
Figure 6.
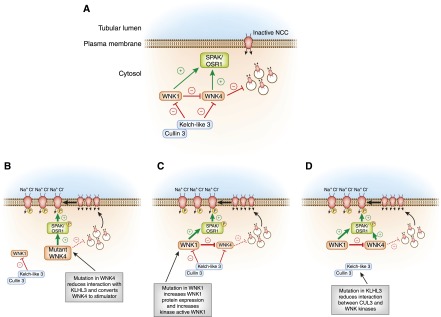
A model of WNK-SPAK/OSR1 regulation of NCC and its role in the pathogenesis of Familial Hyperkalemic Hypertension (FHHt). (A) In the baseline inactive state, WNK4 suppresses NCC trafficking to the plasma membrane, holding the cotransporter in an intracellular storage pool. The kinase active form of WNK1 can reverse this process. The Kelch-like 3/Cullin-3 (KLHL3/CUL3) E3 ubiquitin ligase complex constitutively degrades the WNKs. (B) FHHt-associated mutations in WNK4 reduce binding to KLHL3, increasing WNK4 abundance and triggering NCC activation through the WNK effector kinases SPAK and OSR1. Additionally, FHHt-causing mutations in WNK4 reduce its inhibitory effect on NCC traffic (represented by the hatched bar-headed line), which releases NCC from its intracellular compartment, increasing its trafficking to the cell surface. Thus, FHHt mutations in WNK4 convert it into an NCC stimulator. (C) WNK1 gene mutations increase kinase-active WNK1 expression, which overcomes constitutive degradation by KLHL3/CUL3. Because kinase active WNK1 can inhibit wild-type WNK4 and activate SPAK/OSR1, increased WNK1 expression stimulates NCC surface delivery and phosphorylation. (D) Mutations in KLHL3 either reduce binding of KLHL3/CUL3 to WNK1 and WNK4 or disconnect CUL3 from KLHL3; in either case, the CUL3 E3 ligase is unable to mark WNK signaling complexes for degradation. Increased WNK1 and WNK4 abundance stimulates NCC trafficking to the surface and triggers NCC phosphorylation. FHHt-causing mutations in CUL3 also likely reduce its activity to WNKs, although the mechanism by which this occurs remains unknown. WNK, With-No-Lysine [amino acid=K] kinase.