH, Mitchell JH. Effects of static muscular contraction on impulse activity of groups III and IV afferents in cats. J Appl Physiol Respir Environ Exercise Physiol 55: 105–112, 1983. [DOI] [PubMed] [Google Scholar]
- 17.Kleiger RE, Miller JP, Bigger JT, Jr, Moss AJ. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol 59: 256–262, 1987. [DOI] [PubMed] [Google Scholar]
- 18.Koba S, Xing J, Sinoway LI, Li J. Sympathetic nerve responses to muscle contraction and stretch in ischemic heart failure. Am J Physiol Heart Circ Physiol 294: H311–H321, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Leimbach WN, Jr, Wallin BG, Victor RG, Aylward PE, Sundlof G, Mark AL. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 73: 913–919, 1986. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Sinoway AN, Gao Z, Maile MD, Pu M, Sinoway LI. Muscle mechanoreflex and metaboreflex responses after myocardial infarction in rats. Circulation 110: 3049–3054, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Middlekauff HR, Chiu J, Hamilton MA, Fonarow GC, Maclellan WR, Hage A, Moriguchi J, Patel J. Cyclooxygenase products sensitize muscle mechanoreceptors in humans with heart failure. Am J Physiol Heart Circ Physiol 294: H1956–H1962, 2008. [DOI] [PubMed] [Google Scholar]
- 22.Middlekauff HR, Chiu J, Hamilton MA, Fonarow GC, Maclellan WR, Hage A, Moriguchi J, Patel J. Muscle mechanoreceptor sensitivity in heart failure. Am J Physiol Heart Circ Physiol 287: H1937–H1943, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Middlekauff HR, Nitzsche EU, Hoh CK, Hamilton MA, Fonarow GC, Hage A, Moriguchi JD. Exaggerated muscle mechanoreflex control of reflex renal vasoconstriction in heart failure. J Appl Physiol 90: 1714–1719, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Middlekauff HR, Nitzsche EU, Hoh CK, Hamilton MA, Fonarow GC, Hage A, Moriguchi JD. Exaggerated renal vasoconstriction during exercise in heart failure patients. Circulation 101: 784–789, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Middlekauff HR, Sinoway LI. Increased mechanoreceptor stimulation explains the exaggerated exercise pressor reflex seen in heart failure. J Appl Physiol 102: 492–494; discussion 496, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Middlekauff HR, Vigna C, Verity MA, Fonarow GC, Horwich TB, Hamilton MA, Shieh P, Tupling AR. Abnormalities of calcium handling proteins in skeletal muscle mirror those of the heart in humans with heart failure: a shared mechanism? J Card Fail 18: 724–733, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell JH, Kaufman MP, Iwamoto GA. The exercise pressor reflex: its cardiovascular effects, afferent mechanisms, and central pathways. Annu Rev Physiol 45: 229–242, 1983. [DOI] [PubMed] [Google Scholar]
- 28.Moore AE, Young LE, Dixon DA. MicroRNA and AU-rich element regulation of prostaglandin synthesis. Cancer Metastasis Rev 30: 419–435, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morales A, Gao W, Lu J, Xing J, Li J. Muscle cyclo-oxygenase-2 pathway contributes to the exaggerated muscle mechanoreflex in rats with congestive heart failure. Exp Physiol 97: 943–954, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neves M, Barreto G, Boobis L, Harris R, Roschel H, Tricoli V, Ugrinowitsch C, Negrão C, Gualano B. Incidence of adverse events associated with percutaneous muscular biopsy among healthy and diseased subjects. Scand J Med Sci Sports 22: 175–178, 2012. [DOI] [PubMed] [Google Scholar]
- 31.O'Connor CM, Whellan DJ, Lee KL, Keteyian SJ, Cooper LS, Ellis SJ, Leifer ES, Kraus WE, Kitzman DW, Blumenthal JA, Rendall DS, Miller NH, Fleg JL, Schulman KA, McKelvie RS, Zannad F, Pina IL. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 301: 1439–1450, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piepoli M, Ponikowski P, Clark AL, Banasiak W, Capucci A, Coats AJ. A neural link to explain the “muscle hypothesis” of exercise intolerance in chronic heart failure. Am Heart J 137: 1050–1056, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Pina IL, Apstein CS, Balady GJ, Belardinelli R, Chaitman BR, Duscha BD, Fletcher BJ, Fleg JL, Myers JN, Sullivan MJ. Exercise and heart failure: a statement from the American Heart Association Committee on exercise, rehabilitation, and prevention. Circulation 107: 1210–1225, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Ponikowski PP, Chua TP, Francis DP, Capucci A, Coats AJ, Piepoli MF. Muscle ergoreceptor overactivity reflects deterioration in clinical status and cardiorespiratory reflex control in chronic heart failure. Circulation 104: 2324–2330, 2001. [DOI] [PubMed] [Google Scholar]
- 35.Ray CA, Rea RF, Clary MP, Mark AL. Muscle sympathetic nerve responses to static leg exercise. J Appl Physiol (1985) 73: 1523–1529, 1992. [DOI] [PubMed] [Google Scholar]
- 36.Roveda F, Middlekauff HR, Rondon MU, Reis SF, Souza M, Nastari L, Barretto AC, Krieger EM, Negrao CE. The effects of exercise training on sympathetic neural activation in advanced heart failure: a randomized controlled trial. J Am Coll Cardiol 42: 854–860, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Rusca N, Monticelli S. MiR-146a in immunity and disease. Mol Biol Int 2011: 437301, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skinner JS, McLellan TH. The transition from aerobic to anaerobic metabolism. Res Q Exerc Sport 51: 234–248, 1980. [DOI] [PubMed] [Google Scholar]
- 39.Smith SA, Mitchell JH, Naseem RH, Garry MG. Mechanoreflex mediates the exaggerated exercise pressor reflex in heart failure. Circulation 112: 2293–2300, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Smith SA, Williams MA, Mitchell JH, Mammen PP, Garry MG. The capsaicin-sensitive afferent neuron in skeletal muscle is abnormal in heart failure. Circulation 111: 2056–2065, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Sterns DA, Ettinger SM, Gray KS, Whisler SK, Mosher TJ, Smith MB, Sinoway LI. Skeletal muscle metaboreceptor exercise responses are attenuated in heart failure. Circulation 84: 2034–2039, 1991. [DOI] [PubMed] [Google Scholar]
- 42.Tsatsanis C, Androulidaki A, Venihaki M, Margioris AN. Signalling networks regulating cyclooxygenase-2. Int J Biochem Cell Biol 38: 1654–1661, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Ueno LM, Drager LF, Rodrigues AC, Rondon MU, Braga AM, Mathias W, Krieger EM, Barretto AC, Middlekauff HR, Lorenzi-Filho G, Negrão CE. Effects of exercise training in patients with chronic heart failure and sleep apnea. Sleep 32: 637–647, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ventura-Clapier R, Mettauer B, Bigard X. Beneficial effects of endurance training on cardiac and skeletal muscle energy metabolism in heart failure. Cardiovasc Res 73: 10–18, 2007. [DOI] [PubMed] [Google Scholar]
- 44a.Wallin BG. Assessment of sympathetic mechanisms from recordings of postganglionic efferent nerve traffic. In: Cardiovascular Reflex Control in Health and Disease, edited by Hainsworth R, Mark AL. London, UK: W. B. Saunders, 1993, p. 65–93. [Google Scholar]
- 45.Wang HJ, Li YL, Gao L, Zucker IH, Wang W. Alteration in skeletal muscle afferents in rats with chronic heart failure. J Physiol 588: 5033–5047, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang HJ, Li YL, Zucker IH, Wang W. Exercise training prevents skeletal muscle afferent sensitization in rats with chronic heart failure. Am J Physiol Regul Integr Comp Physiol 302: R1260–R1270, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang HJ, Pan YX, Wang WZ, Gao L, Zimmerman MC, Zucker IH, Wang W. Exercise training prevents the exaggerated exercise pressor reflex in rats with chronic heart failure. J Appl Physiol 108: 1365–1375, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wenceslau CF, Davel AP, Xavier FE, Rossoni LV. Long-term ouabain treatment impairs vascular function in resistance arteries. J Vasc Res 48: 316–326, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Williams MA, Smith SA, O'Brien DE, Mitchell JH, Garry MG. The group IV afferent neuron expresses multiple receptor alterations in cardiomyopathyic rats: evidence at the cannabinoid CB1 receptor. J Physiol 586: 835–845, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winston J, Toma H, Shenoy M, Pasricha PJ. Nerve growth factor regulates VR-1 mRNA levels in cultures of adult dorsal root ganglion neurons. Pain 89: 181–186, 2001. [DOI] [PubMed] [Google Scholar]
- 51.Xue Q, Jong B, Chen T, Schumacher MA. Transcription of rat TRPV1 utilizes a dual promoter system that is positively regulated by nerve growth factor. J Neurochem 101: 212–222, 2007. [DOI] [PubMed] [Google Scholar]
- 52.Yamauchi K, Kim JS, Stone AJ, Ruiz-Velasco V, Kaufman MP. Endoperoxide 4 receptors play a role in evoking the exercise pressor reflex in rats with simulated peripheral artery disease. J Physiol 591: 2949–2962, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J 24: 4211–4223, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zucker IH, Patel KP, Schultz HD. Neurohumoral stimulation. Heart Fail Clin 8: 87–99, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]